

- 1 Department of Anesthesiology, Weill Cornell Medical College, New York, NY, USA
- 2 Department of Pharmacology, Weill Cornell Medical College, New York, NY, USA
The molecular mechanisms of modern inhaled anesthetics are still poorly understood although they are widely used in clinical settings. Considerable evidence supports effects on membrane proteins including ligand- and voltage-gated ion channels of excitable cells. Na+ channels are crucial to action potential initiation and propagation, and represent potential targets for volatile anesthetic effects on central nervous system depression. Inhibition of presynaptic Na+ channels leads to reduced neurotransmitter release at the synapse and could therefore contribute to the mechanisms by which volatile anesthetics produce their characteristic end points: amnesia, unconsciousness, and immobility. Early studies on crayfish and squid giant axon showed inhibition of Na+ currents by volatile anesthetics at high concentrations. Subsequent studies using native neuronal preparations and heterologous expression systems with various mammalian Na+ channel isoforms implicated inhibition of presynaptic Na+ channels in anesthetic actions at clinical concentrations. Volatile anesthetics reduce peak Na+ current (INa) and shift the voltage of half-maximal steady-state inactivation (h∞) toward more negative potentials, thus stabilizing the fast-inactivated state. Furthermore recovery from fast-inactivation is slowed, together with enhanced use-dependent block during pulse train protocols. These effects can depress presynaptic excitability, depolarization and Ca2+ entry, and ultimately reduce transmitter release. This reduction in transmitter release is more potent for glutamatergic compared to GABAergic terminals. Involvement of Na+ channel inhibition in mediating the immobility caused by volatile anesthetics has been demonstrated in animal studies, in which intrathecal infusion of the Na+ channel blocker tetrodotoxin increases volatile anesthetic potency, whereas infusion of the Na+ channels agonist veratridine reduces anesthetic potency. These studies indicate that inhibition of presynaptic Na+ channels by volatile anesthetics is involved in mediating some of their effects.
Background
It has been over 160 years since the use of diethyl ether as a general anesthetic was publicly demonstrated, yet our mechanistic understanding of these vitally important drugs lags far behind that of most other major drug classes. Most modern inhaled anesthetics are derivatives of ether, and over the years have been developed to have improved pharmacokinetics, but they are still plagued by a lack of specificity with significant cardiovascular and respiratory side effects. It remains unclear how these drugs produce general anesthesia, a pharmacologically induced coma characterized by amnesia, unconsciousness, and immobility in response to painful stimuli (Hemmings et al., 2005b). Studies into their molecular mechanisms in the 1960s, which have their origins in the Meyer–Overton correlation of anesthetic potency with lipophilicity from 1900, led to a lipid-based theory involving a unitary mechanism of non-specific actions on the lipid bilayer (Meyer, 1899; Overton, 1901).
With technical advances in biochemistry and biophysics, specific targets were studied and identified. Pioneering studies showed that anesthetic interactions with proteins themselves, not necessarily involving lipid interactions, could explain anesthetic effects at a biochemical level (Franks and Lieb, 1994). Animal studies showed that volatile anesthetics produce their immobilizing effects primarily by actions on the spinal cord (Antognini and Schwartz, 1993; Rampil et al., 1993), whereas unconsciousness and amnesia involve actions at supra-spinal centers (Eger et al., 2008). Membrane proteins including ion channels have been implicated as key mediators of the depressive effects of anesthetics on neuronal function. Many potential targets have been identified, and it has become clear that anesthetics act at multiple distinct targets in the central nervous system to produce the various component effects of the anesthetic state (multi-site hypothesis).
Mechanisms of General Anesthetic Effects on the Central Nervous System
The idea of general anesthetics acting both on excitatory and inhibitory synaptic transmission has lead to many studies pointing out the complexity of anesthetic mechanisms (Rudolph and Antkowiak, 2004; Hemmings et al., 2005b; Franks, 2006). General anesthetics, including both volatile and intravenous anesthetics, enhance synaptic inhibition via postsynaptic γ-aminobutyric acid type A (GABAA) receptor modulation (Nicoll et al., 1975; Zimmerman et al., 1994). More recent studies also point out the importance of extrasynaptic GABAA receptors as a target of anesthetics by potentiating tonic inhibitory currents (Orser, 2006; Rau et al., 2009) and by enhancing the release of GABA by a presynaptic increase in miniature inhibitory postsynaptic current (mIPSC) frequency (Nishikawa and MacIver, 2001). Depression of excitatory transmission by presynaptic effects is another target of anesthetic action (Perouansky et al., 1995; Maclver et al., 1996; Ouanonou et al., 1999; Wakasugi et al., 1999). Both volatile and intravenous anesthetics reduce excitatory postsynaptic potentials (EPSPs) in neurons, an effect most likely due to presynaptic mechanisms (Weakly, 1969; Richards and White, 1975; Kullmann et al., 1989; Berg-Johnsen and Langmoen, 1992). Recent evidence suggests that inhibition of glutamatergic synaptic transmission through N-methyl-D-aspartate (NMDA)-type glutamate receptor blockade by inhaled anesthetics might also contribute to depression of excitatory transmission (Dickinson et al., 2007; Haseneder et al., 2008).
It is now evident that ligand-gated ion channels are major targets for general anesthetics (Franks and Lieb, 1994). Both inhibition of excitatory NMDA receptors and potentiation of inhibitory GABAA and glycine receptors have come under scrutiny as important targets for both intravenous and inhaled anesthetic effects on synaptic transmission (Franks, 2006). These receptors are found throughout the central nervous system and are major transducers of excitatory and inhibitory neurotransmitter signaling.
Second-messenger regulated protein phosphorylation of Na+ channels has been implicated as another possible target of volatile anesthetics. Halothane increases both purified (Hemmings and Adamo, 1994) and endogenous (Hemmings and Adamo, 1996) brain protein kinase C (PKC) activity. Phosphorylation of Na+ channels by PKC and PKA reduces Na+ channel activity by altering channel kinetics, for example by slowing inactivation, and is therefore an important component of neuromodulation (Cantrell and Catterall, 2001). It is possible that some of the inhibitory effects of volatile anesthetics on Na+ channel activity are mediated through PKC phosphorylation.
More recent studies have extended the range of likely anesthetic targets to include neuronal nicotinic acetylcholine receptors (Flood et al., 1997), two pore domain K2P channels and K+ leak channels (Patel and Honore, 2001; Sirois et al., 2002), and presynaptic voltage-gated Na+ channels. This review considers Na+ channels as targets for the effects of volatile anesthetics (inhaled alkane and ether derivatives).
Presynaptic Na+ Channels as Anesthetic Targets
Na+ channels play a crucial role in cell-to-cell communication, as they are involved in initiating and propagating action potentials in excitable cells throughout the nervous system (Hodgkin and Huxley, 1952). Early reports in the 1970s associated the effects of volatile anesthetics on lipid bilayer properties to alterations of certain membrane bound ion channels, in particular voltage-gated Na+ channels (Figure 1).
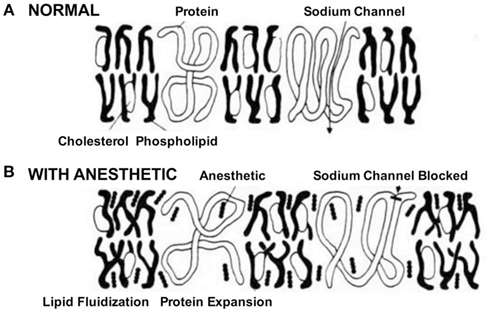
Figure 1. Schematic of the effects of anesthetics on cell membrane and Na+ channels. In the absence of the drug. (A) Na+ channels initiate and propagate electrical signals, i.e., action potentials. (B) The anesthetic was believed to affect Na+ channels by partitioning and interacting with the membrane. This process called lipid fluidification altered the cell membrane and subsequently distorted the channel protein leading to block of channel function (Seeman, 1974).
These reports were among the first to hypothesize a specific ion channel (Na+ channels) as a potential target of volatile anesthetics, though at that time no specific binding site or specific mechanism could be identified. Early studies on the effects of general anesthetics on Na+ and K+ currents in the crayfish or squid giant axon showed inhibition of peak Na+ (INa) current and effects on channel recovery, but in these preparations inhibition occurred at relatively high concentrations (Bean et al., 1981; Haydon and Simon, 1988). Subsequent studies examined the effects of various volatile anesthetics on mammalian brain derived Na+ channels heterologously expressed in mammalian cell lines (Rehberg et al., 1996). Inhibition of peak INa due to stabilization of the inactivated state of Na+ channels was evident as a hyperpolarizing “left-shift” in steady-state (or h∞) inactivation. These experiments were among the first to demonstrate inhibition of neuronal Na+ channels by volatile anesthetics. The sensitivity of Na+ channels to clinically relevant concentrations of volatile anesthetics was confirmed in various in vitro expression systems and was subsequently extended to more physiologically relevant neuronal preparations.
Electrophysiological recordings performed in isolated rat neurohypophysial nerve terminals, an experimentally accessible nerve terminal preparation, showed that clinically relevant concentrations of isoflurane inhibited peak INa in nerve terminals in a concentration- and voltage-dependent manner (Ouyang et al., 2003; Figure 2A, upper panel). Similar to heterologous expression systems, a left-shift in the voltage-dependence of steady-state inactivation demonstrated stabilization of the fast-inactivated state. These results support the hypothesis that volatile anesthetics depress excitatory synaptic transmission by inhibiting presynaptic voltage-gated Na+ channels. In addition, in the rat neurohypophysial nerve terminal preparation, isoflurane inhibited action potential amplitude and increased action potential half-width (Ouyang and Hemmings, 2005; Figure 2A, lower panel). The underlying current mediating the fast and rising depolarizing phase of the action potential is carried by tetrodotoxin (TTX)-sensitive Na+ channels, which were inhibited by isoflurane using a voltage-stimulus based on an averaged action potential. The effects of non-immobilizers (structurally similar compounds without anesthetic properties) in rat dorsal root ganglion neurons showed that compound F3, an anesthetic fluorinated cyclobutane, inhibited Na+ channels similar to the effects of conventional volatile anesthetics, but the non-anesthetic (non-immobilizer) fluorinated cyclobutane F6 had only minimal effects (Ratnakumari et al., 2000; Figure 4A). These findings support the role of Na+ channels as molecular targets for volatile anesthetic action.
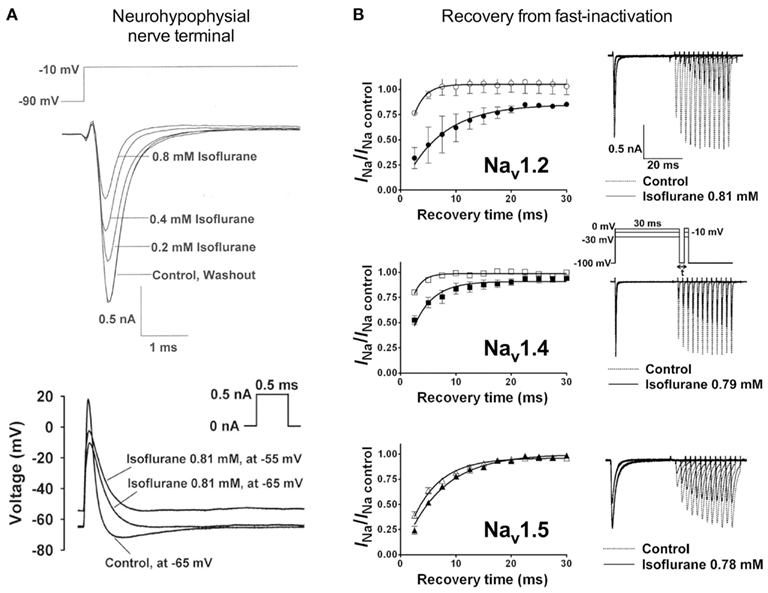
Figure 2. Volatile anesthetics inhibit Na+ channels in various expression systems. [(A), upper panel] Electrophysiological recordings of isolated rat neurohypophysial nerve terminals show a reversible block of Na+ currents and [(A), lower panel] action potentials evoked by small current injections at clinically relevant concentrations of isoflurane (Ouyang et al., 2003; Ouyang and Hemmings, 2005). (B) Effects of isoflurane on channel recovery from fast-inactivation of three different Na+ channel isoforms heterologously expressed in mammalian cells. Recovery was assessed using a two-pulse protocol with a 30-ms conditioning pulse followed by a variable recovery interval of up to 30 ms, and then a 5-ms test pulse to peak activation voltages. The time course of channel recovery from fast-inactivation was well fitted by a monoexponential function [(B), left panels]. Representative current traces for a holding potential (Vh) of −100 mV are shown for all three subtypes [(B), right panels; Ouyang and Hemmings, 2007].
Studies investigating subtype-specific effects of volatile anesthetics revealed small, but potentially significant, differences in isoflurane potency with IC50 values ranging from 0.45 to 0.7 mM (at Vh −70 mV) on Nav1.2, Nav1.4, Nav1.5 expressed in Chinese hamster ovary cells (Ouyang and Hemmings, 2007). Despite the small potency differences, there were differences between isoforms in recovery from fast-inactivation tested by a double-pulse protocol. The effect of isoflurane on channel recovery was greatest in Nav1.2, a major brain isoform (Figure 2B). Another study in which subtypes Nav1.2, Nav1.4, Nav1.6, and TTX-resistant Nav1.8 were expressed (with and without β1 subunit co-expression) in Xenopus oocytes also revealed that Nav1.2, Nav1.4, Nav1.6 were sensitive to isoflurane, whereas the TTX-resistant subtype Nav1.8, which is highly expressed in dorsal root ganglion nociceptive neurons, was insensitive (Shiraishi and Harris, 2004). Nerve terminals of nociceptive sensory neurons are the (main) origin of neuropathic and inflammatory pain signals (Dib-Hajj et al., 2010), but the pro- or anti-nociceptive effects of volatile anesthetics are not clearly defined. It is evident that these nociceptive neurons carry a distinct selection of Na+ channel subtypes related to pain signaling (e.g., Nav1.7, Nav1.8, Nav1.9; see review Dib-Hajj et al., 2010). Subsequently, Nav1.8 expressed in mammalian neuronal cells revealed concentration- and voltage-dependent inhibition of Nav1.8 by clinically relevant concentrations of isoflurane similar to other subtypes (Herold et al., 2009; Figure 3A, upper panel). This demonstrates the importance of choosing a suitable expression system for pharmacological studies of ion channels. In this case the neuronal cell line ND7/23, a hybrid cell line between rat dorsal root ganglion neurons and mouse neuroblastoma cells, may have provided auxiliary β-subunits or other neuron-specific signaling pathways that are important for inhibition by anesthetics. A comparative study showing the effects of several different volatile anesthetics on heterologously expressed Na+ channels in mammalian cells revealed that desflurane, a highly fluorinated inhaled anesthetic, had the strongest effect on peak INa inhibition, but all agents in this class were effective at clinically relevant concentrations (Ouyang et al., 2009; Figure 3B). In contrast, the intravenous anesthetic propofol inhibits Na+ channels only at supratherapeutic concentrations (Rehberg and Duch, 1999).
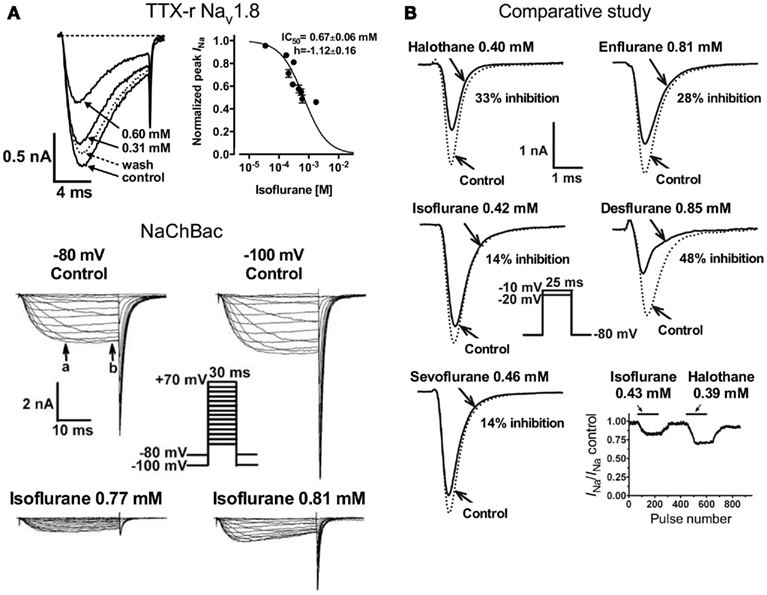
Figure 3. [(A), upper panel] Concentration-dependent inhibition of tetrodotoxin-resistant (TTX-r) Nav1.8 by isoflurane. Current traces of TTX-r Nav1.8 are shown in the absence or presence of two isoflurane concentrations. Normalized peak INa values for TTX-r Nav1.8 were fitted to the Hill equation to yield IC50 values and Hill slopes (Herold et al., 2009). [(A), lower panel] Effects of isoflurane on NaChBac expressed in HEK293 cells. Families of current traces are shown at two different holding potentials (Vh) in the absence or presence of isoflurane. Isoflurane significantly inhibited INa from a Vh of either −80 or −100 mV (Ouyang et al., 2007). (B) Inhibition of Nav1.4 by equipotent concentrations of various inhaled anesthetics. Peak INa were recorded from a holding potential of –80 mV by 25-ms test steps as shown in the inset. The effects of clinically equipotent concentrations of halothane, isoflurane, sevoflurane, enflurane, and desflurane are shown in these representative traces. Desflurane had the greatest effect of peak INa reduction.
The prototypical halogenated ether isoflurane also inhibits the prokaryotic voltage-gated Na+ channel of Bacillus halodurans (NaChBac; Ouyang et al., 2007; Figure 3A, lower panel). This was the first prokaryotic channel shown to be inhibited by an anesthetic, and demonstrates impressive evolutionary conservation of the mechanism responsible for this pharmacological effect. As with mammalian channels, inhibition of peak INa was concentration- and voltage-dependent, and was associated with a positive shift in the voltage-dependence of activation and a negative shift in the voltage-dependence of steady-state fast-inactivation. Furthermore use-dependent block occurred due to slowed recovery from inactivation. Despite the evolutionary difference between prokaryotic and eukaryotic voltage-gated Na+ channels, the mechanisms by which volatile anesthetics act on the channel seem remarkably similar.
Aromatic compounds such as fluorobenzene, hexafluorobenzene, and 1,2-difluorobenzene have been shown to inhibit Nav1.2a expressed in Xenopus oocytes. Inhibition of peak INa as well as a shift in the V1/2 of fast-inactivation occurs in an agent-dependent manner (Horishita et al., 2008). The exact mechanism of the differential effects of these structurally different compounds has yet to be elucidated. Differences also exist in the potency of volatile anesthetic inhibition of specific Na+ channel subtypes (Ouyang et al., 2009), but again the mechanisms for these differences have to be studied in more detail. Such differences might underlie region-specific presynaptic effects of volatile anesthetics on neurotransmitter release (Westphalen et al., 2010, 2011).
Na+ Channel Inhibition Leads to Inhibition of Neurotransmitter Release by Anesthetics
A physiological consequence of presynaptic Na+ channel inhibition is depression of presynaptic action potential generation and conduction. Considerable evidence indicates that volatile anesthetics inhibit neurotransmitter release, and that this is due in part to inhibition of presynaptic Na+ channels. Volatile anesthetics preferentially inhibit 4-aminopyridine (4AP)-evoked release of glutamate compared to GABA from isolated rat cortical nerve terminals (Westphalen and Hemmings, 2006). Action potential-evoked depolarization and release can be pharmacologically mimicked by 4AP, a K+ channel blocker, while Na+ channel independent release can be elicited by depolarization with elevated extracellular K+ (Tibbs et al., 1989). Using this approach, 4AP-evoked release is significantly more sensitive to inhibition by volatile anesthetics as compared to KCl-evoked release, supporting a role for blockade of presynaptic Na+ channels in the inhibitory effects of the anesthetics (Schlame and Hemmings, 1995; Westphalen and Hemmings, 2003). Interestingly, inhibition of glutamate release occurs with about 50% greater potency than inhibition of GABA release, consistent with pharmacologically relevant transmitter-specific specializations in neurotransmitter release regulation, perhaps involving differential coupling to Na+ channels (Westphalen et al., 2010; Figure 4B). There is also evidence that volatile anesthetics inhibit neurotransmitter release in a brain region-specific manner (Westphalen et al., 2011), which suggests diversity in presynaptic Na+ channel subtype expression and/or coupling to release (Westphalen et al., 2010).
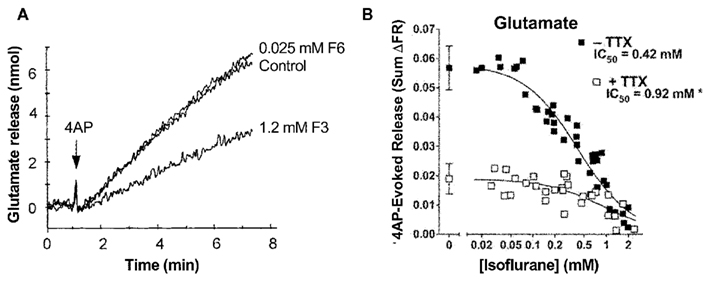
Figure 4. Volatile anesthetics inhibit neurotransmitter release in nerve terminals. (A) Effects of the anesthetic compound F3 and the non-immobilizer F6 on 4-aminopyridine- (4AP) evoked glutamate release from cortical synaptosomes. The anesthetic cyclobutane F3 significantly inhibits glutamate release, whereas the non-anesthetic (non-immobilizer) cyclobutane F6 shown no inhibitory effect (Ratnakumari et al., 2000). (B) Isoflurane inhibition of 4AP-evoked glutamate release from rat cortical nerve terminals in the absence or presence of tetrodotoxin (TTX, 1 μM). The potency of isoflurane inhibition is much greater in the absence of the Na+ channel blocker TTX indicating a strong involvement of Na+ channels in inhibition of neurotransmitter release by volatile anesthetics (Westphalen et al., 2011).
Further experiments have examined the effects of volatile anesthetics on synaptic vesicle exocytosis, detected using fluorescence imaging, in cultured rat hippocampal neurons. This preparation allows electrical stimulation of release, and showed concentration-dependent and reversible inhibition of action potential-evoked exocytosis by isoflurane. Involvement of presynaptic Na+ channels is supported by the observation that exocytosis, evoked by depolarization with elevated extracellular K+ (which is insensitive to TTX), was relatively insensitive to isoflurane (Hemmings et al., 2005a). Isoflurane has also shown to inhibit excitatory postsynaptic currents (EPSCs) in the rat calyx of Held due to inhibition of neurotransmitter release caused by a reduction of presynaptic action potential amplitude (Wu et al., 2004). These effects of volatile anesthetics on synaptic transmission result primarily from inhibition of action potential-evoked synaptic vesicle exocytosis, most likely as a result of Na+ channel blockade upstream of Ca2+ entry and exocytosis.
In vivo studies on rodents have implicated spinal Na+ channels in immobilization, a major component of general anesthesia. Intravenous infusion of lidocaine, a classical local anesthetic, or intrathecal administration of riluzole, another potent Na+ channel inhibitor, significantly increases the potency of volatile anesthetics as immobilizers (Xing et al., 2003; Zhang et al., 2007). The role of Na+ channels in volatile anesthetic-mediated immobility is further supported by the observation that intrathecal infusion of the Na+ channel activator veratridine, a plant neurotoxin that binds to site 2 and stabilizes the open state (Ulbricht, 1998), reduces the potency of isoflurane (Zhang et al., 2008), while intrathecal infusion of TTX increases the potency of isoflurane, and reverses the effect of veratridine (Zhang et al., 2010). Taken together, these results indicate that inhibition of spinal voltage-gated Na+ channels by inhaled anesthetics is likely an important mechanism in anesthetic immobility.
Non-Anesthetic Effects of Volatile Anesthetics
A major side effect of volatile anesthetics is cardiovascular depression. Multiple ion channel types expressed in cardiomyocytes contribute to action potential conduction and myocardial contractility. Inhibition of L-type Ca2+ currents or voltage-gated transient and sustained outward K+ currents by volatile anesthetics can lead to reduced contractility and delayed repolarization with mismatch of action potential duration (Huneke et al., 2004). In cardiac Na+ channels (Nav1.5), volatile anesthetics at clinically relevant concentrations inhibit peak INa and affect steady-state fast- as well as slow-inactivation (Stadnicka et al., 1999; Ouyang and Hemmings, 2007). This can, in combination with other cardiodepressant drugs, slow conduction and lead to tachyarrhythmias. Na+ channels have also been implicated as potential targets for neuroprotection by volatile anesthetics (Hemmings, 2004). The possible role of voltage-gated Na+ channels and other beneficial and detrimental side effects of volatile anesthetics in brain and other organs cannot be excluded.
Conclusion
Both electrophysiological and functional studies indicate that presynaptic voltage-gated Na+ channels are inhibited by clinically used concentrations of volatile anesthetics. This leads to reductions in evoked neurotransmitter release that is both brain region and neurotransmitter selective. The selective inhibition of glutamate release underlies a reduction in excitatory synaptic transmission with resultant nervous system depression. Detailed information regarding the presynaptic localization, function, and regulation of specific Na+ channel subtypes is currently lacking. Further studies are necessary to identify the roles of specific presynaptic Na+ channel subtypes in mediating neurotransmitter release and its inhibition by volatile anesthetics and other Na+ channel inhibitors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by NIH grant GM58055 (Hugh C. Hemmings Jr.) and DFG (German Research Foundation) grant HE4551/5-1 (Karl F. Herold).
References
Antognini, J. F., and Schwartz, K. (1993). Exaggerated anesthetic requirements in the preferentially anesthetized brain. Anesthesiology 79, 1244–1249.
Bean, B. P., Shrager, P., and Goldstein, D. A. (1981). Modification of sodium and potassium channel gating kinetics by ether and halothane. J. Gen. Physiol. 77, 233–253.
Berg-Johnsen, J., and Langmoen, I. A. (1992). The effect of isoflurane on excitatory synaptic transmission in the rat hippocampus. Acta Anaesthesiol. Scand. 36, 350–355.
Cantrell, A., and Catterall, W. (2001). Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat. Rev. Neurosci. 2, 397–407.
Dib-Hajj, S. D., Cummins, T. R., Black, J. A., and Waxman, S. G. (2010). Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 33, 325–347.
Dickinson, R., Peterson, B. K., Banks, P., Simillis, C., Martin, J. C., Valenzuela, C. A., Maze, M., and Franks, N. P. (2007). Competitive inhibition at the glycine site of the N-methyl-D-aspartate receptor by the anesthetics xenon and isoflurane: evidence from molecular modeling and electrophysiology. Anesthesiology 107, 756–767.
Eger, E. I., Raines, D. E., Shafer, S. L., Hemmings, H. C., and Sonner, J. M. (2008). Is a new paradigm needed to explain how inhaled anesthetics produce immobility? Anesth. Analg. 107, 832–848.
Flood, P., Ramirez-Latorre, J., and Role, L. (1997). Alpha 4 beta 2 neuronal nicotinic acetylcholine receptors in the central nervous system are inhibited by isoflurane and propofol, but alpha 7-type nicotinic acetylcholine receptors are unaffected. Anesthesiology 86, 859–865.
Franks, N. P. (2006). Molecular targets underlying general anaesthesia. Br. J. Pharmacol. 147(Suppl. 1), S72–S81.
Franks, N. P., and Lieb, W. R. (1994). Molecular and cellular mechanisms of general anaesthesia. Nature 367, 607–614.
Haseneder, R., Kratzer, S., Kochs, E., Eckle, V. S., Zieglgansberger, W., and Rammes, G. (2008). Xenon reduces N-methyl-D-aspartate and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor-mediated synaptic transmission in the amygdala. Anesthesiology 109, 998–1006.
Haydon, D. A., and Simon, A. J. (1988). Excitation of the squid giant axon by general anaesthetics. J. Physiol. (Lond.) 402, 375–389.
Hemmings, H., Yan, W., Westphalen, R., and Ryan, T. (2005a). The general anesthetic isoflurane depresses synaptic vesicle exocytosis. Mol. Pharmacol. 67, 1591–1599.
Hemmings, H. C. Jr., Akabas, M. H., Goldstein, P. A., Trudell, J. R., Orser, B. A., and Harrison, N. L. (2005b). Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol. Sci. 26, 503–510.
Hemmings, H. C. Jr. (2004). Neuroprotection by Na+ channel blockade. J. Neurosurg. Anesthesiol. 16, 100–101.
Hemmings, H. C., and Adamo, A. I. (1994). Effects of halothane and propofol on purified brain protein kinase C activation. Anesthesiology 81, 147–155.
Hemmings, H. C., and Adamo, A. I. (1996). Activation of endogenous protein kinase C by halothane in synaptosomes. Anesthesiology 84, 652–662.
Herold, K. F., Nau, C., Ouyang, W., and Hemmings, H. C. (2009). Isoflurane inhibits the tetrodotoxin-resistant voltage-gated sodium channel Nav1.8. Anesthesiology 111, 591–599.
Hodgkin, A., and Huxley, A. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. (Lond.) 117, 500–544.
Horishita, T., Eger, E. I., and Harris, R. A. (2008). The effects of volatile aromatic anesthetics on voltage-gated Na+ channels expressed in Xenopus oocytes. Anesth. Analg. 107, 1579–1586.
Huneke, R., Fassl, J., Rossaint, R., and Luckhoff, A. (2004). Effects of volatile anesthetics on cardiac ion channels. Acta Anaesthesiol. Scand. 48, 547–561.
Kullmann, D. M., Martin, R. L., and Redman, S. J. (1989). Reduction by general anaesthetics of group Ia excitatory postsynaptic potentials and currents in the cat spinal cord. J. Physiol. (Lond.) 412, 277–296.
Maclver, M. B., Mikulec, A. A., Amagasu, S. M., and Monroe, F. A. (1996). Volatile anesthetics depress glutamate transmission via presynaptic actions. Anesthesiology 85, 823–834.
Nicoll, R. A., Eccles, J. C., Oshima, T., and Rubia, F. (1975). Prolongation of hippocampal inhibitory postsynaptic potentials by barbiturates. Nature 258, 625–627.
Nishikawa, K., and MacIver, M. B. (2001). Agent-selective effects of volatile anesthetics on GABAA receptor-mediated synaptic inhibition in hippocampal interneurons. Anesthesiology 94, 340–347.
Orser, B. A. (2006). Extrasynaptic GABAA receptors are critical targets for sedative-hypnotic drugs. J. Clin. Sleep. Med. 2, S12–S18.
Ouanonou, A., Zhang, Y., and Zhang, L. (1999). Changes in the calcium dependence of glutamate transmission in the hippocampal CA1 region after brief hypoxia-hypoglycemia. J. Neurophysiol. 82, 1147–1155.
Ouyang, W., and Hemmings, H. (2005). Depression by isoflurane of the action potential and underlying voltage-gated ion currents in isolated rat neurohypophysial nerve terminals. J. Pharmacol. Exp. Ther. 312, 801–808.
Ouyang, W., and Hemmings, H. (2007). Isoform-selective effects of isoflurane on voltage-gated Na+ channels. Anesthesiology 107, 91–98.
Ouyang, W., Herold, K. F., and Hemmings, H. C. (2009). Comparative effects of halogenated inhaled anesthetics on voltage-gated Na+ channel function. Anesthesiology 110, 582–590.
Ouyang, W., Jih, T., Zhang, T., Correa, A., and Hemmings, H. (2007). Isoflurane inhibits NaChBac, a prokaryotic voltage-gated sodium channel. J. Pharmacol. Exp. Ther. 322, 1076–1083.
Ouyang, W., Wang, G., and Hemmings, H. (2003). Isoflurane and propofol inhibit voltage-gated sodium channels in isolated rat neurohypophysial nerve terminals. Mol. Pharmacol. 64, 373–381.
Overton, C. (1901). Studien über die Narkose zugleich ein Beitrag zur allgemeinen Pharmakologie. Jena: Verlag von Gustav Fischer.
Patel, A. J., and Honore, E. (2001). Anesthetic-sensitive 2P domain K+ channels. Anesthesiology 95, 1013–1021.
Perouansky, M., Baranov, D., Salman, M., and Yaari, Y. (1995). Effects of halothane on glutamate receptor-mediated excitatory postsynaptic currents. A patch-clamp study in adult mouse hippocampal slices. Anesthesiology 83, 109–119.
Rampil, I. J., Mason, P., and Singh, H. (1993). Anesthetic potency (MAC) is independent of forebrain structures in the rat. Anesthesiology 78, 707–712.
Ratnakumari, L., Vysotskaya, T., Duch, D., and Hemmings, H. (2000). Differential effects of anesthetic and nonanesthetic cyclobutanes on neuronal voltage-gated sodium channels. Anesthesiology 92, 529–541.
Rau, V., Iyer, S. V., Oh, I., Chandra, D., Harrison, N., Eger, E. I. II, Fanselow, M. S., Homanics, G. E., and Sonner, J. M. (2009). Gamma-aminobutyric acid type A receptor alpha 4 subunit knockout mice are resistant to the amnestic effect of isoflurane. Anesth. Analg. 109, 1816–1822.
Rehberg, B., and Duch, D. (1999). Suppression of central nervous system sodium channels by propofol. Anesthesiology 91, 512–520.
Rehberg, B., Xiao, Y., and Duch, D. (1996). Central nervous system sodium channels are significantly suppressed at clinical concentrations of volatile anesthetics. Anesthesiology 84, 1223–1233; discussion 1227A.
Richards, C. D., and White, A. E. (1975). The actions of volatile anaesthetics on synaptic transmission in the dentate gyrus. J. Physiol. (Lond.) 252, 241–257.
Rudolph, U., and Antkowiak, B. (2004). Molecular and neuronal substrates for general anaesthetics. Nat. Rev. Neurosci. 5, 709–720.
Schlame, M., and Hemmings, H. (1995). Inhibition by volatile anesthetics of endogenous glutamate release from synaptosomes by a presynaptic mechanism. Anesthesiology 82, 1406–1416.
Shiraishi, M., and Harris, R. (2004). Effects of alcohols and anesthetics on recombinant voltage-gated Na+ channels. J. Pharmacol. Exp. Ther. 309, 987–994.
Sirois, J. E., Lynch, C. III, and Bayliss, D. A. (2002). Convergent and reciprocal modulation of a leak K+ current and I(h) by an inhalational anaesthetic and neurotransmitters in rat brainstem motoneurones. J. Physiol. (Lond.) 541, 717–729.
Stadnicka, A., Kwok, W. M., Hartmann, H. A., and Bosnjak, Z. J. (1999). Effects of halothane and isoflurane on fast and slow inactivation of human heart hH1a sodium channels. Anesthesiology 90, 1671–1683.
Tibbs, G. R., Barrie, A. P., Van Mieghem, F. J., Mcmahon, H. T., and Nicholls, D. G. (1989). Repetitive action potentials in isolated nerve terminals in the presence of 4-aminopyridine: effects on cytosolic free Ca2+ and glutamate release. J. Neurochem. 53, 1693–1699.
Ulbricht, W. (1998). Effects of veratridine on sodium currents and fluxes. Rev. Physiol. Biochem. Pharmacol. 133, 1–54.
Wakasugi, M., Hirota, K., Roth, S. H., and Ito, Y. (1999). The effects of general anesthetics on excitatory and inhibitory synaptic transmission in area CA1 of the rat hippocampus in vitro. Anesth. Analg. 88, 676–680.
Weakly, J. N. (1969). Effect of barbiturates on “quantal” synaptic transmission in spinal motoneurons. J. Physiol. (Lond.) 204, 63–77.
Westphalen, R., and Hemmings, H. (2003). Selective depression by general anesthetics of glutamate versus GABA release from isolated cortical nerve terminals. J. Pharmacol. Exp. Ther. 304, 1188–1196.
Westphalen, R., and Hemmings, H. (2006). Volatile anesthetic effects on glutamate versus GABA release from isolated rat cortical nerve terminals: 4-aminopyridine-evoked release. J. Pharmacol. Exp. Ther. 316, 216–223.
Westphalen, R. I., Kwak, N. B., Daniels, K., and Hemmings, H. C. Jr. (2011). Regional differences in the effects of isoflurane on neurotransmitter release. Neuropharmacology 61, 699–706.
Westphalen, R. I., Yu, J., Krivitski, M., Jih, T. Y., and Hemmings, H. C. Jr. (2010). Regional differences in nerve terminal Na+ channel subtype expression and Na+ channel-dependent glutamate and GABA release in rat CNS. J. Neurochem. 113, 1611–1620.
Wu, X., Sun, J., Evers, A., Crowder, M., and Wu, L. (2004). Isoflurane inhibits transmitter release and the presynaptic action potential. Anesthesiology 100, 663–670.
Xing, Y., Zhang, Y., Stabernack, C. R., Eger, E. I. II, and Gray, A. T. (2003). The use of the potassium channel activator riluzole to test whether potassium channels mediate the capacity of isoflurane to produce immobility. Anesth. Analg. 97, 1020–1024.
Zhang, Y., Guzinski, M., Eger, E. I., Laster, M. J., Sharma, M., Harris, R. A., and Hemmings, H. C. (2010). Bidirectional modulation of isoflurane potency by intrathecal tetrodotoxin and veratridine in rats. Br. J. Pharmacol. 159, 872–878.
Zhang, Y., Laster, M. J., Eger, E. I., Sharma, M., and Sonner, J. M. (2007). Lidocaine, MK-801, and MAC. Anesth. Analg. 104, 1098–1102.
Zhang, Y., Sharma, M., Eger, E. I., Laster, M. J., Hemmings, H. C., and Harris, R. A. (2008). Intrathecal veratridine administration increases minimum alveolar concentration in rats. Anesth. Analg. 107, 875–878.
Keywords: sodium channels, volatile anesthetics, presynaptic, anesthetic mechanism
Citation: Herold KF and Hemmings HC Jr (2012) Sodium channels as targets for volatile anesthetics. Front. Pharmacol. 3:50. doi: 10.3389/fphar.2012.00050
Received: 21 December 2011; Paper pending published: 08 January 2012;
Accepted: 07 March 2012; Published online: 30 March 2012.
Edited by:
Mohamed Chahine, Laval University, CanadaReviewed by:
Xander H. T. Wehrens, Baylor College of Medicine, USAClemens Möller, Albstadt-Sigmaringen University, Germany
Copyright: © 2012 Herold and Hemmings Jr. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Hugh C. Hemmings Jr., Departments of Anesthesiology and Pharmacology, Weill Cornell Medical College, Box-50, LC-203, 1300 York Avenue, New York, NY 10065, USA. e-mail:aGNoZW1taUBtZWQuY29ybmVsbC5lZHU=