 Santosh Philips1
Santosh Philips1 James M. Rae2
James M. Rae2 Steffi Oesterreich3 Daniel F. Hayes2 Vered Stearns4 N. Lynn Henry2 Anna M. Storniolo5 David A. Flockhart1
Steffi Oesterreich3 Daniel F. Hayes2 Vered Stearns4 N. Lynn Henry2 Anna M. Storniolo5 David A. Flockhart1 Todd C. Skaar1*
Todd C. Skaar1*- 1 Division of Clinical Pharmacology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, USA
- 2 Department of Internal Medicine, University of Michigan, Ann Arbor, MI, USA
- 3 Department of Pharmacology and Chemical Biology, University of Pittsburgh Cancer Institute, Pittsburgh, PA, USA
- 4 Breast Cancer Program, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University, Baltimore, MD, USA
- 5 Department of Medicine, Indiana University Melvin and Bren Simon Cancer Center, Indianapolis, IN, USA
Whole genome amplification (WGA) technologies can be used to amplify genomic DNA when only small amounts of DNA are available. The Multiple Displacement Amplification Phi polymerase based amplification has been shown to accurately amplify DNA for a variety of genotyping assays; however, it has not been tested for genotyping many of the clinically relevant genes important for pharmacogenetic studies, such as the cytochrome P450 genes, that are typically difficult to genotype due to multiple pseudogenes, copy number variations, and high similarity to other related genes. We evaluated whole genome amplified samples for Taqman™ genotyping of SNPs in a variety of pharmacogenetic genes. In 24 DNA samples from the Coriell human diversity panel, the call rates, and concordance between amplified (∼200-fold amplification) and unamplified samples was 100% for two SNPs in CYP2D6 and one in ESR1. In samples from a breast cancer clinical trial (Trial 1), we compared the genotyping results in samples before and after WGA for three SNPs in CYP2D6, one SNP in CYP2C19, one SNP in CYP19A1, two SNPs in ESR1, and two SNPs in ESR2. The concordance rates were all >97%. Finally, we compared the allele frequencies of 143 SNPs determined in Trial 1 (whole genome amplified DNA) to the allele frequencies determined in unamplified DNA samples from a separate trial (Trial 2) that enrolled a similar population. The call rates and allele frequencies between the two trials were 98 and 99.7%, respectively. We conclude that the whole genome amplified DNA is suitable for Taqman™ genotyping for a wide variety of pharmacogenetically relevant SNPs.
Introduction
Genotyping in pharmacogenetics studies frequently becomes limited due to small DNA quantities. In some studies, DNA yields are low because of the methods of collection. In others, DNA samples are depleted by extensive genotyping and DNA sequencing. Recontacting patients is often not feasible because the most informative patients have often had disease recurrences and died. In addition, some genotyping techniques, including those directed at next generation sequencing, genome wide association studies, and OpenArray™ platforms, require high DNA concentrations that are higher than those obtained from the initial DNA extractions. In some cases, DNA samples can be concentrated to achieve the necessary concentrations; however, sample recovery can be low and concentrating the samples often results in a parallel increase in the concentrations of contaminants.
Whole genome amplification (WGA) protocols are now available that may provide a solution to the deficiencies made inevitable by small yields and low concentrations. WGA using the Multiple Displacement Amplification Phi polymerase was recently shown to accurately amplify DNA samples for genotyping using GWAS chips (Jasmine et al., 2008). Although this study indicated that the MDA worked well for many SNPs across the genome, GWAS chips often have poor coverage of many genes important to pharmacogenetics, such as the cytochrome P450 genes. Consequently, additional validation is required to show the applicability for WGA on samples that will be used for genotyping genes focused on pharmacogenetics. Therefore, we carried out a study to determine the feasibility of WGA for the genotyping of several SNPs in clinically relevant genes.
Methods
DNA Samples
DNA samples used in these studies were obtained from three populations. First, lymphocyte DNA samples from African Americans (n = 7), Caucasians (n = 8), and Asians (n = 9) were obtained directly from the Coriell DNA repository; second, Trial 1 DNA samples from whole blood collected in heparin vacutainers were obtained from a multisite trial carried out by the Consortium on Breast Cancer Pharmacogenomics (COBRA) of patients treated with tamoxifen (Clinicaltrials.gov # NCT00228930); and third, Trial 2 DNA samples from whole blood collected in EDTA vacutainers were obtained from a second multisite COBRA exemestane and letrozole pharmacogenetics (ELPh) clinical trial (Clinicaltrials.gov # NCT00228956). DNA was extracted from whole blood obtained from patients enrolled in Trial 1 and Trial 2 using DNA Blood Maxi extraction kits (Qiagen, Inc., Valencia, CA, USA). All patients involved had signed informed consent for trials, and the studies were approved by the IRBs at the Indiana University, University of Michigan, Johns Hopkins University, and Georgetown University.
Whole Genome Amplification
Whole genome amplification was performed using the REPLI-g mini WGA kit (Qiagen, Inc., Valencia, CA, USA). Twenty-five nanograms of DNA was amplified in a final volume of 50 μl at 30°C for 16 h followed by 3 min at 65°C.
DNA Measurements
DNA concentrations were determined using the Quant-iT dsDNA assay kit (Invitrogen Corporation, Carlsbad, CA, USA). The relative DNA concentrations of amplified and unamplified samples were also determined using the RNase P real-time PCR assay (assay id# 4403326; Applied Biosystems, Inc., Foster City, CA, USA). The size of the whole genome amplified DNA products was assessed using 0.8% agarose gel electrophoresis stained with SYBR Safe DNA gel stain (Invitrogen Corporation, Carlsbad, CA, USA).
Genotyping of the Coriell DNA Samples
The 24 samples from the Coriell human diversity panel were genotyped for the CYP2D6*4 (rs3892097) and the CYP2D6*10 (rs1065852) alleles using Taqman™ assays, assay id# C__27102431_D0 and C__11484460_40, respectively. The ESR1 SNP (rs9340799) was determined using a Taqman™ assay id# C___3163591_10. All Taqman™ assays used in this manuscript were obtained from Applied Biosystems, Inc., Foster City, CA, USA.
Genotyping of the Whole Genome Amplified Samples from Trial 1
The CYP2D6*4, *10, *41, and CYP2C19*2 were genotyped in the amplified samples using the Taqman™ assays, assay id# C__27102431_D0, C__11484460_40, C__34816116_20, and C__25986767_70 respectively. The remaining SNPs were genotyped using the OpenArray™ Taqman™ genotyping platform (Applied Biosystems, Inc., Foster City, CA, USA). We genotyped 143 SNPs on 5 arrays plated in the 32 SNP OpenArray™ formats. The assays for the ESR1, ESR2, and CYP19A1 described above were included on the OpenArrays™. The OpenArrays™ were also used to genotype 468 DNA samples from Trial 2.
Genotyping of the Unamplified Samples from Trial 1 and Trial 2
The genotyping results from unamplified samples from Trial 1 have previously been described (Borges et al., 2006; Jin et al., 2008; Ntukidem et al., 2008). CYP2D6*4, *10, and *41 were genotyped on either the CYP450 Amplichip (Roche Diagnostics, Indianapolis, IN, USA) or the Tag-It Mutation Detection Kit P450-2D6 (Previously Tm Biosciences Corporation, now Luminex Molecular Diagnostics Inc., Toronto, ON, Canada). In addition, the CYP2D6*4 genotype was also confirmed by the Taqman™ assay described above. As previously described for the estrogen receptors (Onitilo et al., 2009), the ESR1 SNPs rs9340799 and rs2234693 were genotyped using Taqman™ assay id# C___3163591_10 and C___3163590_10, respectively, and ESR2 SNPs rs1256049 and rs4986938 using assay id# C____7573265_1_ and C_11462726_10, respectively. CYP19A1 rs4646 was genotyped using assay id# C___8234730_1_.
Results
In our initial study, we performed WGA on 24 DNA samples (25 ng each) from the Human Coriell DNA Diversity panel. After amplification, the DNA yield range, mean, and SD were 1.6–7, 5.4, and 1.2 μg respectively. This was an average amplification of 217-fold. To determine whether we could accurately genotype several genes important in pharmacogenetic studies, the original samples and the whole genome amplified samples were genotyped for rs3892097 (CYP2D6*4), rs1065852 (CYP2D6*10), and rs9340799 (ESR1–XbaI) alleles. The call rates and the concordance with the genotypes from the unamplified samples were both 100% for all three SNPs. These samples were specifically chosen so that our sample set included multiple samples with variant alleles for each SNP. The minor allele frequencies in these samples for the CYP2D6*4, CYP2D6*10, and ESR1–XbaI alleles were 0.08, 0.27, and 0.23, respectively. Since these results indicated that WGA would work for these types of assays, we next amplified DNA samples from a larger clinical trial.
We performed WGA on the DNA samples obtained from 285 breast cancer patients enrolled in Trial 1. The DNA yield range, mean, and SD were 2.5–24.5, 7.2, and 3.7 μg, respectively, as determined by the Picogreen DNA quantification assay. We verified that the DNA was successfully amplified using a quantitative PCR assay for the RNase P gene (Figure 1A). We compared the Ct values for whole genome amplified samples to the Ct values of the same samples that were diluted to the same extent as the dilution resulting from the WGA assay preparation. Thus, the difference in the Ct values represents the increased DNA resulting from the WGA. The average difference between the amplified and unamplified samples was seven cycles. Also, there was clear separation between the amplification curves indicating consistently good amplification (Figure 1A). Using the ΔCt calculation, a seven cycle difference indicates that the DNA was amplified approximately 188-fold. The major bands of the amplified DNA were >12 kb, indicating high quality of DNA amplification (Figure 1B).
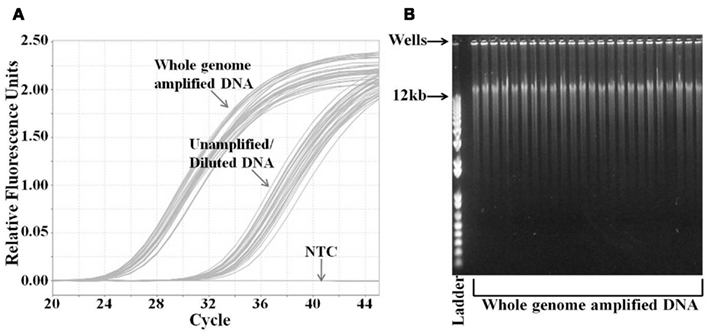
Figure 1. Analysis of whole genome amplified DNA samples. (A) Quantification of DNA concentration by real-time PCR analysis of RNase P gene. DNA from whole genome amplified and unamplified samples were analyzed by RNase P Taqman™ assay. DNA from unamplified samples was diluted equivalently to that amount of dilution by the whole genome amplification. (B) Agarose gel electrophoresis analysis of the whole genome amplified DNA samples. Samples were run on 0.8% agarose gel with the DNA ladder. The highest molecular weight in the ladder is 12 kb. The PCR graph and the gel image are representative of the 285 samples from Trial 1. NTC, no template control.
We tested the functionality of the whole genome amplified DNA from Trial 1 for pharmacogenetics genotyping using two approaches. The first was to genotype nine SNPs in genes that are important in drug pharmacokinetics and pharmacodynamics that were previously genotyped using the unamplified DNA from Trial 1 (Borges et al., 2006; Jin et al., 2008; Ntukidem et al., 2008). These SNPs were rs3892097 (CYP2D6*4), rs1065852 (CYP2D6*10), rs28371725 (CYP2D6*41), rs4244285 (CYP2C19*2), rs4646 (CYP19A1), rs2234693 (ESR1), rs9340799 (ESR1), rs1256049 (ESR2), and rs4986938 (ESR2). The CYP2D6*4, *10, *41, and CYP2C19*2 were genotyped using individual Taqman assays. A representative allelic discrimination plot of CYP2D6*4 is shown in Figure 2A. The other SNPs were genotyped using Taqman assays plated in OpenArrays™. The concordance between the genotyping of the original DNA and the whole genome amplified DNA was >97% for all nine SNPs.
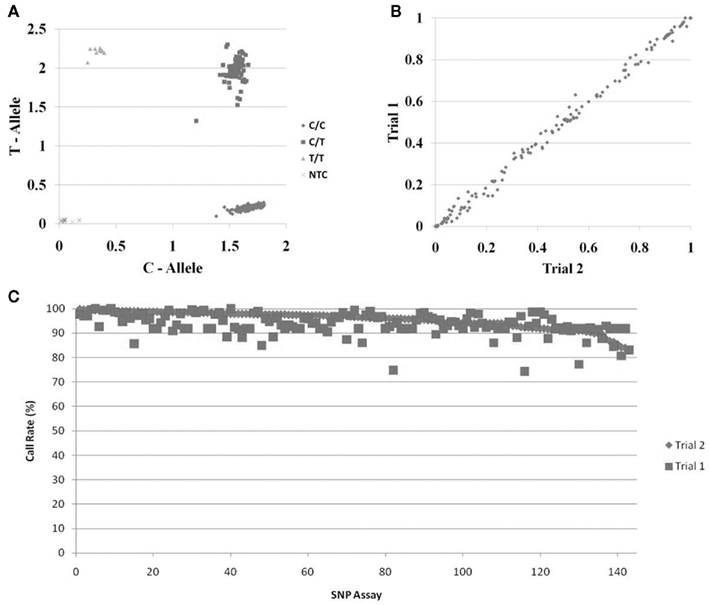
Figure 2. Genotyping of DNA samples following whole genome amplification. (A) Representative Taqman™ allelic discrimination plot from the CYP2D6*4 genotyping of DNA samples following whole genome amplification. (B) Comparison of the allele frequencies of 143 SNPs from DNA samples from two breast cancer clinical trials. The samples from Trial 1 were genotyped following whole genome amplification. The samples from Trial 2 were genotyped directly from DNA isolated from whole blood. Frequencies of the alleles labeled with Taqman™ FAM probe are shown in the plot. (C) Call rates of genotyping assays from samples following whole genome amplification. DNA was obtained from Trial 2 (directly from the DNA isolation; diamonds) or from Trial 1 (following whole genome amplification; squares); samples are ordered from left to right by the call rates in the Trial 2 DNA samples.
The second approach was to compare the allele frequencies of 143 SNPs in the whole genome amplified DNA samples from Trial 1 (n = 285 breast cancer patients) with the allele frequencies in breast cancer subjects from Trial 2 (n = 468 breast cancer patients). These samples were genotyped on OpenArrays™. Since these populations were very similar, we expected the allele frequencies to be very similar if the WGA resulted in accurate and unbiased amplification of these genes. Figure 2B shows the correlation of the frequencies of the FAM linked allele in the two sets of DNA samples. The allele frequencies in both populations were 99.7% identical. Likewise, the call rates for each SNP were also very high and similar between the unamplified and the whole genome amplified samples Figure 2C.
Discussion
Collectively, these studies indicate that WGA is a valuable tool for generating DNA suitable for genotyping a variety of genes in pharmacogenetic studies. This extends studies of others that have shown that the REPLI-g based WGA reliably amplifies DNA for use in large scale genome wide association studies (Jasmine et al., 2008). It was important to validate this approach in studies where genes of importance in pharmacogenetics were the focus, because many of these genes are difficult to genotype due to multiple pseudogenes, copy number variations, and other related genes that have a very high sequence similarity and also because many of these genes are poorly covered on the GWAS chips (Peters and McLeod, 2008). We have shown that SNPs that are commonly genotyped in three of the CYP genes, CYP2D6, CYP2C19, and CYP19A1, were genotyped with high accuracy when whole genome amplified DNA was used. Since this approach appeared to work well with CYP2D6, which is a relatively difficult gene to genotype, we expect that the WGA would also work well with other CYP genes. This was confirmed by the genotyping of the CYP2C19 and CYP19A1 genotyping. Our results also showed that the whole genome amplified DNA is suitable for genotyping with the OpenArray™ genotyping platform.
We sought to validate this approach further by correlating the allele frequencies in two breast cancer patient populations. Although this is not as definitive as genotyping the DNA from the unamplified samples as the control, since the two populations of subjects were quite similar, mostly Caucasian women with estrogen receptor positive breast cancer, we expected that the allele frequencies in the two populations would be very similar. We recognize a potential, but unlikely, caveat is that there could be errors in the genotyping that happen to coincided with true differences in allele frequencies that make them look accurate when they are not. However, it seems unlikely that there would have been such a high correlation across so many SNPs if that were true. Furthermore, direct validation using whole genome amplified versus unamplified samples in both the Coriell sample set and several SNPs from the Trial 1 indicated that the accuracy for these genes is very high. Although the SNP genotyping was successful, we were not able to accurately quantify the CYP2D6 copy numbers due to an apparent overestimation of the copy number in the whole genome amplified samples.
Whole genome amplification could be a valuable tool for generating sufficient quantities of DNA for future pharmacogenetic studies. First, in sample sets for which much of the DNA has been used up, this would provide a mechanism to extend the samples to allow a larger number of studies. Second, in studies in which only a limited amount of blood or sample was collected, it may be useful to make the DNA available for a wider array of genotyping. It should be cautioned, however, that when possible, collection of adequate samples to begin with would still be the preferred choice for most studies, since some assays, such as methylation measurements, will not be possible on the amplified DNA. Furthermore, additional studies will be required for validating the suitability of whole genome amplified DNA for determining of copy number variations.
In conclusion, collectively, our results indicate that the REPLI-g WGA is a reliable method for amplifying DNA that can be used in Taqman™ and OpenArray™ platforms to genotype a variety of SNPs important for pharmacogenetics.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants from the NIH-NIGMS (1R01GM088076, Todd C. Skaar), NIH-PGRN (5U01GM061373, David A. Flockhart), NIH-NCRR M01-RR000042 (University of Michigan), M01-RR020359 (Georgetown University), and M01-RR00750 (Indiana University), and M01-RR00052 (Johns Hopkins University). This project was supported by grant number R01HS019818 from the Agency for Healthcare Research and Quality. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Agency for Healthcare Research and Quality.
References
Borges, S., Desta, Z., Li, L., Skaar, T. C., Ward, B. A., Nguyen, A., Jin, Y., Storniolo, A. M., Nikoloff, D. M., Wu, L., Hillman, G., Hayes, D. F., Stearns, V., and Flockhart, D. A. (2006). Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin. Pharmacol. Ther. 80, 61–74.
Jasmine, F., Ahsan, H., Andrulis, I. L., John, E. M., Chang-Claude, J., and Kibriya, M. G. (2008). Whole-genome amplification enables accurate genotyping for microarray-based high-density single nucleotide polymorphism array. Cancer Epidemiol. Biomarkers Prev. 17, 3499–3508.
Jin, Y., Hayes, D. F., Li, L., Robarge, J. D., Skaar, T. C., Philips, S., Nguyen, A., Schott, A., Hayden, J., Lemler, S., Storniolo, A. M., Flockhart, D. A., and Stearns, V. (2008). Estrogen receptor genotypes influence hot flash prevalence and composite score before and after tamoxifen therapy. J. Clin. Oncol. 26, 5849–5854.
Ntukidem, N. I., Nguyen, A. T., Stearns, V., Rehman, M., Schott, A., Skaar, T., Jin, Y., Blanche, P., Li, L., Lemler, S., Hayden, J., Krauss, R. M., Desta, Z., Flockhart, D. A., and Hayes, D. F. (2008). Estrogen receptor genotypes, menopausal status, and the lipid effects of tamoxifen. Clin. Pharmacol. Ther. 83, 702–710.
Onitilo, A. A., Mccarty, C. A., Wilke, R. A., Glurich, I., Engel, J. M., Flockhart, D. A., Nguyen, A., Li, L., Mi, D., Skaar, T. C., and Jin, Y. (2009). Estrogen receptor genotype is associated with risk of venous thromboembolism during tamoxifen therapy. Breast Cancer Res. Treat. 115, 643–650.
Keywords: whole genome amplification, pharmacogenetics, genotyping, CYP
Citation: Philips S, Rae JM, Oesterreich S, Hayes DF, Stearns V, Henry NL, Storniolo AM, Flockhart DA and Skaar TC (2012) Whole genome amplification of DNA for genotyping pharmacogenetics candidate genes. Front. Pharmacol. 3:54. doi: 10.3389/fphar.2012.00054
Received: 05 January 2012; Accepted: 13 March 2012;
Published online: 30 March 2012.
Edited by:
Colin Palmer, University of Dundee, UKCopyright: © 2012 Philips, Rae, Oesterreich, Hayes, Stearns, Henry, Storniolo, Flockhart and Skaar. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Todd C. Skaar, Division of Clinical Pharmacology, Department of Medicine, Indiana University School of Medicine, 1001 West 10th Street, WD Myers Building W7123, Indianapolis, IN 46202, USA. e-mail:dHNrYWFyQGl1cHVpLmVkdQ==