



95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 17 April 2012
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 3 - 2012 | https://doi.org/10.3389/fphar.2012.00063
This article is part of the Research Topic Mechanisms of ion channels voltage-dependency View all 17 articles
Voltage-activated K+ (KV) channels are important for shaping action potentials and maintaining resting membrane potential in excitable cells. KV channels contain a central pore-gate domain (PGD) surrounded by four voltage-sensing domains (VSDs). The VSDs will change conformation in response to alterations of the membrane potential thereby inducing the opening of the PGD. Many KV channels are heteromeric protein complexes containing auxiliary β subunits. These β subunits modulate channel expression and activity to increase functional diversity and render tissue specific phenotypes. This review focuses on the KV β subunits that contain transmembrane (TM) segments including the KCNE family and the β subunits of large conductance, Ca2+- and voltage-activated K+ (BK) channels. These TM β subunits affect the voltage-dependent activation of KV α subunits. Experimental and computational studies have described the structural location of these β subunits in the channel complexes and the biophysical effects on VSD activation, PGD opening, and VSD–PGD coupling. These results reveal some common characteristics and mechanistic insights into KV channel modulation by TM β subunits.
Cellular electrical signals organize and control activity in the nervous, muscular, and hormonal tissues. The voltage-activated K+ (KV) channels are a large group of transmembrane (TM) proteins that open in response to membrane depolarization to permit the selective efflux of potassium ions across the membrane. KV channels play an important role in shaping the electric signals of excitable tissues and also contribute to the maintenance of ion homeostasis. Although KV channels vary greatly in their activity, conductance and pharmacology, the basic structure of the channel and the mechanism of voltage-dependent activation are well conserved across the KV family. KV channels share a common topology of six TM α helices (S1–S6) that are organized into two structural domains, the voltage-sensing domain (VSD, S1–S4) and the pore-gate domain (PGD, S5 and S6). As seen in the available KV channel crystal structures, four α-subunits coassemble to form a tetrameric complex with a central pore built from the PGDs of all four subunits and with the four VSDs located peripheral to the central pore (Figure 1).
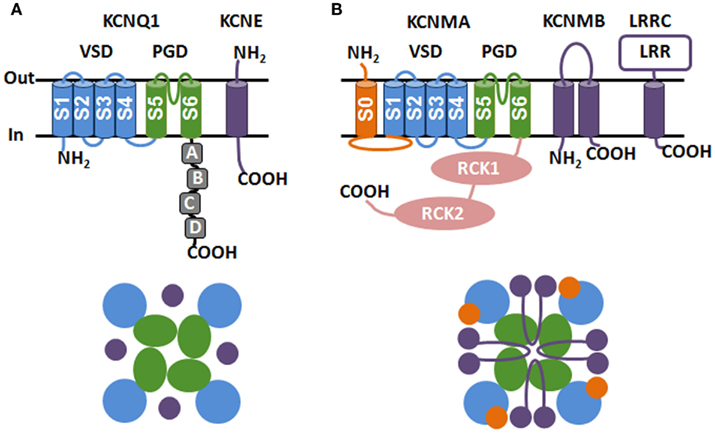
Figure 1. Topology (top) and coassembly (bottom) of KCNQ1 (A) and BK channel (B) α subunits and β subunits. The voltage-sensing domain (VSD, S1–S4), pore-gate domain (PGD, S5 and S6), cytosolic domain (RCK1 and RCK2), S0 segment, and β subunits are colored as blue, green, pink, orange, and purple, respectively. The COOH terminal α-helices (A–D) of KCNQ1 and the leucine-rich-repeat domain (LRR) for LRRC are indicated. The coassembly of the heteromeric channel complex are illustrated as viewed from the extracellular face of the membrane (bottom).
Voltage-dependent activation of KV channels involves three general molecular events (Zagotta et al., 1994). First, depolarization of the membrane potential drives the outward movement of the S4 segment of the VSD that contains conserved basic residues positioned within the electric field (Tombola et al., 2005; Bezanilla, 2008). Second, the conformational change during VSD activation is propagated to the PGD through interactions between the S4/S5 linker and the cytosolic side of S6 (Lu et al., 2001; Tristani-Firouzi et al., 2002; Long et al., 2005), the S4 and S5 helices (Ledwell and Aldrich, 1999; Soler-Llavina et al., 2006; Grabe et al., 2007), and the extracellular side of the S1 and the pore helix (Lee et al., 2009); this event is known as coupling. The third event of KV channel activation is the opening of the PGD to allow ion permeation (Yellen, 1998).
Voltage-activated K+ channel function is modulated by auxiliary β subunits (Pongs and Schwarz, 2010). Of particular interest to this review are the TM β subunits due to their ability to regulate the voltage-dependent activation of KV channels. Here we review the major TM β subunits including the KCNE family and the β subunits of voltage- and Ca2+-activated K+ (BK) channels to summarize our current understanding of the location of β subunits in the channel complex and their impact on the three molecular events of KV channel voltage-dependent activation.
The KCNE family of β subunits consists of five members (KCNE1–5, also known as minK and minK related peptides 1–4) that all contain a single TM domain with an intracellular COOH terminus and an extracellular NH2 terminus (Takumi et al., 1988; Abbott et al., 1999; Piccini et al., 1999; Figure 1A). KCNE subunits coassemble with and modulate KV α subunits resulting in a diverse set of KV channel phenotypes. Not only can one member of the KCNE family regulate multiple different KV channels, but one KV family member can be regulated by different KCNEs. For example, KCNQ1 (KV7.1) can coassemble with all five of the KCNE family peptides; however, the effects of the different KCNEs on KCNQ1 function are very different (Barhanin et al., 1996; Sanguinetti et al., 1996; Schroeder et al., 2000; Tinel et al., 2000; Angelo et al., 2002; Grunnet et al., 2002). These effects range from constitutive, voltage-independent activation with KCNE3 (Schroeder et al., 2000), a shift of voltage-dependent activation to more positive voltages with KCNE1 (Barhanin et al., 1996; Sanguinetti et al., 1996) to inhibition of the ionic current by KCNE4 (Grunnet et al., 2002). Promiscuity in coassembly and diversity of functional regulation allows different tissues to carry unique electrophysiological phenotypes by expressing different KCNE–KV combinations.
Of the many KCNE–KV pairs, the channel formed by coassembly of KCNE1 with the KCNQ1 is the best studied due to the physiological importance of this channel, the dramatic effects of KCNE1 on the function of KCNQ1, and the fact that it was the first identified KCNE–KV partnership. In the heart the heteromeric assembly of KCNQ1 + KCNE1 subunits generates the slow delayed-rectifier current (Barhanin et al., 1996; Sanguinetti et al., 1996), IKs, that is important for termination of the cardiac action potential (Sanguinetti and Jurkiewicz, 1990; Jost et al., 2007). In the inner ear KCNQ1 + KCNE1 channels play a role in the maintenance of endolymph potassium homeostasis (Vetter et al., 1996). Mutations in KCNQ1 (Wang et al., 1996b) or KCNE1 (Splawski et al., 1997) that compromise IKs function can cause long QT syndrome (LQTS) that manifests as prolongation of the QT interval on the surface electrocardiogram and a high risk of ventricular arrhythmias and sudden death. In some cases, the cardiac phenotype can be accompanied by congenital deafness (for review see Hedley et al., 2009). In contrast, IKs gain of function mutations cause premature repolarization and are associated with short QT syndrome (SQTS; Bellocq et al., 2004) and atrial fibrillation (AF; Chen et al., 2003; Hong et al., 2005; Lundby et al., 2007; Das et al., 2009).
The functional importance of KCNE1 effects on KCNQ1 channels is demonstrated by the existence of LQTS mutations in KCNE1 and by the remarkable modulation of KCNQ1 activity by KCNE1 in heterologous expression systems. Exogenous expression of KCNQ1 alone is sufficient to generate voltage-dependent potassium channels; however, coexpression of KCNE1 with KCNQ1 shifts the voltage-dependence of activation toward more depolarized potentials, slows activation and deactivation kinetics (Barhanin et al., 1996; Sanguinetti et al., 1996), removes inactivation (Pusch et al., 1998; Tristani-Firouzi and Sanguinetti, 1998), increases single channel conductance (Sesti and Goldstein, 1998; Yang and Sigworth, 1998), increases PIP2 sensitivity (Li et al., 2011), renders the channel functionally sensitive to PKA phosphorylation (Kurokawa et al., 2003, 2009), and alters the channel pharmacology (Busch et al., 1997; Lerche et al., 2000). These functional changes result in a current that closely recapitulates the properties of the cardiac IKs current that are key to myocyte function in vivo (Silva and Rudy, 2005). In this brief review we will discuss the current evidence regarding the mechanism by which KCNE1 regulates the voltage-dependent activation of KCNQ1 as the best understood example of KCNE regulation of KV channel gating. The study of KCNE3 and KCNE4 regulation of KCNQ1 has also provided similar mechanistic insight; for more details we refer the interested reader to another review to appear in this issue (Wrobel et al., submitted).
Biochemical and cysteine cross-linking assays have established proximity between various locations of the KCNE1 TM segment and KCNQ1 (Xu et al., 2008; Chung et al., 2009; Lvov et al., 2010; Chan et al., 2012; Figure 1A). On the extracellular side of the membrane, proximity between the KCNE1 NH2 terminus (positions 36–43) and the tops of S1, S4, and S6 has been shown through disulfide cross-linking (Wang et al., 2011; Chan et al., 2012). An interaction between the distal KCNE1 COOH terminus and helix C of the KCNQ1 COOH terminus (Figure 1A) has been detected by co-immunoprecipitation (Haitin et al., 2009). However, truncation experiments indicate that removing either the extracellular NH2 terminal domain (Takumi et al., 1991) or the cytosolic distal COOH terminus (Tapper and George, 2000; Chen et al., 2009) of KCNE1 does not prevent the right shift in voltage-dependence and slowing of activation kinetics upon coexpression with KCNQ1, indicating that these structures are not absolutely required for modulation of voltage-dependent activation in exogenous expression systems. Nevertheless, the distal COOH terminus of KCNE1 is required for imparting sensitivity to PKA phosphorylation (Kurokawa et al., 2009) downstream of β adrenergic stimulation, which is vital for the ability of IKs to regulate heart rhythm in vivo (Marx et al., 2002; Kurokawa et al., 2003; Volders et al., 2003). In addition, PKA-dependent phosphorylation can modify voltage-dependent activation by shifting the G–V curve toward less depolarized potentials and slowing deactivation kinetics (Kurokawa et al., 2003, 2009; Li et al., 2011).
In contrast, truncation and chimera studies show that the KCNE1 TM segment and proximal COOH terminus are both absolutely required for right shifting the KCNQ1 G–V and slowing of activation kinetics in heterologous expression systems (Tapper and George, 2000). Metal bridging of G55C in KCNE1 to C331 in KCNQ1 (Tapper and George, 2001) and mutant cycle analyses (Strutz-Seebohm et al., 2011) have provided evidence for an interaction between the KCNE1 TM segment and the outer aspect of the PGD. Cysteine cross-linking indicates that the proximal KCNE1 COOH terminus is in interaction with the KCNQ1 S4/S5 linker and S6 gate, key structures of the gating machinery (Lvov et al., 2010). An early study showed that mutation of the KCNE1 proximal COOH terminus strongly affects the activity of channels consisting of exogenously expressed KCNE1 and the KCNQ1 homolog that is endogenously expressed in the Xenopus oocyte (Takumi et al., 1991). Of note, the proximal COOH terminus is among the most highly conserved regions across the KCNE family, although this region may be of lower importance for the regulation of KCNQ1 activation by other KCNE family members (Melman et al., 2001, 2002; Gage and Kobertz, 2004).
These experimental results have been used to build various models of the KCNQ1 + KCNE1 channel complex using the atomic coordinates of KV1.2 as a template for KCNQ1 (Kang et al., 2008; Xu et al., 2008; Chung et al., 2009; Strutz-Seebohm et al., 2011; Van Horn et al., 2011; Chan et al., 2012). These models consistently locate the KCNE1 TM segment in a cleft that is formed between two VSDs and adjacent to the outer face of the PGD (Figure 1A). From this position, KCNE1 participates in a broad set of interactions with KCNQ1 that span the entire length of the KCNE1 peptide. This interface includes interactions with both the VSD and the PGD, and contacts are made with as many as three KCNQ1 subunits of the tetramer. These structural models do not identify the likely mechanism for how KCNE1 modulates the voltage-dependent activation of KCNQ1 as the interactions with the VSD, PGD, and S4/S5 linker suggest that regulation of VSD movement, PGD opening, and electromechanical coupling are all plausible. These results are consistent with the biophysical experiments as reviewed below.
The biophysical effects of KCNE1 on the KCNQ1 VSD have been explored in several studies. First the state-dependent accessibility of a cysteine engineered into the NH2 terminal part of S4 (positions 226–230) has been probed with MTS reagents to track S4 movement in response to membrane depolarization (Nakajo and Kubo, 2007; Rocheleau and Kobertz, 2008). In these studies, coexpression of KCNE1 with KCNQ1 slowed the rate of S4 cysteine modification. However, this result alone does not conclusively indicate that KCNE1 slows the movement of the KCNQ1 VSD since the kinetics of chemical modification are dependent on the solvent accessibility and chemical environment of the engineered cysteine (Rocheleau and Kobertz, 2008). In our hands, coexpression of KCNE1 with KCNQ1 exposes a cysteine engineered into the KCNQ1 VSD (E160C in S2) to extracellular MTS reagents (Wu et al., 2010a) indicating that KCNE1 can change either the solvent penetration into the VSD or the electrostatic environment within the VSD. Tryptophan and alanine scanning of the S4 segment with and without KCNE1 shows that KCNE1 changes the tolerance to mutation at positions within the VSD (Shamgar et al., 2008; Wu et al., 2010b) suggesting an alteration in the protein packing between S4 and the rest of the VSD or the PGD. These effects on protein packing were most dramatic for mutations of the intracellular half of S4 (Wu et al., 2010b). Changes in the solvent penetration or protein packing within the voltage sensor will change the profile of the electric field surrounding the S4 gating charges and thereby directly alter voltage sensation within the VSD. Recently, the voltage-clamp fluorometry (VCF) technique has been used to directly track VSD movement revealing that the voltage-dependence of VSD activation extends to far more hyperpolarized potentials in the presence of KCNE1 relative to KCNQ1 alone (Osteen et al., 2010). Together these results indicate that KCNE1 changes the structure and movements of the VSD. However, these changes do not offer a clear mechanism for the slow current kinetics and right shifted G–V relationship observed upon coexpression of KCNE1 with KCNQ1. As shown by MTS modification and VCF experiments, the kinetics of VSD activation are much faster than the current activation in the presence of KCNE1 (Rocheleau and Kobertz, 2008; Osteen et al., 2010), and the voltage-dependence of VSD movements is shifted by KCNE1 in the opposite direction (i.e., left) relative to the shift in the G–V relationship (Osteen et al., 2010).
Study of either wild-type or mutant KCNE1 showed that KCNE1 alters single channel conductance (Sesti and Goldstein, 1998; Yang and Sigworth, 1998), ion selectivity (Goldstein and Miller, 1991), and channel block (Goldstein and Miller, 1991; Wang et al., 1996a; Tai and Goldstein, 1998) suggesting that KCNE1 affects the structure of the KCNQ1 channel pore. Transplantation of the KCNQ1 PGD and cytosolic COOH terminus into KV1.4 yielded a chimeric channel that coassembles with and is regulated by KCNE1 (Melman et al., 2004). Similarly, a recent study shows that KCNE1 shifts the G–V and activation kinetics of human and invertebrate KCNQ1 homologs differently, and chimeric channels between the two show that parts of the human KCNQ1 PGD (especially the S5–S6 pore loop) are required to endow the chimeric channel with the human phenotype of KCNE1 regulation (Nakajo et al., 2011). An interaction between KCNE1 and the PGD could modulate steady-state voltage-dependence by changing the energetic gap between the closed and open state of the PGD; also, this interaction could change activation kinetics by changing the energy of the transition between the open and closed state.
As mentioned previously, disulfide cross-linking establishes an interaction between the KCNE1 proximal COOH terminus (residues 70–81) and the KCNQ1 S4/S5 linker (residues 251–257) as well as the S6 gate (residues 342–370; Lvov et al., 2010). The protein contacts between the S4/S5 linker and S6 gate are probably the major structural pathway for coupling VSD activation to PGD opening in KCNQ1, similar to electromechanical coupling in other KV channels (see Introduction). Central to the coupling interface in the KV1.2 crystal structure is a phenylalanine residue in the S6 gate that is highly conserved among KV family channels (Haddad and Blunck, 2011; F481 in shaker, F351 in KCNQ1). Interestingly, an artificial mutation at this position in the KCNQ1 S6 (i.e., F351A; Boulet et al., 2007) as well as two naturally occurring LQTS mutations (R243C and W248R; Franqueza et al., 1999) and several artificial mutations (Labro et al., 2011) in the KCNQ1 S4/S5 linker all result in a right shifted G–V relationship and slow gating kinetics when the mutant α subunits were expressed alone. Thus, perturbing the internal coupling interface in KCNQ1 can mimic the phenotype of KCNE1 modulation of KCNQ1. Decoupling of PGD opening from VSD movements by KCNE1 is shown by the recent KCNQ1 VCF data as KCNE1 coexpression shifts the F–V (VSD activation) toward more hyperpolarized potentials while the G–V (PGD opening) is shifted to more depolarized potentials (Osteen et al., 2010).
A second interaction interface for coupling between the KV VSD and the PGD has been identified between S1 and the pore loop (see Introduction). Interestingly, a cluster of gain of function KCNQ1 mutation that are associated with AF (S140G, V141M, Q147R) are located in this part of S1. Furthermore, these mutations have little impact on the function of homomeric KCNQ1 channels, but cause defective deactivation in the presence of KCNE1 (Chen et al., 2003; Hong et al., 2005; Lundby et al., 2007; Chan et al., 2012). These results indicate that KCNE1 may affect the coupling of the VSD and the PGD at both the intracellular and extracellular face of the membrane.
BK channels have a unitary conductance of ∼100–300 pS (Marty, 1981). The opening of BK channels repolarizes the membrane and shuts down the voltage-dependent Ca2+ channels, thereby reducing Ca2+ influx. Through this negative feedback mechanism, BK channels regulate membrane excitability and intracellular Ca2+ signaling (Lancaster and Nicoll, 1987; Storm, 1987; Brayden and Nelson, 1992). BK channels are composed of pore-forming α subunits (Slo1) and accessory β subunits. The Slo1 subunit contains three structural domains, which are the VSD, the cytosolic domain (CTD) and PGD. In BK channels the opening of the PGD is controlled by voltage-sensing in the VSD, as in other KV channels, and by Ca2+ binding in the CTD. In addition to the canonical VSD (S1–S4) of KV channels, Slo1 contains a unique TM segment (S0; Wallner et al., 1996) which affects VSD activation (Koval et al., 2007; Figure 1B). A prominent feature of BK channel activation is the allosteric coupling between the VSD and the activation gate, i.e., the PGD can open when the VSDs are either in the resting or activated state but the activation of the VSDs promotes channel opening (Horrigan et al., 1999).
Four types of 2TM β subunit (β1–β4, encoded by the genes KCNMB1–4; Knaus et al., 1994; Wallner et al., 1999; Xia et al., 1999, 2000; Behrens et al., 2000; Brenner et al., 2000; Meera, 2000) and one 1TM β subunit (leucine-rich-repeat-containing protein 26, LRRC26; Yan and Aldrich, 2010) have been identified to date. Since the BK channel α subunit is encoded by a single gene (slo1), the tissue specific expression and differential modulation among these β subunits provide a major mechanism for generating a diversity of BK channel phenotypes in different tissues. The effects of β subunits on BK channel function are multifaceted, including altering voltage-dependent activation (Cox and Aldrich, 2000; Wang et al., 2006), changing Ca2+ sensitivity (McManus et al., 1995; Nimigean and Magleby, 1999; Wallner et al., 1999; Xia et al., 1999; Brenner et al., 2000), conferring inactivation (Wallner et al., 1999; Xia et al., 1999, 2000; Brenner et al., 2000), and endowing sensitivity to extracellular ligands (Valverde et al., 1999). Here we focus on the structural and functional interactions between different β subunits and Slo1 to elucidate how BK channel β subunits regulate voltage-dependent activation. Readers may refer to excellent reviews elsewhere for other perspectives of BK β subunits (Orio et al., 2002; Torres et al., 2007; Pongs and Schwarz, 2010; Wu and Marx, 2010).
Among the four 2TM β subunits, β1, β2, and β3 share higher sequence homology (53% similarity between β2 and β1; 37% similarity between β3 and β1), whereas β4 is less conserved (less than 20% similarity with β1; Wallner et al., 1999; Brenner et al., 2000; Xia et al., 2000). However, all four β subunits adopt a similar membrane topology that includes two TM segments (TM1 and TM2), a large extracellular loop (116–128 amino acid residues) and short cytosolic NH2 and COOH termini (Knaus et al., 1994; Xia et al., 1999; Brenner et al., 2000; Orio et al., 2002; Figure 1B).
Different Slo1 orthologs respond to β subunits modulation differently. For instance, the β1 and β2 subunits alter the voltage- and Ca2+-dependent activation of mammalian Slo1 (human hSlo1 and mouse mSlo1) channels more prominently than drosophila Slo1 (dSlo1; Wallner et al., 1996; Lee et al., 2010). Studies of chimerical channels of mammalian Slo1 and dSlo1 revealed that the S0 TM segment is important for the functional effects of β subunits (Wallner et al., 1996; Morrow et al., 2006; Lee et al., 2010). By engineering disulfide linkages, Marx and colleagues model S0 outside of the VSD adjacent to the S3 and S4 helices, while the two TM segments of the β subunits (TM1 and TM2) are packed close to each other at the mouth of the cleft between VSDs of two adjacent Slo1 subunits (Liu et al., 2008a, 2010; Figure 1B). Within this cleft, TM1 is close to S1 of one VSD and TM2 close to S0 of the adjacent VSD (Liu et al., 2008b, 2010; Wu et al., 2009; Zakharov et al., 2009). This model gives rise to the possibility of direct interactions between β subunits and the Slo1 VSD within the membrane. Interestingly, although experimental data suggest that the TM segments of β1, β2, and β4 subunits similarly localize with the Slo1 VSD, the functional effects of these β subunits on the voltage-dependent activation of BK channels differ (see below).
The extracellular loop of β subunits has been shown to alter BK channel block by charybdotoxin (CTX) and iberiotoxin (IbTX; Hanner et al., 1998; Meera, 2000; Chen et al., 2008). These peptide toxins block the channel pore by binding to the extracellular vestibule. The extracellular loop of β subunits interacts with these toxins to either enhance or reduce channel block, suggesting that the extracellular loop may extend from the TM domain at the channel periphery into the external vestibule and alter the entrance of the channel pore (Figure 1B). This picture was supported by another study regarding the rectification mechanism of BK channels in the presence of β3, in which the authors suggested that the extracellular loop of the β3 subunit associates with the Slo1 subunit near the axis of the permeation pathway to form gates blocking ion permeation (Zeng et al., 2003). The extracellular loop of the β1 subunit affects voltage-dependent activation. A recently published work (Gruslova et al., 2012) identified a functional domain in the extracellular loop of β1, consisting of residues Y74, S104, Y105, and I106, that alters voltage-dependent activation, possibly by promoting VSD activation and reducing the intrinsic open probability of the PGD. The extracellular loop connects the channel periphery to the pore so that it may modulate voltage-dependent activation by providing an additional linkage between the VSD and PGD or through direct interactions with these Slo1 structural domains.
The location of the cytosolic termini of β subunits relative to the Slo1 subunit is not clear except that the NH2 terminus of β2 subunits is known to contain an inactivating domain that blocks the entrance to the intracellular pore with a “ball-and-chain” mechanism indicating that the NH2 can reach the internal vestibule (Wallner et al., 1999; Xia et al., 1999, 2003; Bentrop et al., 2001; Zhang et al., 2006, 2009; Li et al., 2007). Nevertheless, deletion mutations of the cytosolic NH2 and COOH termini eliminate the major effects of the β1 subunit on voltage-dependent activation (Wang and Brenner, 2006), indicating that the termini play an important role in Slo1-β subunits interactions. Consistently, a study of the chimeras between the β1 and β2 subunits showed that switching the cytosolic termini between the two β subunits switched the phenotypes of modulation of voltage-dependence and kinetics of activation (Orio et al., 2006).
Among four types of 2TM β subunits, the β1 subunit has the largest effect on voltage-dependent activation. Recordings of macroscopic and single channel currents in the absence of Ca2+ show that the β1 subunit shifts the G–V or Po–V relationship to more positive voltages (∼+20 mV) and decreases the steepness of these curves, indicating that the β1 subunit reduces voltage-sensitivity of the channel (McManus et al., 1995; Cox and Aldrich, 2000; Nimigean, 2000; Orio and Latorre, 2005). The β1 subunit may alter all three general steps of voltage-dependent activation based on the following evidence. First, gating current recordings show that the β1 subunit shifts the voltage-dependence of gating charge movements (Q–V curve) to more negative voltages in the absence of Ca2+ (Bao and Cox, 2005), indicating an impact of the β1 subunit on VSD activation. Second, at negative voltages, where the VSDs are at the resting state, the open probability of the channel is reduced in the presence of the β1 subunit, indicating that the intrinsic opening of the PGD is affected (Orio and Latorre, 2005; Wang and Brenner, 2006). Finally, evidence for the β1 subunit modulating the coupling between the VSD and the activation gate comes from fitting the above data to the HCA model (Horrigan et al., 1999) that describes the allosteric mechanism of voltage-dependent activation of BK channels. In the HCA model the VSDs activate when the channel is either open or closed, but at the open state VSD activation is more likely at any given voltage (the resting-activation equilibrium increases D fold, D is called the allosteric factor describing coupling between VSD activation and PGD opening). Studies from various laboratories show that the β1 subunit alters the allosteric factor D (Cox and Aldrich, 2000; Bao and Cox, 2005; Orio and Latorre, 2005; Wang and Brenner, 2006), indicating changes in the coupling between VSD activation and PGD opening.
The modulation of voltage-dependant activation may be a major mechanism underlying another predominant effect of the β1 subunit: increasing Ca2+ sensitivity, i.e., a Ca2+ concentration change from 0 to 100 μM elicits a larger shift of the G–V relation toward negative voltages in the presence of the β1 subunit (McManus et al., 1995). In studies by a number of laboratories, the effects of the β1 subunit were fitted to the HA model that describes allosteric voltage- and Ca2+-dependent activation of BK channels (Horrigan and Aldrich, 2002). These model fittings show that changes of VSD activation and the intrinsic probability of PGD opening account for a large portion of increased Ca2+ sensitivity observed in experiments, while the changes in Ca2+ binding and its coupling to the activation gate are relatively minor (Orio and Latorre, 2005; Wang and Brenner, 2006). Such a correlation between altered VSD activation and increased Ca2+ sensitivity may not be a coincidence. Mutations in the Slo1 VSD that alter VSD activation (Ma et al., 2006) have been shown to reduce the effect of the β1 subunit on Ca2+ sensitivity; interestingly, the degree to which a mutation alters voltage-sensor activation is inversely correlated with the magnitude of the β1 subunit’s effects on Ca2+ sensitivity of the mutant channel (Yang et al., 2008). These results suggest that the β1 subunit increases Ca2+ sensitivity of BK channels by altering VSD activation. However, the molecular mechanism of how changes in VSD activation alter Ca2+ sensitivity is not clear.
Unlike the β1 subunit, the β2 subunit effects on voltage-dependent activation of BK channels are minimal (Orio and Latorre, 2005). While both β subunits enhance Ca2+ sensitivity, β2 does not shift the G–V curve of BK channels in the absence of Ca2+ (Orio and Latorre, 2005; Lee et al., 2010). Mutations in the Slo1 VSD that alter VSD activation and reduce the β1 subunit modulation of Ca2+ sensitivity have little impact on the β2 subunit modulation of Ca2+ sensitivity (Yang et al., 2008). On the other hand, the NH2 terminus of the CTD (the AC region) and the peptide linker between S6 and the CTD (the C-linker) are important for the modulation of Ca2+ sensitivity by β2 but not β1 (Lee et al., 2010). These results suggest that the β2 subunit targets the CTD, not the VSD, to modulate BK channel Ca2+ sensitivity. Nevertheless, using VCF, Savalli et al. (2007) showed that the β2 subunit shifts the F–V relationship toward hyperpolarized potentials when the S3–S4 linker of the Slo1 was labeled with thiol-reactive fluorescent dyes to track VSD activation. This result suggests that association with β2 subunits alters voltage-sensor activation. A further study comparing the gating current generated in the absence and in the presence of the β2 subunit may shed additional light on whether the β2 subunit alters voltage-dependent activation.
The β4 subunit reduces the intrinsic open probability of the PGD in the absence of Ca2+ binding as well as voltage-sensor activation (Wang et al., 2006), which reduces channel opening and shifts the G–V relation to more positive voltages at [Ca2+] of < ∼6 μM (Brenner et al., 2000; Wang et al., 2006; Lee and Cui, 2009). However, at [Ca2+] > ∼10 μM, the β4 subunit shifts the G–V relation to less positive voltages due to a shift of voltage-sensor activation that compensates for the reduced intrinsic opening of the PGD (Wang et al., 2006). Thus, the β4 subunit seems to modify voltage-dependent activation similarly as the β1 subunit.
Leucine-rich-repeat-containing protein 26 (Yan and Aldrich, 2010) is a newly identified BK channel β subunit; the protein sequence and structure of which are unrelated to the 2TM BK β subunits. LRRC proteins constitute a large protein family. LRRC26 belongs to an extracellular leucine-rich-repeat-only (Elron) cluster (Dolan et al., 2007) that consists of members a single TM segment, an extracellular LRR motif, and a short cytosolic COOH tail containing a stretch of acidic residues (Figure 1B). LRRC proteins have recently begun to gain appreciation as β subunits of KV channels. LRRC52, another member of Elron, is enriched in testis and a β subunit of Slo3 (Yang et al., 2011). Slo3 is activated by voltage and H+ and is the α subunit of Ksper, the voltage and acid activated K+ channel in sperm (Schreiber et al., 1998; Navarro et al., 2007). LRRC52 association shifts the Slo3 G–V to negative voltages that match with the G–V relation of native Ksper in vivo (Yang et al., 2011). Amphoterin-induced gene and ORF (AMIGO) is a LRRC protein belonging to a large LRRC-IG/FN3 protein cluster (Kuja-Panula, 2003). AMIGO has a single TM domain, an extracellular LRR motif and an extracellular immunoglobulin motif. AMIGO associates with and also left shifts the G–V relation of the KV2.1 channel (Peltola et al., 2011).
The leftward shift of the G–V relationship caused by LRRC26 is dramatic, around −140 mV, which makes BK channels activate at negative voltages without rises in Ca2+ concentration. The effect of LRRC26 on channel activation is independent of Ca2+ since mutations of both Ca2+ binding sites of BK channels (Ca2+ bowl deleted and D362A/D367A) have no effect on LRRC26 modulation. Fitting the HCA model showed that LRRC26 modulates BK channel activation mainly through a large enhancement (∼20-fold) of the allosteric factor D, suggesting that LRRC26 affects coupling between the VSD and PGD. The location of LRRC26 relative to Slo1 is not known. However, coexpression of Slo1 with both LRRC26 and the 2TM β1 subunit results in channels with the phenotype of Slo1 + β1, suggesting that the β1 subunit may compete with LRRC26 for a similar association site and prevent LRRC26 from binding. Functional analyses of LRRC26 deletion mutants indicate that except for the fifth leucine-rich-repeat (LRR5) motif and the cytosolic polyPD motif that contains multiple proline and aspartate repeats (residues 304–316), deletion of any region abolishes the function of LRRC26, while only the putative TM segment is necessary for association with Slo1 (Yan and Aldrich, 2010). These results further suggest that the TM segment of LRRC26 may be localized in the cleft between VSDs of the adjacent Slo1 subunits, similar to the 2TM BK channel β subunits and KCNE peptides.
The studies of TM β subunits’ regulation of KV channels reveal common properties. First, the inter VSD cleft provides a docking site for the TM segment(s) offering the starting point for regulation of voltage-dependent activation. This conserved structural feature may be the basis for the promiscuity of some of the TM β subunits. Second, all three domains of the β subunits (extracellular, TM, intracellular) participate in the interactions with the KV α subunit that modulate voltage-dependent activation; although, the specific interactions governing phenotypical changes are not entirely clear. Third, consistent with the structural understanding, β subunit interaction may affect all three general events of voltage-dependent activation; i.e., VSD activation, coupling, and PGD opening. All these results suggest that there is no single deciding interaction, or even a simple additive sum of individual interactions, that is responsible for the phenotype changes that are observed experimentally. Rather, the cooperation among multiple individual interactions may synergistically bring about the unique structure and function of the KV α+β complex.
Mark A. Zaydman wrote the part on KCNEs; Xiaohui Sun wrote the part on BK channel β subunits; Jianmin Cui, Xiaohui Sun, and Mark A. Zaydman integrated the whole paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Yoram Rudy for reading and commenting on the KCNEs part of the manuscript. This work was supported by National Institutes of Health Grants R01-HL70393 and R01-NS060706 (Jianmin Cui). Jianmin Cui is the Professor of Biomedical Engineering on the Spencer T. Olin Endowment.
AF, atrial fibrillation; CTD, cytosolic domain; F–V, fluorescence–voltage relationship; G–V, conductance–voltage relationship; KV channels, voltage-activated K+ channels; LQTS, long QT syndrome; PGD, pore-gate domain; Po–V, open probability–voltage relationship; SQTS, short QT syndrome; TM, transmembrane; VCF, voltage-clamp fluorometry; VSD, voltage-sensing domain.
Abbott, G. W., Sesti, F., Splawski, I., Buck, M. E., Lehmann, M. H., Timothy, K. W., Keating, M. T., and Goldstein, S. A. (1999). MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97, 175–187.
Angelo, K., Jespersen, T., Grunnet, M., Nielsen, M. S., Klaerke, D. A., and Olesen, S.-P. (2002). KCNE5 induces time- and voltage-dependent modulation of the KCNQ1 current. Biophys. J. 83, 1997–2006.
Bao, L., and Cox, D. H. (2005). Gating and ionic currents reveal how the BKCa channel’s Ca2+ sensitivity is enhanced by its beta1 subunit. J. Gen. Physiol. 126, 393–412.
Barhanin, J., Lesage, F., Guillemare, E., Fink, M., Lazdunski, M., and Romey, G. (1996). K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384, 78–80.
Behrens, R., Nolting, A., Reimann, F., Schwarz, M., Waldschutz, R., and Pongs, O. (2000). hKCNMB3 and hKCNMB4, cloning and characterization of two members of the large-conductance calcium-activated potassium channel beta subunit family. FEBS Lett. 474, 99–106.
Bellocq, C., Van Ginneken, A. C. G., Bezzina, C. R., Alders, M., Escande, D., Mannens, M. M. A. M., Baro, I., and Wilde, A. A. M. (2004). Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 109, 2394–2397.
Bentrop, D., Beyermann, M., Wissmann, R., and Fakler, B. (2001). NMR structure of the “ball-and-chain” domain of KCNMB2, the beta 2-subunit of large conductance Ca2+- and voltage-activated potassium channels. J. Biol. Chem. 276, 42116–42121.
Boulet, I. R., Labro, A. J., Raes, A. L., and Snyders, D. J. (2007). Role of the S6 C-terminus in KCNQ1 channel gating. J. Physiol. 585, 325–337.
Brayden, J. E., and Nelson, M. T. (1992). Regulation of arterial tone by activation of calcium-dependent potassium channels. Science 256, 532–535.
Brenner, R., Jegla, T. J., Wickenden, A., Liu, Y., and Aldrich, R. W. (2000). Cloning and functional characterization of novel large conductance calcium-activated potassium channel beta subunits, hKCNMB3 and hKCNMB4. J. Biol. Chem. 275, 6453–6461.
Busch, A. E., Busch, G. L., Ford, E., Suessbrich, H., Lang, H. J., Greger, R., Kunzelmann, K., Attali, B., and Stühmer, W. (1997). The role of the IsK protein in the specific pharmacological properties of the IKs channel complex. Br. J. Pharmacol. 122, 187–189.
Chan, P. J., Osteen, J. D., Xiong, D., Bohnen, M. S., Doshi, D., Sampson, K. J., Marx, S. O., Karlin, A., and Kass, R. S. (2012). Characterization of KCNQ1 atrial fibrillation mutations reveals distinct dependence on KCNE1. J. Gen. Physiol. 139, 135–144.
Chen, J., Zheng, R., Melman, Y. F., and McDonald, T. V. (2009). Functional interactions between KCNE1 C-terminus and the KCNQ1 channel. PLoS ONE 4, e5143. doi:10.1371/journal.pone.0005143
Chen, M., Gan, G., Wu, Y., Wang, L., and Ding, J. (2008). Lysine-rich extracellular rings formed by hβ2 subunits confer the outward rectification of BK channels. PLoS ONE 3, e2114. doi:10.1371/journal.pone.0002114
Chen, Y.-H., Xu, S.-J., Bendahhou, S., Wang, X.-L., Wang, Y., Xu, W.-Y., Jin, H.-W., Sun, H., Su, X.-Y., Zhuang, Q.-N., Yang, Y.-Q., Li, Y.-B., Liu, Y., Xu, H.-J., Li, X.-F., Ma, N., Mou, C.-P., Chen, Z., Barhanin, J., and Huang, W. (2003). KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 299, 251–254.
Chung, D. Y., Chan, P. J., Bankston, J. R., Yang, L., Liu, G., Marx, S. O., Karlin, A., and Kass, R. S. (2009). Location of KCNE1 relative to KCNQ1 in the I(KS) potassium channel by disulfide cross-linking of substituted cysteines. Proc. Natl. Acad. Sci. U.S.A. 106, 743–748.
Cox, D. H., and Aldrich, R. W. (2000). Role of the beta1 subunit in large-conductance Ca(2+)-activated K (+) channel gating energetics. Mechanisms of enhanced Ca(2+) sensitivity. J. Gen. Physiol. 116, 411–432.
Das, S., Makino, S., Melman, Y. F., Shea, M. A., Goyal, S. B., Rosenzweig, A., Macrae, C. A., and Ellinor, P. T. (2009). Mutation in the S3 segment of KCNQ1 results in familial lone atrial fibrillation. Heart rhythm 6, 1146–1153.
Dolan, J., Walshe, K., Alsbury, S., Hokamp, K., O’Keeffe, S., Okafuji, T., Miller, S. F. C., Tear, G., and Mitchell, K. J. (2007). The extracellular leucine-rich repeat superfamily; a comparative survey and analysis of evolutionary relationships and expression patterns. BMC Genomics 8, 320. doi:10.1186/1471-2164-10-230
Franqueza, L., Lin, M., Shen, J., Splawski, I., Keating, M. T., and Sanguinetti, M. C. (1999). Long QT syndrome-associated mutations in the S4–S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J. Biol. Chem. 274, 21063–21070.
Gage, S. D., and Kobertz, W. R. (2004). KCNE3 truncation mutants reveal a bipartite modulation of KCNQ1 K+ channels. J. Gen. Physiol. 124, 759–771.
Goldstein, S. A., and Miller, C. (1991). Site-specific mutations in a minimal voltage-dependent K+ channel alter ion selectivity and open-channel block. Neuron 7, 403–408.
Grabe, M., Lai, H. C., Jain, M., Jan, Y. N., and Jan, L. Y. (2007). Structure prediction for the down state of a potassium channel voltage sensor. Nature 445, 550–553.
Grunnet, M., Jespersen, T., Rasmussen, H. B., Ljungstrøm, T., Jorgensen, N. K., Olesen, S.-P., and Klaerke, D. A. (2002). KCNE4 is an inhibitory subunit to the KCNQ1 channel. J. Physiol. 542, 119–130.
Gruslova, A., Semenov, I., and Wang, B. (2012). An extracellular domain of the accessory beta1 subunit is required for modulating BK channel voltage sensor and gate. J. Gen. Physiol. 139, 57–67.
Haddad, G. A., and Blunck, R. (2011). Mode shift of the voltage sensors in Shaker K+ channels is caused by energetic coupling to the pore domain. J. Gen. Physiol. 137, 455–472.
Haitin, Y., Wiener, R., Shaham, D., Peretz, A., Cohen, E. B.-T., Shamgar, L., Pongs, O., Hirsch, J. A., and Attali, B. (2009). Intracellular domains interactions and gated motions of I(KS) potassium channel subunits. EMBO J. 28, 1994–2005.
Hanner, M., Vianna-Jorge, R., Kamassah, A., Schmalhofer, W. A., Knaus, H. G., Kaczorowski, G. J., and Garcia, M. L. (1998). The beta subunit of the high conductance calcium-activated potassium channel. Identification of residues involved in charybdotoxin binding. J. Biol. Chem. 273, 16289–16296.
Hedley, P. L., Jørgensen, P., Schlamowitz, S., Wangari, R., Moolman-Smook, J., Brink, P. A., Kanters, J. K., Corfield, V. A., and Christiansen, M. (2009). The genetic basis of long QT and short QT syndromes: a mutation update. Hum. Mutat. 30, 1486–1511.
Hong, K., Piper, D. R., Diaz-Valdecantos, A., Brugada, J., Oliva, A., Burashnikov, E., Santos-De-Soto, J., Grueso-Montero, J., Diaz-Enfante, E., Brugada, P., Sachse, F., Sanguinetti, M. C., and Brugada, R. (2005). De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc. Res. 68, 433–440.
Horrigan, F. T., and Aldrich, R. W. (2002). Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 120, 267–305.
Horrigan, F. T., Cui, J., and Aldrich, R. W. (1999). Allosteric voltage gating of potassium channels I. Mslo ionic currents in the absence of Ca(2+). J. Gen. Physiol. 114, 277–304.
Jost, N., Papp, J. G., and Varró, A. (2007). Slow delayed rectifier potassium current (IKs) and the repolarization reserve. Ann. Noninvasive Electrocardiol. 12, 64–78.
Kang, C., Tian, C., Sönnichsen, F. D., Smith, J. A., Meiler, J., George, A. L., Vanoye, C. G., Kim, H. J., and Sanders, C. R. (2008). Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry 47, 7999–8006.
Knaus, H. G., Folander, K., Garcia-Calvo, M., Garcia, M. L., Kaczorowski, G. J., Smith, M., and Swanson, R. (1994). Primary sequence and immunological characterization of beta-subunit of high conductance Ca(2+)-activated K+ channel from smooth muscle. J. Biol. Chem. 269, 17274–17278.
Koval, O. M., Fan, Y., and Rothberg, B. S. (2007). A role for the S0 transmembrane segment in voltage-dependent gating of BK channels. J. Gen. Physiol. 129, 209–220.
Kuja-Panula, J. (2003). AMIGO, a transmembrane protein implicated in axon tract development, defines a novel protein family with leucine-rich repeats. J. Cell Biol. 160, 963–973.
Kurokawa, J., Bankston, J. R., Kaihara, A., Chen, L., Furukawa, T., and Kass, R. S. (2009). KCNE variants reveal a critical role of the beta subunit carboxyl terminus in PKA-dependent regulation of the IKs potassium channel. Channels (Austin) 3, 16–24.
Kurokawa, J., Chen, L., and Kass, R. S. (2003). Requirement of subunit expression for cAMP-mediated regulation of a heart potassium channel. Proc. Natl. Acad. Sci. U.S.A. 100, 2122–2127.
Labro, A. J., Boulet, I. R., Choveau, F. S., Mayeur, E., Bruyns, T., Loussouarn, G., Raes, A. L., and Snyders, D. J. (2011). The S4–S5 linker of KCNQ1 channels forms a structural scaffold with the S6 segment controlling gate closure. J. Biol. Chem. 286, 717–725.
Lancaster, B., and Nicoll, R. A. (1987). Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J. Physiol. 389, 187–203.
Ledwell, J. L., and Aldrich, R. W. (1999). Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 113, 389–414.
Lee, S.-Y., Banerjee, A., and Mackinnon, R. (2009). Two separate interfaces between the voltage sensor and pore are required for the function of voltage-dependent K+ channels. PLoS Biol. 7, e47. doi:10.1371/journal.pbio.1000047
Lee, U. S., and Cui, J. (2009). {beta} subunit-specific modulations of BK channel function by a mutation associated with epilepsy and dyskinesia. J. Physiol. 587, 1481–1498.
Lee, U. S., Shi, J., and Cui, J. (2010). Modulation of BK channel gating by the ss2 subunit involves both membrane-spanning and cytoplasmic domains of Slo1. J. Neurosci. 30, 16170–16179.
Lerche, C., Seebohm, G., Wagner, C. I., Scherer, C. R., Dehmelt, L., Abitbol, I., Gerlach, U., Brendel, J., Attali, B., and Busch, A. E. (2000). Molecular impact of MinK on the enantiospecific block of I(Ks) by chromanols. Br. J. Pharmacol. 131, 1503–1506.
Li, H., Yao, J., Tong, X., Guo, Z., Wu, Y., Sun, L., Pan, N., Wu, H., Xu, T., and Ding, J. (2007). Interaction sites between the Slo1 pore and the NH2 terminus of the beta2 subunit, probed with a three-residue sensor. J. Biol. Chem. 282, 17720–17728.
Li, Y., Zaydman, M. A., Wu, D., Shi, J., Guan, M., Virgin-Downey, B., and Cui, J. (2011). KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proc. Natl. Acad. Sci. U.S.A. 108, 9095–9100.
Liu, G., Niu, X., Wu, R. S., Chudasama, N., Yao, Y., Jin, X., Weinberg, R., Zakharov, S. I., Motoike, H., Marx, S. O., and Karlin, A. (2010). Location of modulatory beta subunits in BK potassium channels. J. Gen. Physiol. 135, 449–459.
Liu, G., Zakharov, S. I., Yang, L., Deng, S. X., Landry, D. W., Karlin, A., and Marx, S. O. (2008a). Position and role of the BK channel alpha subunit S0 helix inferred from disulfide crosslinking. J. Gen. Physiol. 131, 537–548.
Liu, G., Zakharov, S. I., Yang, L., Wu, R. S., Deng, S. X., Landry, D. W., Karlin, A., and Marx, S. O. (2008b). Locations of the 1 transmembrane helices in the BK potassium channel. Proc. Natl. Acad. Sci. U.S.A. 105, 10727–10732.
Long, S. B., Campbell, E. B., and Mackinnon, R. (2005). Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 309, 903–908.
Lu, Z., Klem, A. M., and Ramu, Y. (2001). Ion conduction pore is conserved among potassium channels. Nature 413, 809–813.
Lundby, A., Ravn, L. S., Svendsen, J. H., Olesen, S.-P., and Schmitt, N. (2007). KCNQ1 mutation Q147R is associated with atrial fibrillation and prolonged QT interval. Heart rhythm 4, 1532–1541.
Lvov, A., Gage, S. D., Berrios, V. M., and Kobertz, W. R. (2010). Identification of a protein–protein interaction between KCNE1 and the activation gate machinery of KCNQ1. J. Gen. Physiol. 135, 607–618.
Ma, Z., Lou, X. J., and Horrigan, F. T. (2006). Role of charged residues in the S1–S4 voltage sensor of BK channels. J. Gen. Physiol. 127, 309–328.
Marty, A. (1981). Ca-dependent K channels with large unitary conductance in chromaffin cell membranes. Nature 291, 497–500.
Marx, S. O., Kurokawa, J., Reiken, S., Motoike, H., D’Armiento, J., Marks, A. R., and Kass, R. S. (2002). Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 295, 496–499.
McManus, O. B., Helms, L. M., Pallanck, L., Ganetzky, B., Swanson, R., and Leonard, R. J. (1995). Functional role of the beta subunit of high conductance calcium-activated potassium channels. Neuron 14, 645–650.
Meera, P. (2000). A neuronal beta subunit (KCNMB4) makes the large conductance, voltage- and Ca2+-activated K+ channel resistant to charybdotoxin and iberiotoxin. Proc. Natl. Acad. Sci. U.S.A. 97, 5562–5567.
Melman, Y. F., Domènech, A., De La Luna, S., and McDonald, T. V. (2001). Structural determinants of KvLQT1 control by the KCNE family of proteins. J. Biol. Chem. 276, 6439–6444.
Melman, Y. F., Krumerman, A., and McDonald, T. V. (2002). A single transmembrane site in the KCNE-encoded proteins controls the specificity of KvLQT1 channel gating. J. Biol. Chem. 277, 25187–25194.
Melman, Y. F., Um, S. Y., Krumerman, A., Kagan, A., and McDonald, T. V. (2004). KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron 42, 927–937.
Morrow, J. P., Zakharov, S. I., Liu, G., Yang, L., Sok, A. J., and Marx, S. O. (2006). Defining the BK channel domains required for beta1-subunit modulation. Proc. Natl. Acad. Sci. U.S.A. 103, 5096–5101.
Nakajo, K., and Kubo, Y. (2007). KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. J. Gen. Physiol. 130, 269–281.
Nakajo, K., Nishino, A., Okamura, Y., and Kubo, Y. (2011). KCNQ1 subdomains involved in KCNE modulation revealed by an invertebrate KCNQ1 orthologue. J. Gen. Physiol. 138, 521–535.
Navarro, B., Kirichok, Y., and Clapham, D. E. (2007). KSper, a pH-sensitive K+ current that controls sperm membrane potential. Proc. Natl. Acad. Sci. U.S.A. 104, 7688–7692.
Nimigean, C. M. (2000). Functional coupling of the beta1 subunit to the large conductance Ca2+-activated K+ channel in the absence of Ca2+: increased Ca2+ sensitivity from a Ca2+-independent mechanism. J. Gen. Physiol. 115, 719–736.
Nimigean, C. M., and Magleby, K. L. (1999). The beta subunit increases the Ca2+ sensitivity of large conductance Ca2+-activated potassium channels by retaining the gating in the bursting states. J. Gen. Physiol. 113, 425–440.
Orio, P., and Latorre, R. (2005). Differential effects of beta 1 and beta 2 subunits on BK channel activity. J. Gen. Physiol. 125, 395–411.
Orio, P., Rojas, P., Ferreira, G., and Latorre, R. (2002). New disguises for an old channel: MaxiK channel beta-subunits. News Physiol. Sci. 17, 156–161.
Orio, P., Torres, Y., Rojas, P., Carvacho, I., Garcia, M. L., Toro, L., Valverde, M. A., and Latorre, R. (2006). Structural determinants for functional coupling between the beta and alpha subunits in the Ca2+-activated K+ (BK) channel. J. Gen. Physiol. 127, 191–204.
Osteen, J. D., Gonzalez, C., Sampson, K. J., Iyer, V., Rebolledo, S., Larsson, H. P., and Kass, R. S. (2010). KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc. Natl. Acad. Sci. U.S.A. 107, 22710–22715.
Peltola, M. A., Kuja-Panula, J., Lauri, S. E., Taira, T., and Rauvala, H. (2011). AMIGO is an auxiliary subunit of the Kv2.1 potassium channel. EMBO Rep. 12, 1293–1299.
Piccini, M., Vitelli, F., Seri, M., Galietta, L. J., Moran, O., Bulfone, A., Banfi, S., Pober, B., and Renieri, A. (1999). KCNE1-like gene is deleted in AMME contiguous gene syndrome: identification and characterization of the human and mouse homologs. Genomics 60, 251–257.
Pongs, O., and Schwarz, J. R. (2010). Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 90, 755–796.
Pusch, M., Magrassi, R., Wollnik, B., and Conti, F. (1998). Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 75, 785–792.
Rocheleau, J. M., and Kobertz, W. R. (2008). KCNE peptides differently affect voltage sensor equilibrium and equilibration rates in KCNQ1 K+ channels. J. Gen. Physiol. 131, 59–68.
Sanguinetti, M. C., Curran, M. E., Zou, A., Shen, J., Spector, P. S., Atkinson, D. L., and Keating, M. T. (1996). Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83.
Sanguinetti, M. C., and Jurkiewicz, N. K. (1990). Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 96, 195–215.
Savalli, N., Kondratiev, A., De Quintana, S. B., Toro, L., and Olcese, R. (2007). Modes of operation of the BKCa channel beta2 subunit. J. Gen. Physiol. 130, 117–131.
Schreiber, M., Wei, A., Yuan, A., Gaut, J., Saito, M., and Salkoff, L. (1998). Slo3, a novel pH-sensitive K+ channel from mammalian spermatocytes. J. Biol. Chem. 273, 3509–3516.
Schroeder, B. C., Waldegger, S., Fehr, S., Bleich, M., Warth, R., Greger, R., and Jentsch, T. J. (2000). A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 403, 196–199.
Sesti, F., and Goldstein, S. A. (1998). Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J. Gen. Physiol. 112, 651–663.
Shamgar, L., Haitin, Y., Yisharel, I., Malka, E., Schottelndreier, H., Peretz, A., Paas, Y., and Attali, B. (2008). KCNE1 constrains the voltage sensor of Kv7.1 K+ channels. PLoS ONE 3, e1943. doi:10.1371/journal.pone.0001943
Silva, J., and Rudy, Y. (2005). Subunit interaction determines IKs participation in cardiac repolarization and repolarization reserve. Circulation 112, 1384–1391.
Soler-Llavina, G. J., Chang, T. H., and Swartz, K. J. (2006). Functional interactions at the interface between voltage-sensing and pore domains in the Shaker K(v) channel. Neuron 52, 623–634.
Splawski, I., Tristani-Firouzi, M., Lehmann, M. H., Sanguinetti, M. C., and Keating, M. T. (1997). Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat. Genet. 17, 338–340.
Storm, J. F. (1987). Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J. Physiol. 385, 733–759.
Strutz-Seebohm, N., Pusch, M., Wolf, S., Stoll, R., Tapken, D., Gerwert, K., Attali, B., and Seebohm, G. (2011). Structural basis of slow activation gating in the cardiac I Ks channel complex. Cell. Physiol. Biochem. 27, 443–452.
Tai, K. K., and Goldstein, S. A. (1998). The conduction pore of a cardiac potassium channel. Nature 391, 605–608.
Takumi, T., Moriyoshi, K., Aramori, I., Ishii, T., Oiki, S., Okada, Y., Ohkubo, H., and Nakanishi, S. (1991). Alteration of channel activities and gating by mutations of slow ISK potassium channel. J. Biol. Chem. 266, 22192–22198.
Takumi, T., Ohkubo, H., and Nakanishi, S. (1988). Cloning of a membrane protein that induces a slow voltage-gated potassium current. Science 242, 1042–1045.
Tapper, A. R., and George, A. L. (2000). MinK subdomains that mediate modulation of and association with KvLQT1. J. Gen. Physiol. 116, 379–390.
Tapper, A. R., and George, A. L. (2001). Location and orientation of minK within the I(Ks) potassium channel complex. J. Biol. Chem. 276, 38249–38254.
Tinel, N., Diochot, S., Borsotto, M., Lazdunski, M., and Barhanin, J. (2000). KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 19, 6326–6330.
Tombola, F., Pathak, M. M., and Isacoff, E. Y. (2005). How far will you go to sense voltage? Neuron 48, 719–725.
Torres, Y. P., Morera, F. J., Carvacho, I., and Latorre, R. (2007). A marriage of convenience: beta-subunits and voltage-dependent K+ channels. J. Biol. Chem. 282, 24485–24489.
Tristani-Firouzi, M., Chen, J., and Sanguinetti, M. C. (2002). Interactions between S4–S5 linker and S6 transmembrane domain modulate gating of HERG K+ channels. J. Biol. Chem. 277, 18994–19000.
Tristani-Firouzi, M., and Sanguinetti, M. C. (1998). Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J. Physiol. 510(Pt 1), 37–45.
Valverde, M. A., Rojas, P., Amigo, J., Cosmelli, D., Orio, P., Bahamonde, M. I., Mann, G. E., Vergara, C., and Latorre, R. (1999). Acute activation of Maxi-K channels (hSlo) by estradiol binding to the beta subunit. Science 285, 1929–1931.
Van Horn, W. D., Vanoye, C. G., and Sanders, C. R. (2011). Working model for the structural basis for KCNE1 modulation of the KCNQ1 potassium channel. Curr. Opin. Struct. Biol. 21, 283–291.
Vetter, D. E., Mann, J. R., Wangemann, P., Liu, J., Mclaughlin, K. J., Lesage, F., Marcus, D. C., Lazdunski, M., Heinemann, S. F., and Barhanin, J. (1996). Inner ear defects induced by null mutation of the isk gene. Neuron 17, 1251–1264.
Volders, P. G. A., Stengl, M., Van Opstal, J. M., Gerlach, U., Spätjens, R. L. H. M. G., Beekman, J. D. M., Sipido, K. R., and Vos, M. A. (2003). Probing the contribution of IKs to canine ventricular repolarization: key role for beta-adrenergic receptor stimulation. Circulation 107, 2753–2760.
Wallner, M., Meera, P., and Toro, L. (1996). Determinant for beta-subunit regulation in high-conductance voltage-activated and Ca(2+)-sensitive K+ channels: an additional transmembrane region at the N terminus. Proc. Natl. Acad. Sci. U.S.A. 93, 14922–14927.
Wallner, M., Meera, P., and Toro, L. (1999). Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: a transmembrane beta-subunit homolog. Proc. Natl. Acad. Sci. U.S.A. 96, 4137–4142.
Wang, B., and Brenner, R. (2006). An S6 mutation in BK channels reveals beta1 subunit effects on intrinsic and voltage-dependent gating. J. Gen. Physiol. 128, 731–744.
Wang, B., Rothberg, B. S., and Brenner, R. (2006). Mechanism of beta4 subunit modulation of BK channels. J. Gen. Physiol. 127, 449–465.
Wang, K. W., Tai, K. K., and Goldstein, S. A. (1996a). MinK residues line a potassium channel pore. Neuron 16, 571–577.
Wang, Q., Curran, M. E., Splawski, I., Burn, T. C., Millholland, J. M., Vanraay, T. J., Shen, J., Timothy, K. W., Vincent, G. M., De Jager, T., Schwartz, P. J., Toubin, J. A., Moss, A. J., Atkinson, D. L., Landes, G. M., Connors, T. D., and Keating, M. T. (1996b). Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 12, 17–23.
Wang, Y. H., Jiang, M., Xu, X. L., Hsu, K.-L., Zhang, M., and Tseng, G.-N. (2011). Gating-related molecular motions in the extracellular domain of the IKs channel: implications for IKs channelopathy. J. Membr. Biol. 239, 137–156.
Wu, D., Delaloye, K., Zaydman, M. A., Nekouzadeh, A., Rudy, Y., and Cui, J. (2010a). State-dependent electrostatic interactions of S4 arginines with E1 in S2 during Kv7.1 activation. J. Gen. Physiol. 135, 595–606.
Wu, D., Pan, H., Delaloye, K., and Cui, J. (2010b). KCNE1 remodels the voltage sensor of Kv7.1 to modulate channel function. Biophys. J. 99, 3599–3608.
Wu, R. S., Chudasama, N., Zakharov, S. I., Doshi, D., Motoike, H., Liu, G., Yao, Y., Niu, X., Deng, S. X., Landry, D. W., Karlin, A., and Marx, S. O. (2009). Location of the beta 4 transmembrane helices in the BK potassium channel. J. Neurosci. 29, 8321–8328.
Wu, R. S., and Marx, S. O. (2010). The BK potassium channel in the vascular smooth muscle and kidney: alpha- and beta-subunits. Kidney Int. 78, 963–974.
Xia, X. M., Ding, J. P., and Lingle, C. J. (1999). Molecular basis for the inactivation of Ca2+- and voltage-dependent BK channels in adrenal chromaffin cells and rat insulinoma tumor cells. J. Neurosci. 19, 5255–5264.
Xia, X. M., Ding, J. P., and Lingle, C. J. (2003). Inactivation of BK channels by the NH2 terminus of the beta2 auxiliary subunit: an essential role of a terminal peptide segment of three hydrophobic residues. J. Gen. Physiol. 121, 125–148.
Xia, X. M., Ding, J. P., Zeng, X. H., Duan, K. L., and Lingle, C. J. (2000). Rectification and rapid activation at low Ca2+ of Ca2+-activated, voltage-dependent BK currents: consequences of rapid inactivation by a novel beta subunit. J. Neurosci. 20, 4890–4903.
Xu, X., Jiang, M., Hsu, K.-L., Zhang, M., and Tseng, G.-N. (2008). KCNQ1 and KCNE1 in the IKs channel complex make state-dependent contacts in their extracellular domains. J. Gen. Physiol. 131, 589–603.
Yan, J., and Aldrich, R. W. (2010). LRRC26 auxiliary protein allows BK channel activation at resting voltage without calcium. Nature 466, 513–516.
Yang, C., Zeng, X.-H., Zhou, Y., Xia, X.-M., and Lingle, C. J. (2011). LRRC52 (leucine-rich-repeat-containing protein 52), a testis-specific auxiliary subunit of the alkalization-activated Slo3 channel. Proc. Natl. Acad. Sci. U.S.A. 108, 19419–19424.
Yang, H., Zhang, G., Shi, J., Lee, U. S., Delaloye, K., and Cui, J. (2008). Subunit-specific effect of the voltage sensor domain on Ca2+ sensitivity of BK channels. Biophys. J. 94, 4678–4687.
Yang, Y., and Sigworth, F. J. (1998). Single-channel properties of IKs potassium channels. J. Gen. Physiol. 112, 665–678.
Zagotta, W. N., Hoshi, T., and Aldrich, R. W. (1994). Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J. Gen. Physiol. 103, 321–362.
Zakharov, S. I., Wu, R. S., Liu, G., Motoike, H., Karlin, A., and Marx, S. O. (2009). “Locations of the Beta2 transmembrane helices in the BK potassium channel,” in 53rd Annual Biophysical Society Meeting, Boston, MA.
Zeng, X. H., Xia, X. M., and Lingle, C. J. (2003). Redox-sensitive extracellular gates formed by auxiliary beta subunits of calcium-activated potassium channels. Nat. Struct. Biol. 10, 448–454.
Zhang, Z., Zeng, X. H., Xia, X. M., and Lingle, C. J. (2009). N-terminal inactivation domains of beta subunits are protected from trypsin digestion by binding within the antechamber of BK channels. J. Gen. Physiol. 133, 263–282.
Keywords: channel, β subunit, KV, KCNQ1, BK, KCNE, KCNMB, LRRC
Citation: Sun X, Zaydman MA and Cui J (2012) Regulation of voltage-activated K+ channel gating by transmembrane β subunits. Front. Pharmacol. 3:63. doi: 10.3389/fphar.2012.00063
Received: 09 February 2012; Accepted: 29 March 2012;
Published online: 17 April 2012.
Edited by:
Gildas Loussouarn, University of Nantes, FranceReviewed by:
Peter Larsson, University of Miami School of Medicine, USACopyright: © 2012 Sun, Zaydman and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jianmin Cui, Department of Biomedical Engineering, Washington University, One Brookings Drive, Saint Louis, MO 63130, USA. e-mail:amN1aUB3dXN0bC5lZHU=
†Xiaohui Sun and Mark A. Zaydman have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.