
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 22 March 2012
Sec. Drug Metabolism and Transport
Volume 3 - 2012 | https://doi.org/10.3389/fphar.2012.00048
This article is part of the Research Topic The role of bile pigments in health and disease: effects on cell signaling, cytotoxicity and cytoprotection View all 20 articles
 Kelly J. Peyton1 Ahmad R. Shebib1 Mohammad A. Azam1 Xiao-ming Liu1
Kelly J. Peyton1 Ahmad R. Shebib1 Mohammad A. Azam1 Xiao-ming Liu1 David A. Tulis2
David A. Tulis2 William Durante1*
William Durante1*Bilirubin is a heme metabolite generated by the concerted action of the enzymes heme oxygenase and biliverdin reductase. Although long considered a toxic byproduct of heme catabolism, recent preclinical, and clinical studies indicate the bilirubin exerts beneficial effects in the circulation. In the present study, we determined whether local administration of bilirubin attenuates neointima formation following injury of rat carotid arteries. In addition, the ability of bilirubin to regulate the proliferation and migration of human arterial smooth muscle cells (SMCs) was investigated. Local perivascular administration of bilirubin immediately following balloon injury of rat carotid arteries significantly attenuated neointima formation. Bilirubin-mediated inhibition of neointimal thickening was associated with a significant decrease in ERK activity and cyclin D1 and A protein expression, and an increase in p21 and p53 protein expression in injured blood vessels. Treatment of human aortic SMCs with bilirubin inhibited proliferation and migration in a concentration-dependent manner without affecting cell viability. In addition, bilirubin resulted in a concentration-dependent increase in the percentage of cells in the G0/G1 phase of the cell cycle and this was paralleled by a decrease in the fraction of cells in the S and G2M phases of the cell cycle. Finally, bilirubin had no effect on mitochondrial function and ATP content of vascular SMCs. In conclusion, these studies demonstrate that bilirubin inhibits neointima formation after arterial injury and this is associated with alterations in the expression of cell cycle regulatory proteins. Furthermore, bilirubin blocks proliferation and migration of human arterial SMCs and arrests SMCs in the G0/G1 phase of the cell cycle. Bilirubin represents an attractive therapeutic agent in treating occlusive vascular disease.
Bilirubin is a heme metabolite that is formed by the sequential action of heme oxygenase (HO) and biliverdin reductase. HO oxidatively degrades heme into equimolar amounts of biliverdin, iron, and carbon monoxide while biliverdin reductase rapidly metabolizes biliverdin to bilirubin. Bilirubin circulates in blood bound to albumin and eventually enters the liver where it is glucuronidated by UDP-glucuronyltransferase 1A1 (UGT1A1) and excreted into the bile ducts for elimination. Although long considered a toxic waste product of heme catabolism, bilirubin has emerged as an important protective molecule in the circulation. Early in vitro studies demonstrated that bilirubin is an efficient antioxidant under physiological conditions (Stocker et al., 1987a). Both free and albumin-bound bilirubin scavenges peroxyl radicals and protects low-density lipoprotein against peroxidation (Stocker et al., 1987b; Wu et al., 1996). These findings were subsequently extended to show that bilirubin reacts with other reactive oxygen and nitrogen species including hypochlorous acid, singlet oxygen, hydroxyl radical, nitric oxide, and peroxynitrite (see Stocker, 2004). Indeed, numerous studies have demonstrated a cytoprotective role for bilirubin in response to various oxidative insults (Yamaguchi et al., 1996; Clark et al., 2000; Dore et al., 2009; Wei et al., 2009; Jansen et al., 2010). In addition, bilirubin exerts critical anti-inflammatory actions in the vasculature. It attenuates endothelial expression of leukocyte adhesion molecules and prevents the rolling, adhesion, and infiltration of leukocytes into the vessel wall (Vachharajan et al., 2000; Soares et al., 2004). Given the fundamental role of oxidative stress and inflammation in the pathogenesis of vascular disease, the antioxidant, and anti-inflammatory actions of bilirubin may serve to defend against this disorder. Consistent with this notion, several observational studies have demonstrated an inverse correlation between serum bilirubin levels and coronary artery disease in humans (Schwertner et al., 1994; Hopkins et al., 1996). Similarly, high serum bilirubin is associated with a reduced incidence of atherosclerosis and hypertension (Papadakis et al., 1999; Novotny and Vitek, 2003). Remarkably, the Framingham Offspring Study revealed that men with greater serum bilirubin levels have a lower risk of cardiovascular mortality (Djousse et al., 2001). Furthermore, subjects with mild life-long hyperbilirubinemia arising from Gilbert Syndrome, a genetic disorder where the conjugation and excretion of bilirubin is impaired due to an inactivating polymorphism in the promoter region of the gene coding for UGT1A, results in a significant reduction of risk for cardiovascular death compared to individuals without the syndrome (Hirschfield and Alexander, 2006; Lin et al., 2006).
Preclinical studies also support a beneficial role for bilirubin in vascular disease. Elevations in endogenous circulating levels of bilirubin attenuate hypertension in mice and rats while the exogenous administration of bilirubin ameliorates ischemia–reperfusion injury in various animal models (Pflueger et al., 2005; Ollinger et al., 2007b; Vera et al., 2009). Parenteral treatment with bilirubin improves endothelial function in atherogenic mice (Kawamura et al., 2005). Interestingly, neointimal lesions that develop following carotid artery balloon injury are dramatically reduced in Gunn rats, a congenital model of hyperbilirubinemia arising from defective UGT1A1 activity (Ollinger et al., 2005). Furthermore, local or systemic administration of the bilirubin precursor, biliverdin, suppresses neointima formation following injury of rat carotid arteries (Nakao et al., 2005; Ollinger et al., 2005) while ex vivo application of biliverdin to veins prior to grafting abolishes intimal hyperplasia in a rat arterialized vein-graft model (Nakao et al., 2005). However, it remains to be established whether the exogenous delivery of bilirubin is also able to block intimal thickening following vascular damage. Accordingly, the present study determined whether local administration of bilirubin attenuates neointima formation and the expression of cell cycle regulatory proteins in rat injured carotid arteries. In addition, this study investigated the effect of bilirubin on the proliferation and migration of human arterial smooth muscle cells (SMCs).
Sodium dodecyl sulfate (SDS), Tris, dimethyl sulfoxide (DMSO), penicillin, Tes, streptomycin, neomycin, trypan blue, propidium iodide, L-glutamine, trypsin, RNase, ethylenediaminetetraacetic acid (EDTA), bromophenol blue, and glycerol were from Sigma-Aldrich (St. Louis, MO, USA). M199 media, bovine calf serum, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were from Invitrogen (Carlsbad, CA, USA). Ketamine and xylazine were purchased from Butler Schein Animal Health Corporation (Dublin, OH, USA). Unconjugated bilirubin was from Frontier Scientific (Logan, UT, USA). Antibodies against cyclin D1, cyclin E, cyclin A, p21, p27, p53, and β-actin were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). A polyclonal antibody against HO-1 was from Assay Designs (Ann Arbor, MI, USA) while antibodies against phospho-ERK1/2 and ERK1/2 were from Cell Signaling (Beverly, MA, USA).
Male Sprague Dawley rats (400–500 g) were obtained from Charles River Laboratories, (Wilmington, MA, USA) and maintained on standard rat chow and water ad libitum with 12 h light–dark cycles. All experimental procedures conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996) and were approved by the institutional animal use and care committee. Animals were anesthetized with an intravenous injection of ketamine (100 mg/kg) and xylazine (7.5 mg/kg), and a Fogarty 2F embolectomy catheter (Baxter Healthcare Corporation, Deerfield, IL, USA) was introduced into the external carotid branch through an arteriotomy site and advanced to the aortic arch. The balloon was then inflated and withdrawn three times with rotation along the length of the common carotid artery, as we previously described (Tulis et al., 2001a; Granada et al., 2005; Peyton et al., 2009, 2011). The catheter was then removed, the external carotid artery ligated, overlying tissue sutured, and the skin closed with rodent wound clips. In some cases, a local polymer-based delivery system was used to administer bilirubin to the vessel wall immediately after injury and closure of the external carotid arteriotomy. Bilirubin (1 mg) was dissolved in 50 μL DMSO and mixed into 200 μL of a 25% solution of copolymer gel (Pluronic F-127; BASF Corporation, Mount Olive, NJ, USA). The bilirubin-containing gel was applied topically and circumferentially on the exposed adventitia of the carotid artery. A separate control group of animals received a vehicle (DMSO)-containing gel. At various times after arterial injury, animals were sacrificed by an anesthetic overdose followed by pneumothorax and exsanguination, and carotid arteries collected for analysis.
Following sacrifice, the thoracic aorta was clamped and normal saline was perfused trans-cardially followed by 10% buffered formalin in phosphate-buffered saline. The carotid arteries were then excised, paraffin-embedded, and sections (5 μm) stained with Verhoeff–Van Gieson for measurement of vessel dimensions. Microscopic quantification of vessel dimensions was performed using Image-Pro Plus (Media Cybernetics Incorporated, Bethesda, MD, USA) and Adobe Photoshop software (Adobe Systems, Mountain View, CA, USA) linked through a digital camera (QICAM Fast 1394; Hitschfel Instruments Incorporated) to an Olympus model BX41TF light microscope (Olympus America Inc., Center Valley, PA, USA), as previously described (Tulis et al., 2001a; Granada et al., 2005; Peyton et al., 2009, 2011).
Blood vessels were excised, pulverized in liquid nitrogen, and lysed in electrophoresis buffer [125 mM Tris (pH 6.8), 12.5% glycerol, 2% SDS, and trace bromophenol blue] and proteins separated by SDS-PAGE. Following transfer to nitrocellulose membranes, blots were blocked with PBS and non-fat milk (5%) and then incubated with antibodies directed against cyclin D1 (1:500), cyclin E (1:500), cyclin A (1:500), p27 (1:300), p21 (1:500), p53 (1:100), phospho-ERK(1:250), ERK (1;1000), HO-1 (1:1000), or β-actin (1:200). Blots were washed in PBS, incubated with horseradish peroxidase-conjugated goat anti-rabbit or rabbit anti-goat antibodies and developed with commercial chemiluminescence reagents. Protein expression was quantified by scanning densitometry and normalized with respect to β-actin or ERK1/2.
Human aortic SMCs were obtained commercially (Lonza Inc., Allendale, NJ, USA) and cultured serially in M199 media containing 20% bovine calf serum, 2 mM L-glutamine, 20 mM TES, and 20 mM HEPES, as previously described (Liu et al., 2011b). Culture media were supplemented with 100 units/ml penicillin and streptomycin, and cells propagated in an atmosphere of 95% air – 5% CO2.
Vascular SMCs were seeded (2 × 104 cells) onto six-well plates and grown overnight. The next day cells were incubated with fresh culture media in the absence or presence of bilirubin. Media with appropriate additions were replenished every 2 days. Cell number determinations were made after 4 days by dissociating cells with trypsin (0.05%):EDTA (0.53 mM) and counting cells in a Beckman Z1 Coulter Counter (Beckman Coulter, Fullerton, CA, USA).
Cell cycle progression was monitored by flow-activated cell sorting (Peyton et al., 2009). Vascular SMCs were grown in culture media in the absence or presence of bilirubin for 24 h. Cells were then collected, washed in PBS, pelleted by centrifugation (1000 × g for 5 min), suspended in 70% ethanol, and fixed overnight at 4°C. Fixed cells were then incubated with propidium iodide (50 μg/ml) and RNAse A (100 μg/ml) for 1 h at room temperature. DNA fluorescence was measured in a Becton Dickinson FACScan flow cytometer (Franklin Lakes, NJ, USA) and histograms of DNA content analyzed using Modfit (Verity Software) to determine the fractions of the population in each phase of the cell cycle (Peyton et al., 2009).
Vascular SMC migration was assessed using a scratch wound assay (Peyton et al., 2011). Confluent cell monolayers were scratched with a sterile 200 μl pipette tip and treated with culture media in the presence or absence of bilirubin. Cells were photographed immediately after injury and 18 h later with a digital camera (QImaging QICAM; Hitschfel Instruments, Inc., St. Louis, MO, USA). The wound area was then measured to determine the extent of cell migration.
Vascular cell viability was assessed by measuring the uptake of the membrane-impermeable dye trypan blue. Cells were incubated with trypsin (0.25%), collected, diluted (1:4) with trypan blue, and examined by microscopy. Viability was assessed by the fraction of cells that excluded the dye, as previously reported (Wei et al., 2009).
Mitochondrial function was assessed using the tetrazolium salt MTT, which is efficiently reduced by mitochondrial dehydrogenases to a purple formazan product (Liu et al., 2011a). Cells were incubated with MTT (0.5 mg/ml) at 37°C for 4 h to allow the formation of formazan crystals. Residual MTT was removed and crystals dissolved by incubation with DMSO for 10 min. Absorbance was measured by spectrophotometry at 540 nm wavelength using a plate reader (μQuant spectrophotometer, Bio-Tek Instruments, Winooski, VT, USA). Mitochondrial activity was normalized for cell number and expressed relative to control, untreated cells.
ATP levels were determined using a CellTiter-Glo luciferase/luciferin reaction assay (Promega, Madison, WI, USA). Cells were incubated with the ATP Assay Mix Reagent containing luciferin and luciferase for 10 min at room temperature. Luminescence was measured using a GloMax™ Luminometer (Promega, Madison, WI, USA) and ATP levels normalized for cell number and expressed relative to control, untreated cells.
Results are expressed as mean ± SEM. Statistical analyses were performed with the use of a Student’s two-tailed t-test and an analysis of variance with the Bonferroni post hoc test when more than two treatment regimens were compared. P values < 0.05 were considered statistically significant.
Balloon injury of rat carotid arteries resulted in significant neointima formation. Figure 1A shows representative cross-sections of perfusion-fixed, Verhoeff–Van Gieson-stained tissues obtained from animals 2 weeks after balloon injury. Animals treated with an empty gel exhibited a substantial concentric neointima but a markedly diminished neointima was observed in vessels exposed to a gel containing bilirubin. Morphometric measurements indicate that neointimal area, neointimal thickness, and neointimal/medial wall area ratio were all significantly reduced by bilirubin (Figure 1B). Despite a 65% reduction in neointima/medial wall ratio in bilirubin-treated vessels, medial wall area was unaffected by bilirubin. The decrease in neointima formation following the administration of bilirubin was accompanied by pronounced alterations in the expression of cell cycle regulatory proteins (Figure 2A). Arterial injury resulted in a significant increase in the levels of cyclin D1, cyclin A, and p21 protein, and a decrease in the expression of p27 protein in the vessel wall. However, bilirubin-treated vessels exhibited diminished expression of cyclin D1 and cyclin A protein following arterial injury whereas p27 protein expression was unaffected. Interestingly, bilirubin exposed vessels demonstrated a dramatic increase in the expression of the transcription factor p53, a moderate yet significant increase in p21 expression, and a significant decrease in ERK activation after vessel injury (Figures 2A,B). Arterial injury also caused a pronounced increase in the expression of HO-1 that was unaffected by the administration of bilirubin (Figure 2C).
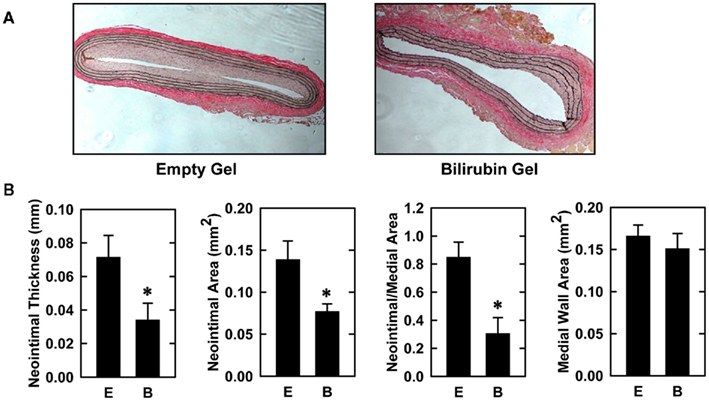
Figure 1. Bilirubin inhibits neointima formation in injured rat carotid arteries. (A) Representative cross-sections of injured carotid arteries treated with an empty gel (E) or a gel containing bilirubin (B) 2 weeks after injury (original magnification, 100×). (B) Morphometric analysis of neointima formation 2 weeks after arterial injury. Results are means ± SEM (n = 7). *Statistically significant effect of bilirubin.
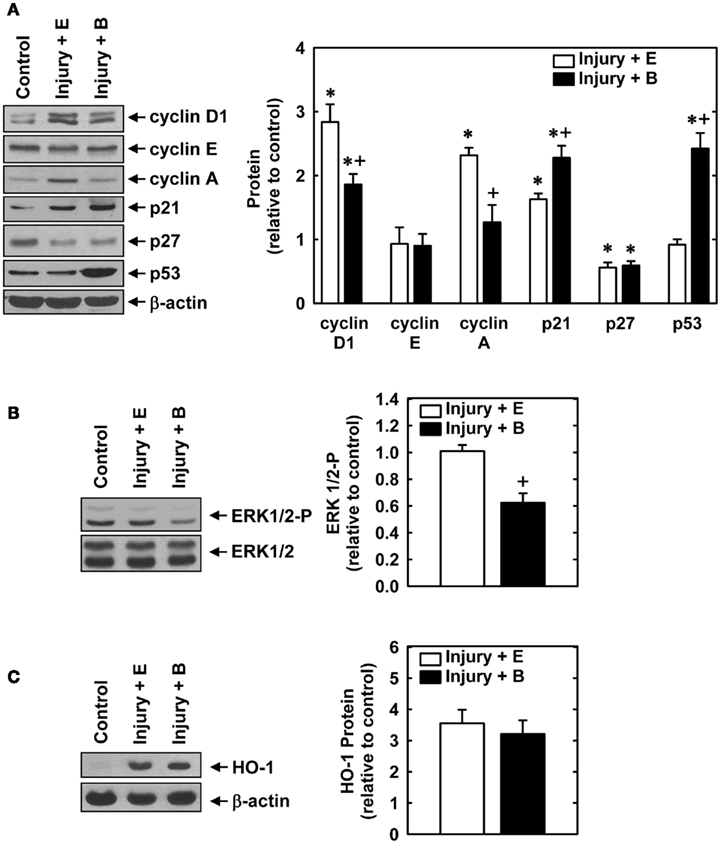
Figure 2. Effect of bilirubin on the expression or phosphorylation of cell cycle regulatory proteins, ERK1/2, or HO-1 following rat carotid artery balloon injury. (A) Expression of cyclin D1, cyclin E, cyclin A, p21, p27, p53, and β-actin protein in control and injured arteries treated with an empty (E) gel or a gel containing bilirubin (B) 2 days after injury. (B) Expression of phospho-ERK1/2 and total ERK1/2 in control and injured arteries treated with an empty (E) gel or a gel containing bilirubin (B) 2 days after injury. (C) Expression of HO-1 in control and injured arteries treated with an empty (E) gel or a gel containing bilirubin (B) 2 days after injury. Quantification of relative protein levels was achieved by scanning densitometry. Results are means ± SEM (n = 3–5). *Statistically significant effect of arterial injury. +Statistically significant effect of bilirubin.
In order to understand the underlying mechanism by which bilirubin inhibits neointima formation, the effect of bilirubin on the function of vascular SMCs was examined. Treatment of human aortic SMCs with bilirubin resulted in a concentration-dependent decrease in cell number beginning at 10 μM (Figure 3A). In contrast, bilirubin had no effect on the viability of SMCs, as assessed by trypan blue exclusion (Figure 3B). The antiproliferative action of bilirubin was associated with the arrest of SMCs in the G0/G1 phase of the cell cycle. Incubation of human aortic SMCs with bilirubin resulted in a concentration-dependent increase in the percentage of cells in the G0/G1 phase of the cell cycle and this was paralleled by a decrease in the fraction of cells in the S and G2M phases of the cell cycle (Figure 4). Finally, bilirubin also retarded the migration of SMCs after scratch wounding. Treatment of human aortic SMCs with bilirubin caused a concentration-dependent inhibition of cell migration starting at a concentration of 20 μM (Figure 5). In contrast, bilirubin had no effect on mitochondrial function or cellular ATP levels (Figures 6A,B).
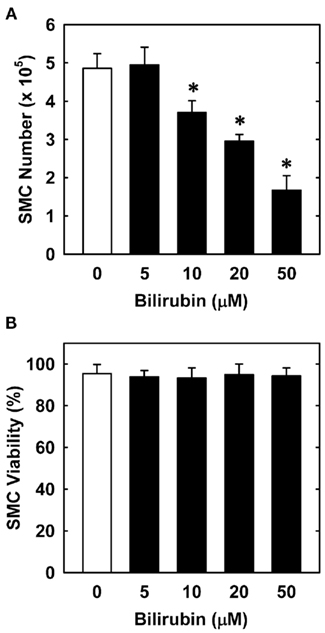
Figure 3. Bilirubin inhibits the proliferation of human vascular SMCs without affecting cell viability. (A) Effect of bilirubin on the proliferation of SMCs. (B) Effect of bilirubin on SMC viability. Cells were treated with serum (20%) in the absence or presence of bilirubin (0–50 μM) for 4 days. Results are means ± SEM (n = 4–5). *Statistically significant effect of bilirubin.
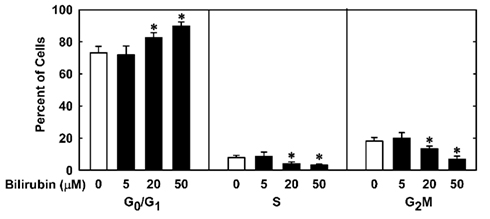
Figure 4. Bilirubin inhibits cell cycle progression of human vascular SMCs. Cells were treated with serum (20%) in the absence or presence of bilirubin (0–50 μM) for 24 h. Results are means ± SEM (n = 4). *Statistically significant effect of bilirubin.
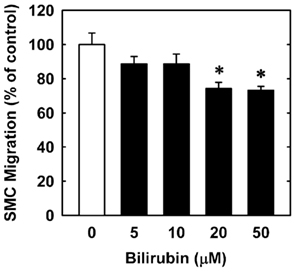
Figure 5. Bilirubin inhibits the migration of human vascular SMCs. Freshly scraped cell monolayers were treated with serum (20%) in the absence or presence of bilirubin (0–50 μM) and cell migration was determined 18 h later. Results are means ± SEM (n = 3). *Statistically significant effect of bilirubin.
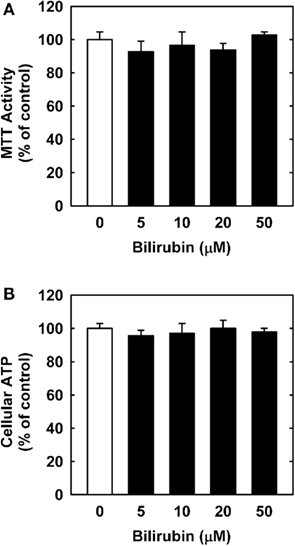
Figure 6. Bilirubin has no effect on mitochondrial function or ATP content in human vascular SMCs. (A) Effect of bilirubin on mitochondrial function. (B) Effect of bilirubin on ATP content. Cells were treated with serum (20%) in the absence or presence of bilirubin (0–50 μM) for 24 h. Results are means ± SEM (n = 3).
The present study identifies bilirubin as a critical regulator of neointima formation following arterial injury. Topical, local administration of bilirubin inhibits intimal thickening after vascular damage and this is associated with alterations in the expression of several cell cycle regulatory proteins. In addition, bilirubin blocks the proliferation and migration of human arterial SMCs in a concentration-dependent manner that is associated with the arrest of vascular SMCs in the G0/G1 phase of the cell cycle, independent of any change in cell viability, mitochondrial function, or intracellular ATP concentration. These findings underscore a potentially important therapeutic role for bilirubin in ameliorating occlusive vascular disease.
The current study is the first to demonstrate that exogenous application of bilirubin attenuates vascular remodeling following arterial injury. Bilirubin was applied topically to the adventitia of blood vessels using a specific local copolymer that has been successfully employed by our laboratory and others (Hu et al., 1999; Granada et al., 2005; Peyton et al., 2009, 2011). This drug delivery approach allows for the sustained release of drugs while circumventing possible non-specific or toxic effects associated with the systemic administration of drugs. This may be especially relevant in the case of bilirubin where high circulating levels of the bile pigment can lead to neurotoxicity (Ostrow et al., 2004). We found that local administration of bilirubin markedly suppresses neointima formation following arterial injury, demonstrating that this bile pigment is capable of blocking the vascular response to injury. The attenuation of lesion formation by bilirubin is associated with diminished expression of the G1 cyclins, cyclin D1 and A, which are critical for entry and progression of cells through S-phase of the cell cycle and DNA synthesis (Sherr, 1994). Interestingly, bilirubin also reduces ERK activity in injured blood vessels. Since the activation of ERK is linked with the induction of cyclin D1 expression following mitogenic stimulation (Weber et al., 1997; Villanueva et al., 2007), the ability of bilirubin to inhibit ERK activity may contribute to the downregulation of cyclin D1 expression observed with this bile pigment after arterial injury. In addition, bilirubin induces the expression of the cyclin-dependent kinase inhibitor p21 and the transcription factor p53, which are established inhibitors of SMC replication and neointimal hyperplasia (Yang et al., 1996; Sanz-Gonzalez et al., 2007). Since p53 has recently been demonstrated to directly induce the expression of HO-1 (Nam and Sabapathy, 2011), which is a key modulator of vascular remodeling (Aizawa et al., 1999; Togane et al., 2000; Duckers et al., 2001; Tulis et al., 2001a), we examined whether bilirubin-mediated increases in p53 expression in damaged blood vessels are associated with elevations in HO-1 expression. Consistent with our previous work (Tulis et al., 2001b), we found that arterial injury results in a marked elevation in HO-1 protein expression. However, this arterial injury-induced rise in HO-1 protein is unaffected by the administration of bilirubin despite a prominent increase in p53 expression, reinforcing the notion that the ability of p53 to stimulate HO-1 gene expression is both stimulus- and cell-specific (Yu et al., 1999; Nam and Sabapathy, 2011). Our in vivo findings showing the bilirubin disrupts ERK activity and the expression of cell cycle regulatory proteins following vascular injury are in-line with earlier studies in cultured vascular SMCs and suggest that bilirubin retards injury-induced arterial lesion formation by repressing cell cycle progression and SMC proliferation (Ollinger et al., 2005, 2007c; Stoeckius et al., 2012).
The present study also found that bilirubin is a robust inhibitor of human vascular SMC growth. Bilirubin blocks the proliferation of human aortic SMCs in a concentration-dependent manner. Significantly, bilirubin inhibits vascular SMC growth in the absence of cell death indicating that bilirubin acts via a cytostatic rather than cytotoxic mechanism. Furthermore, analysis of cell cycle distribution reveals that bilirubin inhibits cell cycle progression by specifically arresting human aortic SMCs in the G0/G1 phase of the cell cycle. These findings complement previous reports showing that physiologically relevant concentrations of bilirubin attenuates cell cycle progression and DNA synthesis in arterial SMCs derived from different animal species or blood vessels (Ollinger et al., 2005, 2007c; Stoeckius et al., 2012). Growth inhibitory effects of bilirubin have also been reported in human airway smooth muscle and various tumor cell lines demonstrating that the antimitogenic effect of this bile pigment is not unique to vascular SMCs (Taille et al., 2003; Rao et al., 2006; Ollinger et al., 2007a). The antiproliferative action of bilirubin likely involves multiple mechanisms. Consistent with our in vivo findings, bilirubin inhibits the activity of ERK, a crucial kinase involved in S-phase entry, in cultured vascular SMCs (Stoeckius et al., 2012). Moreover, inhibition of ERK activation by bilirubin has been implicated in its ability to block airway SMC growth (Taille et al., 2003). However, the transcription factor p53 appears to play an essential role in mediating the antiproliferative action of bilirubin in vascular SMCs. We found that bilirubin is a potent inducer of p53 expression in injured blood vessels. In addition, bilirubin promotes the expression of p53 in numerous cell types and deletion of p53 abrogates the antiproliferative action of bilirubin in murine SMCs (Ollinger et al., 2007a,c). Interestingly, p53 also stimulates apoptotic signaling pathways and may contribute to the apoptotic actions of bilirubin in SMCs grown in serum-free or serum-restricted conditions (Liu et al., 2002; Ollinger et al., 2007c).
Since vascular SMC proliferation and migration are tightly coupled processes (Fukui et al., 2000), we also investigated whether bilirubin influences vascular SMC migration. Indeed, we found that bilirubin inhibits human aortic SMC migration in a concentration-dependent manner. However, the blockade of SMC migration is not as pronounced as the inhibition of SMC proliferation and is only observed at high bilirubin concentrations. Failure of low concentrations of bilirubin to repress SMC migration is consistent with a recently published study in rat aortic SMCs (Rodriguez et al., 2010). Although the mechanism by which bilirubin attenuates SMC migration is not known it may also involve the induction of p53 since this transcription factor has been demonstrated to block SMC migration (Rodriguez-Compos et al., 2001). In addition, p21 may also contribute to the antimigratory action of bilirubin since overexpression of p21 in vascular SMCs has been shown to inhibit their migration (Fukui et al., 1997). However, it does not appear that alterations in mitochondrial function or ATP production contribute to the antiproliferative or antimigratory effect of bilirubin.
The ability of bilirubin to inhibit neointima formation by blocking vascular SMC proliferation and migration identifies this bile pigment as an attractive agent in preventing stenosis of blood vessels following surgical interventions to treat atherosclerosis. In particular, perivascular application of bilirubin to veins or arteries prior to or immediately after grafting may promote graft survival following coronary artery bypass surgery. Alternatively, the use of bilirubin-eluting stents may reduce the incidence of restenosis following percutaneous coronary balloon angioplasty. Interestingly, a recent clinical study found that serum bilirubin is an independent predictor of coronary in-stent stenosis (Kuwano et al., 2011). Patients exhibiting elevated levels of bilirubin have a lower rate of stenosis compared to patients with low serum bilirubin, providing additional support for the use of bilirubin-coated stents in patients undergoing coronary stenting. Finally, since clinical studies show that elevations in serum bilirubin protect against coronary and peripheral artery disease and improve clinical outcomes following surgical interventions to treat vascular disease, the development of pharmacological inhibitors that raise circulating levels of bilirubin may provide a useful strategy in preventing occlusive vascular disorders (Schwertner et al., 1994; Hopkins et al., 1996; Perlstein et al., 2008; Kuwano et al., 2011). In this regard, the design of specific pharmacological inhibitors that target the conjugating enzyme UGTTA1 offers a promising approach in raising endogenous bilirubin levels (Vera et al., 2009; Durante, 2010).
In summary, we demonstrated that bilirubin inhibits neointima formation after arterial injury in the rat and the proliferation and migration of human SMCs. The antiproliferative and antimigratory actions of bilirubin are concentration-dependent and associated with the arrest of SMCs in the G0/G1 phase of the cell cycle. Thus, local delivery of bilirubin to blood vessels or augmentation of endogenous circulating levels of bilirubin represent promising therapeutic approaches in treating arterial disease.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the National Institutes of Health National Heart, Lung and Blood Institute Grant R01-HL059976 (William Durante) and R01-HL081720 (David A. Tulis).
Aizawa, T., Ishizaka, N., Taguchi, J., Kimura, S., Kurokawa, K., and Ohno, M. (1999). Balloon injury does not induce heme oxygenase-1 gene expression, but administration of hemin inhibits neointimal formation in balloon injured rat carotid arteries. Biochem. Biophys. Res. Commun. 261, 302–307.
Clark, J. E., Foresti, R., Green, C. J., and Motterlini, R. (2000). Dynamics of haem oxygenase-1 expression and bilirubin production in cellular protection against oxidative stress. Biochem. J. 348, 615–619.
Djousse, L., Levy, D., Cupples, L. A., Evans, J. C., D’Agostino, R. B., and Ellison, R. C. (2001). Total serum bilirubin and risk of cardiovascular disease in the Framingham offspring study. Am. J. Cardiol. 87, 1196–1200.
Dore, S., Takahashi, M., Ferris, C. D., Zakhary, R., Hester, L. D., Guastella, D., and Snyder, S. H. (2009). Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 96, 2445–2450.
Duckers, H. J., Boehm, M., True, A. L., Yet, S.-F., Park, J. L., Webb, R. C., Lee, M.-E., Nabel, G. J., and Nabel, E. G. (2001). Heme oxygenase-1 protects against vascular constriction and proliferation. Nat. Med. 7, 693–698.
Durante, W. (2010). Targeting heme oxygenase-1 in vascular disease. Curr. Drug Targets 11, 1504–1516.
Fukui, R., Amakawa, M., Hoshiga, M., Shibata, N., Kohbayashi, E., Seto, M., Sasaki, Y., Ueno, T., Negoro, N., Nakakoji, T., Li, M., Nishiguchi, F., Ishihara, T., and Ohsawa, N. (2000). Increased migration in late G1 phase in cultured smooth muscle cells. Am. J. Physiol. 279, C999–C1007.
Fukui, R., Shibata, N., Kohbayashi, E., Amakawa, M., Furutama, D., Hoshiga, M., Negoro, N., Nakakouji, T., Ii, M., Ishitara, T., and Ohsawa, N. (1997). Inhibition of smooth muscle cell migration by the p21 cyclin-dependent kinase inhibitor (Cip1). Atherosclerosis 132, 53–59.
Granada, J. F., Ensenat, D., Keswani, A. N., Kaluza, G. L., Raizner, A. E., Liu, X. M., Peyton, K. J., Azam, M. A., Wang, H., and Durante, W. (2005). Single perivascular delivery of mitomycin C stimulates p21 expression and inhibits neointima formation in rat arteries. Arterioscler. Thromb. Vasc. Biol. 25, 2343–2348.
Hirschfield, G. M., and Alexander, G. J. (2006). Gilbert’s syndrome: an overview for clinical biochemists. Ann. Clin. Biochem. 43, 340–343.
Hopkins, P. N., Wu, L. L., Hunt, S. C., James, B. C., Vincent, G. M., and Williams, R. R. (1996). Higher serum bilirubin is associated with decreased risk for early familial coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 16, 250–255.
Hu, Y., Zou, Y., Dietrich, H., Wick, G., and Xu, Q. (1999). Inhibition of neointima hyperplasia in mouse vein grafts by locally applied suramin. Circulation 100, 861–868.
Institute of Laboratory Animal Resources. (1996). Guide for the Care and use of Laboratory Animals. Washington: National Academy Press.
Jansen, T., Hortmann, M., Oelze, M., Opitz, B., Steven, S., Schell, R., Knorr, M., Karbach, S., Wenzel, P., Munzel, T., and Daiber, A. (2010). Conversion of biliverdin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1-evidence for direct and indirect antioxidant actions of biliverdin. J. Mol. Cell. Cardiol. 49, 186–195.
Kawamura, K., Ishikawa, K., Wada, Y., Kimura, S., Matsumoto, H., Kohro, T., Itabe, H., Kodama, T., and Maruyama, Y. (2005). Bilirubin from heme oxygenase-1 attenuates vascular endothelial activation and dysfunction. Arterioscler. Thromb. Vasc. Biol. 25, 155–160.
Kuwano, T., Miura, S., Shirai, K., Ike, A., Mori, K., Shimizu, T., Zhang, B., Iwata, A., Nishikawa, H., Kawamura, A., and Saku, K. (2011). Serum levels of bilirubin as an independent predictor of coronary in-stent restenosis: a new look at an old molecule. J. Atheroscler. Thromb. 18, 574–583.
Lin, J. P., O’Donnell, C. J., Schwaiger, J. P., Cupples, L. A., Lingenhel, A., Hunt, S. C., Yang, S., and Kronenberg, F. (2006). Association between the UGT1A1*28 allele, bilirubin levels, and coronary heart disease in the Framingham Heart Study. Circulation 114, 1476–1481.
Liu, X. M., Chapman, G. B., Wang, H., and Durante, W. (2002). Adenovirus-mediated heme oxygenase-1 gene expression stimulates apoptosis in vascular smooth muscle cells. Circulation 105, 79–84.
Liu, X. M., Peyton, K. J., Shebib, A. R., Wang, H., and Durante, W. (2011a). Compound C stimulates heme oxygenase-1 gene expression via the Nrf2-ARE pathway to preserve human endothelial cell survival. Biochem. Pharmacol. 82, 371–379.
Liu, X. M., Peyton, K. J., Shebib, A. R., Wang, H., Korthuis, R. J., and Durante, W. (2011b). Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am. J. Physiol. Heart Circ. Physiol. 300, H84–H93.
Nakao, A., Murase, N., Ho, C., Toyokawa, H., Billiar, T. R., and Kanno, S. (2005). Biliverdin administration prevents the formation of intimal hyperplasia induced by vascular injury. Circulation 112, 587–591.
Nam, S. Y., and Sabapathy, K. (2011). P53 promotes cellular survival in a context-dependent manner by directly inducing the expression of haeme-oxygenase-1. Oncogene 30, 4476–4486.
Novotny, L., and Vitek, L. (2003). Inverse relationship between serum bilirubin and atherosclerosis in men: a meta-analysis of published studies. Exp. Biol. Med. (Maywood) 228, 568–571.
Ollinger, R., Bilban, M., Erat, A., Froio, A., McDaid, J., Tyagi, S., Csizmadia, E., Graca-Souza, A. V., Liloia, A., Soares, M. P., Otterbein, L. E., Usheva, A., Yamashita, K., and Bach, F. H. (2005). Bilirubin; a natural inhibitor of vascular smooth muscle cell proliferation. Circulation 112, 1030–1039.
Ollinger, R., Kogler, P., Troppmair, J., Hermann, M., Wurm, M., Drasche, A., Konigsrainer, I., Amberger, A., Weiss, H., Ofner, D., Bach, F. H., and Margreiter, R. (2007a). Bilirubin inhibits tumor cell growth via activation of ERK. Cell Cycle 6, 3078–3085.
Ollinger, R., Wang, H., Yamashita, K., Wegiel, B., Thomas, M., Margreiter, R., and Bach, F. H. (2007b). Therapeutic applications of bilirubin and biliverdin in transplantation. Antioxid. Redox Signal. 9, 2175–2185.
Ollinger, R., Yamashita, K., Bilban, M., Erat, A., Kogler, P., Thomas, M., Csizmadia, E., Usheva, A., Margreiter, R., and Bach, F. H. (2007c). Bilirubin and bilirubin treatment of atherosclerotic disease. Cell Cycle 6, 39–43.
Ostrow, J. D., Pascolo, L., Brites, D., and Tiribelli, C. (2004). Molecular basis of bilirubin-induced neurotoxicity. Trends Mol. Med. 10, 65–70.
Papadakis, J. A., Ganotakis, E. S., Jagroop, I. A., Mikhailidis, D. P., and Winder, A. F. (1999). Effect of hypertension and its treatment on lipid, lipoprotein(a), fibrinogen, and bilirubin levels in patients referred for dyslipidemia. Am. J. Hypertens. 12, 673–681.
Perlstein, T. S., Pande, R. L., Beckman, J. A., and Creager, M. A. (2008). Serum total bilirubin level and prevelant lower-extremity peripheral artery disease. Arterioscler. Thromb. Vasc. Biol. 28, 166–172.
Peyton, K. J., Ensenat, D., Azam, M. A., Keswani, A. N., Kannan, S., Liu, X. M., Wang, H., Tulis, D. A., and Durante, W. (2009). Arginase promotes neointima formation in rat injured carotid arteries. Arterioscler. Thromb. Vasc. Biol. 29, 488–494.
Peyton, K. J., Yu, Y., Yates, B., Shebib, A. R., Liu, X. M., Wang, H., and Durante, W. (2011). Compound C inhibits vascular smooth muscle cell proliferation and migration in an AMP-activated protein kinase-independent fashion. J. Pharmacol. Exp. Ther. 338, 476–484.
Pflueger, A., Croatt, A. J., Peterson, T. E., Smith, L. A., d’Uscio, L. V., Katusic, Z. S., and Nath, K. A. (2005). The hyperbilirubinemic Gunn rat is resistant to the pressor effects of angiotensin II. Am. J. Physiol. Renal Physiol. 288, F552–F558.
Rao, P., Suzuki, R., Mizobuchi, S., Yamaguchi, T., and Sasaguri, S. (2006). Bilirubin exhibits a novel anti-cancer effect on human adenocarcinoma. Biochem. Biophys. Res. Commun. 342, 1279–1283.
Rodriguez, A. I., Gangopadhyay, A., Kelley, E. E., Pagano, P. J., Zuckerbraun, B. S., and Bauer, P. M. (2010). HO-1 and CO decrease platelet-derived growth factor-induced vascular smooth muscle cell migration via inhibition of Nox1. Arterioscler. Thromb. Vasc. Biol. 30, 98–104.
Rodriguez-Compos, A., Ruiz-Enriquez, P., Faraudo, S., and Badimon, L. (2001). Mitogen-induced p53 downregulation precedes vascular smooth muscle cell migration from healthy tunica media and proliferation. Arteriosler. Thromb. Vasc. Biol. 21, 214–219.
Sanz-Gonzalez, S. M., Barquin, L., Garcia-Cao, I., Roque, M., Gonzalez, J. M., Fuster, J. J., Castells, M. T., Flores, J. M., Serrano, M., and Andres, V. (2007). Increased p53 gene dosase reduced neointimal thickening induced by mechanical injury but has no effect on native atherosclerosis. Cardiovasc. Res. 75, 803–812.
Schwertner, H. A., Jackson, W. G., and Tolan, G. (1994). Association of low serum concentration of bilirubin with increased risk of coronary artery disease. Clin. Chem. 40, 18–23.
Soares, M. P., Seldon, M. P., Gregoire, I. P., Vssilevskaia, T., Berberat, P. O., Yu, J., and Bach, F. H. (2004). Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J. Immunol. 172, 3553–3563.
Stocker, R., Glazer, A. N., and Ames, B. N. (1987a). Antioxidant activity of albumin-bound bilirubin. Proc. Natl. Acad. Sci. U.S.A. 84, 5918–5922.
Stocker, R., Yamamato, Y., McDonagh, A. F., Glazer, A. N., and Ames, B. N. (1987b). Bilirubin is an antioxidant of possible physiological importance. Science 235, 1043–1046.
Stoeckius, M., Erat, A., Fujikawa, T., Hiromura, M., Koulova, A., Otterbein, L., Bianchi, C., Tobiasch, E., Dagon, Y., Sellke, F. W., and Usheva, A. (2012). Essential roles of Raf/ERK/MAPK pathway,YY1, and Ca2+ influx in the growth arrest of human vascular smooth muscle cells by bilirubin. J. Biol. Chem. doi:10.1074/jbc.M111.266510
Taille, C., Almolki, A., Benhamed, M., Zedda, C., Megret, J., Berger, P., Leseche, G., Fadel, E., Yamaguchi, T., Marthan, R., Aubier, M., and Boczkowski, J. (2003). Hemeoxygenase inhibits human airway smooth muscle proliferation via a bilirubin-dependent modulation of ERK1/2 phosphorylation. J. Biol. Chem. 278, 27160–27168.
Togane, Y., Toshisuki, M., Suematsu, M., Ishimura, Y., Yamazaki, J., and Katayama, S. (2000). Protective roles of endogenous carbon monoxide in neointimal development elicited by arterial injury. Am. J. Physiol. Heart Circ. Physiol. 278, H623–H632.
Tulis, D. A., Durante, W., Peyton, K. J., Evans, A. J., and Schafer, A. I. (2001a). Adenovirus-mediated heme oxygenase-1 gene delivery inhibits injury-induced vascular neointima formation. Circulation 104, 2710–2715.
Tulis, D. A., Durante, W., Peyton, K. J., Evans, A. J., and Schafer, A. I. (2001b). Heme oxygenase-1 attenuates vascular remodeling following balloon injury in rat carotid arteries. Atherosclerosis 155, 113–122.
Vachharajan, T. J., Work, J., Issekutz, A. C., and Granger, D. N. (2000). Heme oxygenase modulates selectin expression in different vascular beds. Am. J. Physiol. Heart Circ. Physiol. 278, H1613–H1617.
Vera, T., Granger, J. P., and Stec, D. E. (2009). Inhibition of bilirubin metabolism induces moderate hyperbilirubinemia and attenuates angiotensin II-dependent hypertension in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R738–R743.
Villanueva, J., Yung, Y., Walker, J. L., and Assoian, R. K. (2007). ERK activity and G1 phase progression: identifying dispensable versus essential activities and primary versus secondary targets. Mol. Biol. Cell 18, 1457–1463.
Weber, J. D., Raben, D. M., Phillips, P. J., and Baldassare, J. J. (1997). Sustained activation of extracellular-signal regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem. J. 326, 61–68.
Wei, Y., Liu, X. M., Peyton, K. J., Wang, H., Johnson, F. K., Johnson, R. A., and Durante, W. (2009). Hypochlorous acid-induced heme oxygenase-1 gene expression promotes endothelial cell survival. Am. J. Physiol. Cell Physiol. 297, C907–C915.
Wu, T., Fung, K. P., Wu, J., Yang, C. C., and Weisel, R. D. (1996). Antioxidation of human low density lipoprotein by unconjugated and conjugated bilirubins. Biochem. Pharmacol. 51, 859–862.
Yamaguchi, T., Terakado, M., Horio, F., Aoki, K., Tanaka, M., and Nakajima, H. (1996). Role of bilirubin as an antioxidant in an ischemia-reperfusion of rat liver and induction of heme oxygenase. Biochem. Biophys. Res. Commun. 223, 129–135.
Yang, Z. Y., Simari, R. D., Perkins, N. D., San, H., Gordon, D., Nabel, G. J., and Nabel, E. G. (1996). Role of the p21 cyclin-dependent kinase inhibitor in limiting intimal cell proliferation in response to arterial injury. Proc. Natl. Acad. Sci. U.S.A. 93, 7905–7910.
Keywords: bilirubin, stenosis, vascular smooth muscle cells, proliferation, migration
Citation: Peyton KJ, Shebib AR, Azam MA, Liu X-m, Tulis DA and Durante W (2012) Bilirubin inhibits neointima formation and vascular smooth muscle cell proliferation and migration. Front. Pharmacol. 3:48. doi: 10.3389/fphar.2012.00048
Received: 11 January 2012; Accepted: 04 March 2012;
Published online: 22 March 2012.
Edited by:
Jaime Kapitulnik, The Hebrew University of Jerusalem, IsraelReviewed by:
Giovanni E. Mann, King’s College London, UKCopyright: © 2012 Peyton, Shebib, Azam, Liu, Tulis and Durante. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: William Durante, Department of Medical Pharmacology and Physiology, University of Missouri-Columbia School of Medicine, M409 Medical Sciences Building, One Hospital Drive, Columbia, MO 65212, USA. e-mail:durantew@health.missouri.edu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.