


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 14 March 2012
Sec. Drug Metabolism and Transport
Volume 3 - 2012 | https://doi.org/10.3389/fphar.2012.00045
This article is part of the Research Topic The role of bile pigments in health and disease: effects on cell signaling, cytotoxicity and cytoprotection View all 20 articles
Oxidative stress is detrimental to life process and is particularly responsible for aging and age-related diseases. Thus, most organisms are well equipped with a spectrum of biological defense mechanisms against oxidative stress. The major efficient antioxidative mechanism is the glutathione system, operating a redox cycling mechanism for glutathione utilization, which consists of glutathione and its peroxidase and reductase. However, this system is mainly effective for hydrophilic oxidants, while lipophilic oxidants require another scavenging system. Since many age-related pathological conditions are related to lipid peroxidation, especially in association with the aging process, the physiological role of the scavenging system for lipophilic oxidants should be considered. In this regard, the biliverdin to bilirubin conversion pathway, via biliverdin reductase (BVR), is suggested to be another major protective mechanism that scavenges lipophilic oxidants because of the lipophilic nature of bilirubin. The efficiency of this bilirubin system might be potentiated by operation of the intertwined bicyclic systems of the suggested redox metabolic cycle of biliverdin and bilirubin and the interactive control cycle of BVR and heme oxygenase. In order to combat oxidative stress, both antioxidative systems against hydrophilic and lipophilic oxidants are required to work cooperatively. In this regard, the roles of the bilirubin system in aging and age-related diseases are reassessed in this review, and their interacting networks are evaluated.
Oxidative stress is the result of an imbalance between generation and scavenging of reactive oxygen species (ROS). Old cells have higher levels of ROS than young cells (Hagen et al., 1997; Lee et al., 1999). Senescent states of cells can be readily induced by sub-lethal doses of pro-oxidants (Chen and Ames, 1994; Chen et al., 1998). Thus, any adjustment of the ROS balance is expected to prevent or restore cellular senescence.
Biliverdin reductase (BVR) is an evolutionarily conserved enzyme, converting biliverdin to bilirubin, the potent physiologic antioxidant (Schluchter and Glazer, 1997; Baranano et al., 2002; Sedlak et al., 2009). Bilirubin protects cells against high concentrations of hydrogen peroxide (H2O2), for which a redox cycle that amplifies the oxidation of bilirubin into biliverdin, which is then recycled back into bilirubin by BVR, has been proposed (Baranano et al., 2002; Sedlak and Snyder, 2004). Biliverdin is a cleavage product of heme by heme oxygenase (HO). The HOs are heat-shock protein-32 family proteins, comprising a constitutive isoform (HO-2) and an inducible isoform (HO-1). Since BVR can regulate HO-1/HO-2 expression, these intertwined relationships among enzymes of HO and BVR and their metabolites, biliverdin, and bilirubin, may potentiate the efficiency of the cellular physiological defense capacity against oxidative stress. Therefore, it is necessary to assess the physiological role of BVR and the heme degradation pathway in oxidative stress-related phenomena and related diseases. Furthermore, it has recently been reported that knock-down of BVR could induce cellular senescence (Kim et al., 2011a). This relationship of BVR with the aging process, as well as other oxidative stress-associated disorders, led us to consider its roles in age-related diseases as well.
The HO/BVR pathway is the main process for heme degradation and is evolutionarily conserved for control of oxidative stress. Two isozymes of BVR have been characterized in humans: the fetal form, biliverdin reductase B, and the adult form, biliverdin reductase A. Biliverdin reductase A (the major enzyme and hereafter, abbreviated as BVR) is an abundant and ubiquitously expressed enzyme with a high turnover rate. BVR can protect cells against oxidative stress by two different mechanisms: by converting biliverdin to bilirubin and by regulating HO expression (Kravets et al., 2004; Sedlak and Snyder, 2004; Ding et al., 2011). Bilirubin has a strong antioxidant activity, protecting cells against 10- to 1000-fold higher concentrations of H2O2 (Sedlak et al., 2009). This high efficiency of bilirubin as a potent antioxidant is proposed to be amplified by the cyclic conversion of bilirubin and biliverdin: the reduction of biliverdin to bilirubin by BVR and the oxidation of bilirubin to biliverdin by lipophilic ROS (Baranano et al., 2002; Sedlak and Snyder, 2004). Despite the nature of recycling bilirubin to biliverdin by peroxyradicals has been questioned recently, the strong antioxidant effects of biliverdin and bilirubin are evident (McDonagh, 2010).
The major antioxidative defense system in vivo is the glutathione redox cycling system, comprised of glutathione and its two redox enzymes, glutathione peroxidase and glutathione reductase. This system is primarily effective for hydrophilic ROS, because of the hydrophilic nature of glutathione. In contrast, the biliverdin and bilirubin pathway is expected to be effective against lipophilic ROS, because of the lipophilic nature of bilirubin. Therefore, this biliverdin to bilirubin pathway system is presumed to be complementary to the glutathione redox cycling system for scavenging both hydrophilic and lipophilic ROS.
The heme degradation pathway provides two enzymatic antioxidants (i.e., HO and BVR) and two hydrophobic antioxidants (i.e., bilirubin and biliverdin) that contribute to the powerful antioxidative mechanism of an organism. The lipophilic nature of bilirubin alone would provide specific protective activity against lipid soluble metabolites, such as stability of phospholipids in multi-lamellar liposomes and rat liver microsomes (Bliuger et al., 1985; Stocker and Ames, 1987; Stocker et al., 1990). Exogenous bilirubin can reduce lipid peroxidation in the heart and kidney and can protect thymus cells against ultraviolet- or sphingosine-mediated apoptosis (Dudnik et al., 2001). Both unconjugated and conjugated bilirubin can protect low density lipoprotein (LDL) against peroxidative attack by scavenging peroxyl radicals (Wu et al., 1996).
Besides its function as a reductase, BVR has pleiotropic functions in cell signaling, cell metabolism, and gene control. BVR is a dual-specificity (serine/threonine/tyrosine) kinase, involved in various cellular functions (Maines, 2005; Kapitulnik and Maines, 2009). One of the pathways modulated by BVR is the insulin signaling pathway. BVR is a substrate for insulin receptor tyrosine kinase (IRK) activity as well as a kinase for serine phosphorylation of insulin receptor substrate-1 (IRS-1; Lerner-Marmarosh et al., 2005; Wu et al., 2008; Maines, 2010). BVR can also enhance phosphatidyl inositol 3-kinase (PI3K)/Akt activity by binding to SH2 domains, which has been considered a new mechanism of insulin resistance (Lerner-Marmarosh et al., 2005). In addition, BVR plays a role as a carrier protein for nuclear signal transduction, as it has a nuclear localization signal within the carboxy terminal end of its reductase domain. Since BVR has mitogen activated protein kinase (MAPK) docking consensus sequences, it interacts with the MAPK family, in particular, the extracellular signal-regulated kinases 1/2 (ERK1/2), and functions as a nuclear transporter of ERK (Lerner-Marmarosh et al., 2008). BVR is activated and localizes into the nucleus in response to various stress signals, such as bacterial lipopolisaccharide (LPS) and bromobenzene (Maines et al., 2001). In the nucleus, BVR, being a leucine zipper-like DNA binding protein, binds not only to activator protein-1 (AP-1) but also to cyclic adenosine monophosphate (cAMP) response element sites (Kravets et al., 2004; Tudor et al., 2008). Thereby, BVR can bind to the HO-1 promoter, an AP-1 regulated gene, regulating HO-1 induction in response to oxidative stress (Tudor et al., 2008). BVR is also involved in the inflammatory response via adjusting the levels of PI3K and Akt (Wegiel et al., 2009).
Since the structure of polyunsaturated fatty acids (PUFAs) is of a bis-allylic nature, lipids are prone to oxidation. Once lipid peroxidation is initiated, a chain reaction is propagated until terminal products are produced. Peroxyl radicals are rearranged via cyclic reactions to endoperoxides (Marnett, 1999). Scission of the oxidized PUFA results in formation of two aldehyde products: phospholipid aldehydes, such as oxidized phosphatidylcholine (OxPC), and α,β-unsaturated aldehyde cleavage fragments, including malondialdehyde (MDA), 4-hydroxynon-enal (HNE), and 4-oxo-2-non-enal (ONE; Kadl et al., 2004; Adibhatla and Hatcher, 2010). Compared to free radicals, the aldehydes, such as MDA, 4-HNE, and other aldehydes, are moderately stable and can diffuse within the cell or be extruded extracellularly to attack distant targets. They show a very high reactivity toward biomolecules, such as proteins, phospholipids and DNA, leading to a variety of intramolecular and intermolecular covalent adducts. At the membrane level, proteins and amino lipids can be covalently modified by lipid peroxidation products, resulting in damages of membrane structure. In addition, these aldehydes can also act as bioactive molecules in physiological and/or pathological events (Catalá, 2009). Therefore, the cumulative damages of these aldehyde products deteriorate cell integrity and function which may eventually result in cellular senescence. Higher levels of lipid peroxidation have been observed in plasma and tissues of aged organisms, including liver, brain, lung, kidney, and muscle (Bourre, 1988; Mizuno, 1990; Pansarasa et al., 1999; Poon et al., 2004; Ward et al., 2005; Gil et al., 2006).
Cardiovascular diseases (CVD), a family of diseases that include atherosclerosis, coronary heart disease, hypertension, and stroke, are the main causes of death in developed countries. The thickening and stiffening of the arteries in atherosclerosis are due to fatty plaques and mineral deposits, resulting in a shortage of blood supply to the myocardium. Lipid peroxidation markers, such as oxidized low density cholesterol, MDA, 4-HNE, dienoic acid, hydroxyoctadecadienoic acids, and oxidized cholesterol linoleate, are increased in the plaques and blood of atherosclerotic patients (Kuhn et al., 1992; Ahotupa and Asankari, 1999; Valko et al., 2007; Ravandi et al., 2011). A progressive increase of lipid peroxidation in the heart is also observed with aging (Miro et al., 2000). Oxidative stress in cardiac and vascular myocytes has been suggested to be linked with cardiovascular tissue injury (Dhalla et al., 2000). Lipid peroxidation and protein carbonylation lead to disruption of the membrane lipid bilayer and functional deterioration of cellular proteins, eventually resulting in abnormalities of subcellular organelles (Kaneko et al., 1989; Stoyanovsky et al., 1997; Molavi and Mehta, 2004; Valko et al., 2007). In addition, the significant pooling of iron in atherosclerotic lesions implies that the iron-catalyzed formation of free radicals may take place in the development process of atherosclerosis (Yuan and Li, 2003). Therefore, the role of lipophilic antioxidative mechanism for preventing age-related CVD should be considered and evaluated.
The brain is vulnerable to oxidative damage due to its high lipid content, high oxygen consumption, high concentration of redox-active metals (e.g., Cu and Fe), and relatively low antioxidant capacity. Therefore, this organ is susceptible to lipid peroxidation (Markesbery and Lovell, 2007). As a consequence, the increased oxidative stress that occurs with aging in the brain may be responsible for the pathogenesis of age-related neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD) Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), Niemann–Pick’s disease (NPC), and brain traumas (Klein and Ackerman, 2003; Adibhatla and Hatcher, 2007; Zhu et al., 2007). AD, a neurodegenerative disorder of cognitive and memory decline, is represented by a marked accumulation of amyloid-β peptide (Aβ), the main constituent of senile plaques, and deposition of neurofibrillary tangles (Butterfield and Lauderback, 2002; Butterfield et al., 2006). Formation of Aβ is partially induced by oxidative stress, as illustrated by the increased lipid peroxidation, the decreased PUFA content, and the increased MDA and 4-HNE in AD ventricular fluid (Butterfield and Lauderback, 2002; Butterfield et al., 2006; Recuero et al., 2010). The increased lipid peroxidation precedes amyloid plaque formation in an AD animal model (Pratico et al., 2001). Apolipoprotein E (ApoE) is subject to free radical attack and a direct correlation between ApoE peroxidation and AD has been reported (Butterfield and Lauderback, 2002). PD is the second most prevalent age-related neurodegenerative disease after AD. PD is characterized by the selective loss of dopaminergic neurons in the substantia nigra (Lotharius and Brundin, 2002). Oxidative stress-related dopaminergic neuronal damage is the leading theory of PD pathogenesis (Gonzalez-Fraguela et al., 1998; Miller et al., 2009). The important sources of ROS in PD are activation of phospholipases, including cPLA2, and induction of NADPH oxidase in activated microglia (Miller et al., 2009). HD is a rare, inherited neurological disorder, characterized by abnormal body movements and lack of coordination. Although the effect of lipid peroxidation is debatable with respect to the model of HD and type of tissue under consideration, elevated levels of 8-OHdG and HNE in plasma and MDA in striatal and cortical tissues, as well as increased F2-isoprostane, are present in HD (Mariani et al., 2005). In addition, many other neurological diseases are associated with accumulated lipid peroxidation in specific tissues: the spinal cord motor neurons of sporadic ALS patients (Simpson et al., 2004), spinal cord injury, and central nervous system (CNS) injuries, such as stroke, traumatic brain injury (Adibhatla et al., 2006; Adibhatla and Hatcher, 2008), autoimmune diseases, such as MS, all the inflammatory CNS tissues (Muralikrishna Adibhatla and Hatcher, 2006; Qin et al., 2007), and CA1 hippocampal neurons after transient cerebral ischemia (Muralikrishna Adibhatla and Hatcher, 2006). Therefore, in order to develop any strategy to prevent age-related neurological disorders, it is necessary to activate or potentiate the defense mechanism against lipophilic oxidants.
Involvement of lipid peroxidation in cardiovascular or neurodegenerative diseases is well established, but the connection of lipid peroxidation with other age-related diseases, such as type II diabetes mellitus and cancer, has not been properly elucidated. Increased oxidative stress has been proposed to be one of the major causes of the hyperglycemia-induced trigger of diabetic complications (Maritim et al., 2003). Hyperglycemia triggers ROS formation from a variety of sources, such as glucose autoxidation, oxidative phosphorylation, NAD(P)H oxidase, lipoxygenase, cytochrome P450 monooxygenases, xanthine oxidase (XO), and nitric oxide synthase (Valko et al., 2007). Among them, XO is one of the major sources of ROS in diabetes mellitus, since treatment of non-insulin dependent diabetic patients with allopurinol, an XO inhibitor, reduces the level of oxidized lipids in plasma and improves blood flow (Butler et al., 2000). Lipoxygenase catalyzes conversion of arachidonic acid into a broad class of signaling molecules, such as leukotrienes, lipoxins, and hydroxyeicosatetraenoic acid. Diabetes is associated with increased lipoxygenase expression, resulting in increased eicosanoid formation (Brash, 1999). β-cells are particularly sensitive to ROS, because they are low in antioxidant enzymes, such as glutathione peroxidase, catalase, and superoxide dismutase, which leads to β-cell dysfunction (Kaneto et al., 1999; Evans et al., 2003). Cancer is a disease that involves a multi-step process of mutations and preferential clonal expansion of highly neoplastic, mutated cells. It is clear that ROS is associated with genomic instability, which predisposes to mutagenesis and carcinogenesis. It has been reported that MDA is mutagenic in bacterial and mammalian cells and carcinogenic in rats, and HNE is weakly mutagenic, but appears to be the major toxic product of lipid peroxidation (Basu and Marnett, 1984; Plastaras et al., 2000; Riggins and Marnett, 2001). In addition, MDA correlates with the extent of primary tumor and predicts poor prognosis of oropharyngeal cancer (Salzman et al., 2009). Thereby, in order to prevent these degenerative diseases, the protective mechanism of lipophilic antioxidants should be effectively activated as well.
Several lines of evidences have demonstrated the negative relationship of BVR activity and bilirubin concentration with the aging process. An age-dependent decrease in the activity of BVR was detected (Maines, 1990). Together with BVR, HO forms a powerful protective system against various oxidative stresses. But HO-1 induction by various oxidative stressors, such as H2O2 and hemin, is markedly impaired in senescent human fibroblasts (Kim et al., 2011a). Serum bilirubin concentration is highest in the 19- to 24-year-old age group, after which it declines, which might be related to the age-related decrease in BVR activity (Rosenthal et al., 1984). Moreover, since oxidized BVR was associated with impairment of its function (Barone et al., 2011a), the age-related increase in ROS generation might influence BVR activity.
With regard to age-related diseases, bilirubin has a significant and beneficial role in preventing oxidative changes in a number of diseases, including atherosclerosis, neurodegenerative disease, diabetes mellitus, and cancer as well as a number of inflammatory and autoimmune diseases. The mild to moderately elevated levels of serum bilirubin are positively related to better outcomes of CVD. For example, the male air force pilot study showed that a 50% decrease in total bilirubin was associated with a 47% increase in the more severe coronary artery diseases (Schwertner et al., 1994). Low serum bilirubin in patients with CVD was also detected in the Framingham Offspring Study (Djousse et al., 2001, 2003). In addition, positive effects of serum bilirubin were illustrated in the study of Gilbert’s syndrome. Gilbert’s syndrome is the most common hereditary genetic disorder of impaired glucuronyl transferase activity causing mild to moderate elevations of serum bilirubin and is found in up to 5% of the population. The prevalence of the ischemic heart diseases in Gilbert’s syndrome was found to be only 2%, compared with 12% in control individuals (Vitek et al., 2002). It is interesting to note that cigarette smoking, a major risk factor for CVD, was associated with significantly lowered serum bilirubin content (Van Hoydonck et al., 2001). Both unconjugated and conjugated bilirubin can protect human LDL against oxidation by oxy-radicals generated by 2,2′-azo-bis(2-amidinopropane) or Cu (Wu et al., 1996). It seems like BVR/HO-2 link plays an important role in cardiac protection. Recently, it is suggested that BVR would be an upstream stabilizer of HO-2, which prominently expressed in the cardiovascular system, and both entities are intimately linked to cardiomyocyte survival (Ding et al., 2011). There are numerous possible mechanisms by which the HO-1/HO-2 pathway may improve vascular function. It has been reported that HO-2 activation occurs in ischemic hearts in dogs and that inhibition of the HO system reduces vasodilation during ischemia in the presence of NO and COX inhibitors (Nishikawa et al., 2004).
With regard to neurodegenerative disorders, bilirubin is a potent antioxidant against the cellular damage elicited by ROS and contributes to the overall antioxidant network of the brain (Takahashi et al., 2000; Mancuso, 2004). Impairment of the HO/BVR system in the hippocampus of AD, as well as a linkage of BVR with an improvement of cognitive function following atorvastatin treatment, have been reported (Barone et al., 2011b). All these effects contribute to the neuroprotective role of BVR in the brain. In addition, a low concentration of serum bilirubin is related to an increased risk of ALS and mental illnesses, such as winter depression and schizophrenia (Oren et al., 2002; Ilzecka and Stelmasiak, 2003; Iwasaki et al., 2005). HO-2 is selectively enriched in neurons and it is becoming apparent that HO-2 play an important role in cytoprotection in neural tissues. HO-2 expression has been shown to be protective against apoptotic cell death in cortical, hippocampal, and cerebellar granule cultures and an in vivo model of ischemic injury (Dore and Snyder, 1999; Dore et al., 1999a, 2000). In the brain, it is suggested that HO-derived CO would function as a neurotransmitter/neuromodulator (Maines et al., 1993; Seki et al., 2000). HO-2 protects against lipid peroxidation mediated cell loss as well as the impaired motor recovery after traumatic brain injury (Chang et al., 2003). Furthermore, a link has been proposed between HO activities and neurodegenerative conditions such as familial AD. Single point mutations in amyloid precursor proteins, binding to HO-1/HO-2, have been associated with a significant reduction in HO activities, resulting in greatly reduced bilirubin levels and increased neurotoxicity (Takahashi et al., 2000). The neuroprotective effects of HO-2 seem to be attributed to the generation of bilirubin (Dore et al., 1999b, 2000).
In diabetes mellitus, a higher level of serum bilirubin is associated with a lower risk of the disease (Fukui et al., 2008, 2011; Han et al., 2010; Dekker et al., 2011). Using the National Health and Nutrition Examination Survey (NHANES) data, an inverse relationship of serum total bilirubin and the incidence of diabetes has been suggested (Cheriyath et al., 2010).
Although controversial, there are some evidences that serum bilirubin may be protective against some forms of cancer. Baseline serum bilirubin concentration was inversely associated with the risk of cancer development (Ko et al., 1994). Cross-sectional analysis demonstrated that an increase in serum bilirubin was associated with a lower rate of colorectal cancer (Zucker et al., 2006). In addition, a possible beneficial role of bilirubin was also described in several diseases, illustrated by the low serum bilirubin levels detected in various oxidative stress-mediated diseases, such as rheumatoid arthritis, systemic lupus nephritis, idiopathic pulmonary fibrosis, and chronic obstructive pulmonary disease (Ohrui et al., 2001; Fischman et al., 2010; Horsfall et al., 2011).
The Hayflick limit describes the phenomenon of normal mitotic cells undergoing a finite number of cell divisions before entering replicative senescence, after which they cannot divide further and become unresponsive to mitogenic stimuli (Hayflick, 1965). In addition to replicative senescence, cells can also be induced to become prematurely senescent by exposure to oxidants, DNA damaging agents, histone deacetylase inhibitors, or by overexpression of certain oncogenes, which is referred to as stress-induced premature senescence (Chen et al., 1995; Serrano et al., 1997; Robles and Adami, 1998; Zhu et al., 1998). Regardless of the induction mode, senescent cells generally acquire the enlarged and flattened morphology with high expression of senescence-associated β-galactosidase and lose the ability to proliferate. Senescent cells contain higher levels of oxidative DNA lesions than early passage cells with increased p21, hypo-phosphorylated Rb, reduced E2F activity, and G1 stage cell cycle arrest (Stein et al., 1990; Noda et al., 1994; Dimri et al., 1995). Over the past few decades, ROS was presumed to be the pivotal determinant factor underlying age-associated decline of physiological functions. ROS levels increase with age in major organs, such as liver, heart, and brain (Gomi et al., 1993; Bejma et al., 2000; Driver et al., 2000; Zhang et al., 2003), and overproduction of pro-oxidants has been reported in many species during aging (Sohal and Weindruch, 1996; Barja, 2002; Zanetti et al., 2010). Oxidative stress has also been implicated in various age-related pathological conditions involving CVD, cancers, neurological disorders, diabetes, and other diseases (Valko et al., 2007). The primary physiological function of BVR is the production of bilirubin, a major natural and potent antioxidant. Moreover, the suggested redox cyclic nature of the bilirubin to biliverdin pathway and induction of HO by BVR would potentiate the physiological antioxidant capacity of this system (Baranano et al., 2002; Sedlak and Snyder, 2004; Sedlak et al., 2009; Ding et al., 2011). Recent studies have revealed that administration of BVR ameliorates the signs of oxidative stress-mediated diseases more efficiently than administration of other antioxidant enzymes, and when cellular BVR activity is suppressed using siRNA, the levels of ROS and cell death markedly increase (Baranano et al., 2002; Sedlak et al., 2009).
When the effects of BVR knock-down on cell viability and cell cycle progression are monitored, human diploid fibroblasts become growth-arrested for a prolonged period after depletion of BVR, resulting in cellular senescence with decreased levels of cyclin D1, increased levels of p16/INK4a, decreased phosphorylation of Rb, and high expression of senescence-associated β-galactosidase (Kim et al., 2011a). This BVR knock-down-induced cellular senescence could be effectively suppressed by treatment with the ROS scavenger, N-acetyl cysteine (NAC). Since simple depletion of BVR can decrease cell viability and induce senescence, BVR might be suggested as the essential component for cellular survival under normal oxidative stress conditions. In addition, overexpression of BVR partially restored young cell-like morphology in senescent fibroblasts, but without resumption of mitogenic potential, implying that BVR overexpression alone was not sufficient to prevent senescence-related cell cycle arrest (Kim et al., 2011a).
Recently, a number of studies have demonstrated that cellular senescence provides an important barrier to genomic instability and tumorigenesis (Campisi and d’Adda di Fagagna, 2007; Schmitt, 2007). Senescent cells also express high levels of p16 (Alcorta et al., 1996), which might be responsible for blocking tumorigenesis. Interestingly, BVR activity is elevated in tumor tissues (Maines et al., 1999), and serum bilirubin levels are inversely related to cancer mortality (McCarty, 2007). Moreover, knock-down of BVR induces cell death in HeLa and A498 cells and sensitizes these cancer cells to oxidative stress (Kim et al., 2011a). Therefore, it can be assumed that down-regulation of BVR might be an important additive strategy for the successful treatment of cancer.
Biliverdin reductase activity is reduced with aging (Maines, 1990). HO-1 induction by BVR, as well as by oxidative stressors, is markedly impaired in senescent human fibroblasts (Kim et al., 2011a). These aging-related decreased responses to oxidative stress might be due to age-dependent differences in the efficiency of oxidative stress-related signal trafficking into the nucleus. We have demonstrated that the nucleocytoplasmic trafficking system was markedly impaired in senescent human diploid fibroblasts (Kim et al., 2010a,b, 2011b). In addition, the nuclear barrier hypothesis of aging has been proposed, in which the aging phenotype might be induced by the inefficiency of nucleocytoplasmic trafficking of a variety of mitogenic or apoptotic signals due to suppression of nuclear pore complex functions (Park, 2011). BVR is a leucine zipper-like DNA binding protein, which serves as a transcription factor for HO-1 (Kravets et al., 2004; Tudor et al., 2008). To perform its activities, such as signal transduction and induction of HO-1 in response to oxidative stress, the nuclear translocation of BVR is prerequisite. In this regard, nuclear trafficking of BVR and subsequent signaling events may be impaired in senescent human diploid fibroblasts, which might help to explain the age-dependent differences in response to oxidative stress.
Biliverdin reductase in young cells effectively protects the cells from various oxidative stresses through generation of bilirubin, a potent lipophilic ROS scavenger, and by regulating the expression of HO, another potent antioxidant. In this situation, the two cycles of the bilirubin system for ROS scavenging are supposed to be intertwined. The first cycle is the suggested redox cycle of biliverdin to bilirubin by BVR and the cycling back of bilirubin to biliverdin probably by lipophilic oxidants. The second cycle is the induction of HO by BVR and the degradation of heme to biliverdin, which is again metabolized to bilirubin by BVR (Figure 1). These two metabolic and transcriptional cycles are intertwined and operate in response to oxidative stress, providing highly efficient ROS scavenging. This intertwined bicyclic bilirubin system, comprised of BVR and HO for heme degradation to biliverdin and bilirubin, could play a significant role in maintaining cellular integrity against oxidative stress. Senescent cells have increased ROS generation and decreased BVR activity due to its oxidative modification, resulting in reduced bilirubin formation. Furthermore, the senescence-mediated impairment of nucleocytoplasmic trafficking prevents the nuclear translocation of BVR, resulting in decreased HO induction (Figure 1). Therefore, it is natural to assume that dysfunction of this bicyclic bilirubin system would be responsible for aging and, subsequently, for age-related disease.
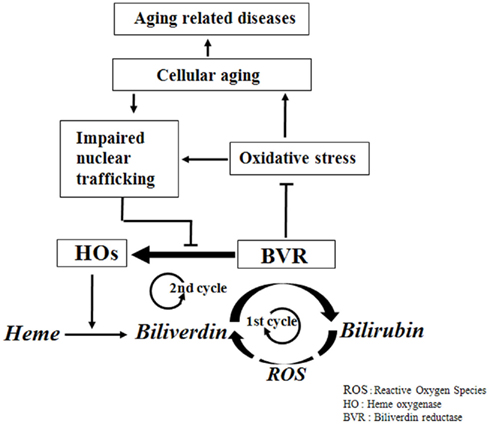
Figure 1. Physiological network of the bicyclic bilirubin system in aging and age-related disease. In normal physiological condition of oxidative stress, BVR catalyzes biliverdin to bilirubin and induces HO, which further provides more biliverdin from heme. And the resulting bilirubin may turn back to biliverdin partially by oxidants. This bicyclic nature of bilirubin system provides the efficient physiological antioxidative capacity. But in harsh conditions of high oxidative stress, BVR is catalytically inactivated and is blocked for nuclear translocation, which inhibits the induction of HO, resulting in inhibition of catalytic turnover of heme to biliverdin and also biliverdin to bilirubin, leading to oxidative damages of the biomolecules and finally to aging and age-related diseases.
Although it is claimed that lipid peroxidation and impaired bilirubin system are related to age-related diseases, several important issues still remain to be resolved. The exact role of bilirubin system in the progression of age-related diseases is not fully understood. Further randomized trial data and molecular studies are necessary to evaluate the potential role of bilirubin system in aging and age-related disease progression. Though the causal links between changes in HO/BVR activity, bilirubin/biliverdin level and upstream/downstream changes in aging process have not been clearly established, the lipid peroxide damages especially at all the cellular membrane fractions would be readily expected to impair cellular functions and its integrity. Therefore, in order to protect the membrane lipid damages from the lipid soluble oxidants, the antioxidants of lipophilic nature rather than hydrophilic nature are expected to play the dominant roles. Therefore, it can be assumed that the bilirubin system might protect the organs effectively from the oxidative stress, especially of lipophilic oxidants, via its potential amplifying antioxidative capacity. If this hypothesis were true, aging and age-related diseases might be prevented by modulating BVR system to a certain degree. In this regard, it can be a novel promising strategy to adjust HO/BVR activity and/or exogenous administration of bilirubin for control of aging process and its related diseases. But it should be reminded that bilirubin possesses both antioxidant and prooxidant properties. Bilirubin shows hormeric dose responses, as illustrated by that it protects red blood cells against oxidative stress at physiologic concentrations, while it is associated with significant cytotoxicity at concentrations of 30 mg/dL or higher (Mireles et al., 1999). The similar results were shown when neuronal cells were exposed to low and high concentrations of bilirubin (Chen et al., 2003). Moreover, since several toxic effects of bilirubin have been demonstrated on erythrocytes, lymphocytes, renal cells, lung, and brain in a dose and time dependent manner (Elias et al., 1987; Alexandra Brito et al., 2006), the specific safe dose response should be considered for its use in regulation of aging process and its related diseases.
Since impaired redox homeostasis and defective oxidative stress defense mechanisms are the key contributors to the aging process, the endogenous antioxidative systems have been presumed to play important roles in the control of age-related diseases as well as aging itself. In addition to the glutathione system of hydrophilic nature, when oxidative stress and lipid peroxidation are issued, the another antioxidative mechanism consisting of BVR and the biliverdin to bilirubin pathway should be concerned against lipophilic oxidants. This bilirubin system is comprised of the intertwined bicyclic control mechanisms: the first, the metabolic redox cycle of biliverdin and bilirubin, and the second, the transcriptional control cycle of HO by BVR. The efficiency of this bilirubin system, amplified by the cyclic nature of the control mechanism, enables it to protect cells effectively against oxidative stress. Aging is associated with decreased BVR activity due to oxidative damages, which results in decreased induction of HO, leading to further reduction of bilirubin generation. Prolonged knock-down of BVR in young cells would result in premature senescence. These results provide the strong supports for the concept that a rise in ROS by way of inefficient BVR activity, either by oxidative damage or by impaired nucleocytoplasmic trafficking, could be an intracellular trigger of cellular senescence. Moreover, the inverse relationship of BVR activity and bilirubin status observed in a variety of diseases implies a significant role of the bilirubin system in the pathogenesis of many age-related diseases as well as aging itself. Therefore, it can be suggested that the bilirubin system might provide new opportunities for drug development and therapy for aging and a variety of age-related disorders.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by National Research Foundation (NRF) grants provided by the Korean Government (MEST; No. 2010-0029150) and the Research Program of Cancer and Aging.
Adibhatla, R. M., and Hatcher, J. F. (2007). Role of lipids in brain injury and diseases. Future Lipidol. 2, 403–422.
Adibhatla, R. M., and Hatcher, J. F. (2008). Phospholipase A(2), reactive oxygen species, and lipid peroxidation in CNS pathologies. BMB Rep. 41, 560–567.
Adibhatla, R. M., and Hatcher, J. F. (2010). Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 12, 125–169.
Adibhatla, R. M., Hatcher, J. F., and Dempsey, R. J. (2006). Lipids and lipidomics in brain injury and diseases. AAPS J. 8, E314–E321.
Ahotupa, M., and Asankari, T. J. (1999). Baseline diene conjugation in LDL lipids: an indicator of circulating oxidized LDL. Free Radic. Biol. Med. 27, 1141–1150.
Alcorta, D. A., Xiong, Y., Phelps, D., Hannon, G., Beach, D., and Barrett, J. C. (1996). Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 93, 13742–13747.
Alexandra Brito, M., Silva, R. F., and Brites, D. (2006). Bilirubin toxicity to human erythrocytes: a review. Clin. Chim. Acta 374, 46–56.
Baranano, D. E., Rao, M., Ferris, C. D., and Snyder, S. H. (2002). Biliverdin reductase: a major physiologic cytoprotectant. Proc. Natl. Acad. Sci. U.S.A. 99, 16093–16098.
Barja, G. (2002). Endogenous oxidative stress: relationship to aging, longevity and caloric restriction. Ageing Res. Rev. 1, 397–411.
Barone, E., Di Domenico, F., Cenini, G., Sultana, R., Coccia, R., Preziosi, P., Perluigi, M., Mancuso, C., and Butterfield, D. A. (2011a). Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer’s disease and amnestic mild cognitive impairment. J. Alzheimers Dis. 25, 623–633.
Barone, E., Mancuso, C., Di Domenico, F., Sultana, R., Paul Murphy, M., Head, E., and Allan Butterfield, D. (2011b). Biliverdin reductase-A: a novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J. Neurochem. 120, 135–146.
Basu, A. K., and Marnett, L. J. (1984). Molecular requirements for the mutagenicity of malondialdehyde and related acroleins. Cancer Res. 44, 2848–2854.
Bejma, J., Ramires, P., and Ji, L. L. (2000). Free radical generation and oxidative stress with ageing and exercise: differential effects in the myocardium and liver. Acta Physiol. Scand. 169, 343–351.
Bliuger, A. F., Dudnik, L. B., Maiore, A., and Mieze, I. E. (1985). Role of bilirubin as a natural antioxidant in regulating lipid peroxidation intensity in acute viral hepatitis. Biull. Eksp. Biol. Med. 99, 166–168.
Bourre, J. M. (1988). Free radicals, polyunsaturated fatty acids, cell death, brain aging. C. R. Seances Soc. Biol. Fil. 182, 5–36.
Brash, A. R. (1999). Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J. Biol. Chem. 274, 23679–23682.
Butler, R., Morris, A. D., Belch, J. J., Hill, A., and Struthers, A. D. (2000). Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension 35, 746–751.
Butterfield, D. A., and Lauderback, C. M. (2002). Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 32, 1050–1060.
Butterfield, D. A., Reed, T., Perluigi, M., De Marco, C., Coccia, R., Cini, C., and Sultana, R. (2006). Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 397, 170–173.
Campisi, J., and d’Adda di Fagagna, F. (2007). Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740.
Catalá, A. (2009). Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 157, 1–11.
Chang, E. F., Wong, R. J., Vreman, H. J., Igarashi, T., Galo, E., Sharp, F. R., Stevenson, D. K., and Noble-Haeusslein, L. J. (2003). Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J. Neurosci. 23, 3689–3696.
Chen, J., Tu, Y., Moon, C., Nagata, E., and Ronnett, G. V. (2003). Heme oxygenase-1 and heme oxygenase-2 have distinct roles in the proliferation and survival of olfactory receptor neurons mediated by cGMP and bilirubin, respectively. J. Neurochem. 85, 1247–1261.
Chen, Q., and Ames, B. N. (1994). Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl. Acad. Sci. U.S.A. 91, 4130–4134.
Chen, Q., Fischer, A., Reagan, J. D., Yan, L. J., and Ames, B. N. (1995). Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. U.S.A. 92, 4337–4341.
Chen, Q. M., Bartholomew, J. C., Campisi, J., Acosta, M., Reagan, J. D., and Ames, B. N. (1998). Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem. J. 332(Pt 1), 43–50.
Cheriyath, P., Gorrepati, V. S., Peters, I., Nookala, V., Murphy, M. E., Srouji, N., and Fischman, D. (2010). High total bilirubin as a protective factor for diabetes mellitus: an analysis of NHANES data from 1999–2006. J. Clin. Med. Res. 2, 201–206.
Dekker, D., Dorresteijn, M. J., Pijnenburg, M., Heemskerk, S., Rasing-Hoogveld, A., Burger, D. M., Wagener, F. A., and Smits, P. (2011). The bilirubin-increasing drug atazanavir improves endothelial function in patients with type 2 diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 31, 458–463.
Dhalla, N. S., Temsah, R. M., and Netticadan, T. (2000). Role of oxidative stress in cardiovascular diseases. J. Hypertens. 18, 655–673.
Dimri, G. P., Lee, X., Basile, G., Acosta, M., Scott, G., Roskelley, C., Medrano, E. E., Linskens, M., Rubelj, I., Pereira-Smith, O., et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 9363–9367.
Ding, B., Gibbs, P. E., Brookes, P. S., and Maines, M. D. (2011). The coordinated increased expression of biliverdin reductase and heme oxygenase-2 promotes cardiomyocyte survival: a reductase-based peptide counters beta-adrenergic receptor ligand-mediated cardiac dysfunction. FASEB J. 25, 301–313.
Djousse, L., Levy, D., Cupples, L. A., Evans, J. C., D’Agostino, R. B., and Ellison, R. C. (2001). Total serum bilirubin and risk of cardiovascular disease in the Framingham offspring study. Am. J. Cardiol. 87, 1196–1200.
Djousse, L., Rothman, K. J., Cupples, L. A., Levy, D., and Ellison, R. C. (2003). Effect of serum albumin and bilirubin on the risk of myocardial infarction (the Framingham Offspring Study). Am. J. Cardiol. 91, 485–488.0
Dore, S., Goto, S., Sampei, K., Blackshaw, S., Hester, L. D., Ingi, T., Sawa, A., Traystman, R. J., Koehler, R. C., and Snyder, S. H. (2000). Heme oxygenase-2 acts to prevent neuronal death in brain cultures and following transient cerebral ischemia. Neuroscience 99, 587–592.
Dore, S., Sampei, K., Goto, S., Alkayed, N. J., Guastella, D., Blackshaw, S., Gallagher, M., Traystman, R. J., Hurn, P. D., Koehler, R. C., and Snyder, S. H. (1999a). Heme oxygenase-2 is neuroprotective in cerebral ischemia. Mol. Med. 5, 656–663.
Dore, S., and Snyder, S. H. (1999). Neuroprotective action of bilirubin against oxidative stress in primary hippocampal cultures. Ann. N. Y. Acad. Sci. 890, 167–172.
Dore, S., Takahashi, M., Ferris, C. D., Zakhary, R., Hester, L. D., Guastella, D., and Snyder, S. H. (1999b). Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. U.S.A. 96, 2445–2450.
Driver, A. S., Kodavanti, P. R., and Mundy, W. R. (2000). Age-related changes in reactive oxygen species production in rat brain homogenates. Neurotoxicol. Teratol. 22, 175–181.
Dudnik, L. B., Tsyupko, A. N., Khrenov, A. V., and Alessenko, A. V. (2001). Effect of bilirubin on lipid peroxidation, sphingomyelinase activity, and apoptosis induced by sphingosine and UV irradiation. Biochemistry Mosc. 66, 1019–1027.
Elias, M. M., Comin, E. J., Grosman, M. E., Galeazzi, S. A., and Rodriguez Garay, E. A. (1987). Possible mechanism of unconjugated bilirubin toxicity on renal tissue. Comp. Biochem. Physiol. A Comp. Physiol. 87, 1003–1007.
Evans, J. L., Goldfine, I. D., Maddux, B. A., and Grodsky, G. M. (2003). Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 52, 1–8.
Fischman, D., Valluri, A., Gorrepati, V. S., Murphy, M. E., Peters, I., and Cheriyath, P. (2010). Bilirubin as a protective factor for rheumatoid arthritis: an NHANES study of 2003–2006 data. J. Clin. Med. Res. 2, 256–260.
Fukui, M., Tanaka, M., Shiraishi, E., Harusato, I., Hosoda, H., Asano, M., Hasegawa, G., and Nakamura, N. (2008). Relationship between serum bilirubin and albuminuria in patients with type 2 diabetes. Kidney Int. 74, 1197–1201.
Fukui, M., Tanaka, M., Yamazaki, M., Hasegawa, G., Nishimura, M., Iwamoto, N., Ono, T., Imai, S., and Nakamura, N. (2011). Low serum bilirubin concentration in haemodialysis patients with Type 2 diabetes. Diabet. Med. 28, 96–99.
Gil, L., Siems, W., Mazurek, B., Gross, J., Schroeder, P., Voss, P., and Grune, T. (2006). Age-associated analysis of oxidative stress parameters in human plasma and erythrocytes. Free Radic. Res. 40, 495–505.
Gomi, F., Utsumi, H., Hamada, A., and Matsuo, M. (1993). Aging retards spin clearance from mouse brain and food restriction prevents its age-dependent retardation. Life Sci. 52, 2027–2033.
Gonzalez-Fraguela, M. E., Cespedes, E. M., Arencibia, R., Broche, F., Gomez, A. A., Castellano, O., and Garcia, J. C. (1998). Indicators of oxidative stress and the effect of antioxidant treatment in patients with primary Parkinson disease. Rev. Neurol. 26, 28–33.
Hagen, T. M., Yowe, D. L., Bartholomew, J. C., Wehr, C. M., Do, K. L., Park, J. Y., and Ames, B. N. (1997). Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proc. Natl. Acad. Sci. U.S.A. 94, 3064–3069.
Han, S. S., Na, K. Y., Chae, D. W., Kim, Y. S., Kim, S., and Chin, H. J. (2010). High serum bilirubin is associated with the reduced risk of diabetes mellitus and diabetic nephropathy. Tohoku J. Exp. Med. 221, 133–140.
Hayflick, L. (1965). The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 37, 614–636.
Horsfall, L. J., Rait, G., Walters, K., Swallow, D. M., Pereira, S. P., Nazareth, I., and Petersen, I. (2011). Serum bilirubin and risk of respiratory disease and death. JAMA 305, 691–697.
Ilzecka, J., and Stelmasiak, Z. (2003). Serum bilirubin concentration in patients with amyotrophic lateral sclerosis. Clin. Neurol. Neurosurg. 105, 237–240.
Iwasaki, Y., Igarashi, O., Iwasa, Y., Hirano, K., Satoh, R., Iwamoto, K., Kawase, Y., Aoyagi, J., Ichikawa, Y., Kawabe, K., and Ikeda, K. (2005). Bilirubin and amyotrophic lateral sclerosis. Clin. Neurol. Neurosurg. 107, 160.
Kadl, A., Bochkov, V. N., Huber, J., and Leitinger, N. (2004). Apoptotic cells as sources for biologically active oxidized phospholipids. Antioxid. Redox Signal. 6, 311–320.
Kaneko, M., Elimban, V., and Dhalla, N. S. (1989). Mechanism for depression of heart sarcolemmal Ca2+ pump by oxygen free radicals. Am. J. Physiol. 257, H804–H811.
Kaneto, H., Kajimoto, Y., Fujitani, Y., Matsuoka, T., Sakamoto, K., Matsuhisa, M., Yamasaki, Y., and Hori, M. (1999). Oxidative stress induces p21 expression in pancreatic islet cells: possible implication in beta-cell dysfunction. Diabetologia 42, 1093–1097.
Kapitulnik, J., and Maines, M. D. (2009). Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol. Sci. 30, 129–137.
Kim, S. Y., Kang, H. T., Choi, H. R., and Park, S. C. (2010a). Reduction of Nup107 attenuates the growth factor signaling in the senescent cells. Biochem. Biophys. Res. Commun. 401, 131–136.
Kim, S. Y., Ryu, S. J., Ahn, H. J., Choi, H. R., Kang, H. T., and Park, S. C. (2010b). Senescence-related functional nuclear barrier by down-regulation of nucleo-cytoplasmic trafficking gene expression. Biochem. Biophys. Res. Commun. 391, 28–32.
Kim, S. Y., Kang, H. T., Choi, H. R., and Park, S. C. (2011a). Biliverdin reductase A in the prevention of cellular senescence against oxidative stress. Exp. Mol. Med. 43, 15–23.
Kim, S. Y., Ryu, S. J., Kang, H. T., Choi, H. R., and Park, S. C. (2011b). Defective nuclear translocation of stress-activated signaling in senescent diploid human fibroblasts: a possible explanation for aging-associated apoptosis resistance. Apoptosis 16, 795–807.
Klein, J. A., and Ackerman, S. L. (2003). Oxidative stress, cell cycle, and neurodegeneration. J. Clin. Invest. 111, 785–793.
Ko, W. F., Helzlsouer, K. J., and Comstock, G. W. (1994). Serum albumin, bilirubin, and uric acid and the anatomic site-specific incidence of colon cancer. J. Natl. Cancer Inst. 86, 1874–1875.
Kravets, A., Hu, Z., Miralem, T., Torno, M. D., and Maines, M. D. (2004). Biliverdin reductase, a novel regulator for induction of activating transcription factor-2 and heme oxygenase-1. J. Biol. Chem. 279, 19916–19923.
Kuhn, H., Belkner, J., Wiesner, R., Schewe, T., Lankin, V. Z., and Tikhaze, A. K. (1992). Structure elucidation of oxygenated lipids in human atherosclerotic lesions. Eicosanoids 5, 17–22.
Lee, A. C., Fenster, B. E., Ito, H., Takeda, K., Bae, N. S., Hirai, T., Yu, Z. X., Ferrans, V. J., Howard, B. H., and Finkel, T. (1999). Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 274, 7936–7940.
Lerner-Marmarosh, N., Miralem, T., Gibbs, P. E., and Maines, M. D. (2008). Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc. Natl. Acad. Sci. U.S.A. 105, 6870–6875.
Lerner-Marmarosh, N., Shen, J., Torno, M. D., Kravets, A., Hu, Z., and Maines, M. D. (2005). Human biliverdin reductase: a member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 7109–7114.
Lotharius, J., and Brundin, P. (2002). Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 3, 932–942.
Maines, M. D. (1990). Multiple forms of biliverdin reductase: age-related change in pattern of expression in rat liver and brain. Mol. Pharmacol. 38, 481–485.
Maines, M. D. (2005). New insights into biliverdin reductase functions: linking heme metabolism to cell signaling. Physiology (Bethesda) 20, 382–389.
Maines, M. D. (2010). Potential application of biliverdin reductase and its fragments to modulate insulin/IGF-1/MAPK/PI3-K signaling pathways in therapeutic settings. Curr. Drug Targets 11, 1586–1594.
Maines, M. D., Ewing, J. F., Huang, T. J., and Panahian, N. (2001). Nuclear localization of biliverdin reductase in the rat kidney: response to nephrotoxins that induce heme oxygenase-1. J. Pharmacol. Exp. Ther. 296, 1091–1097.
Maines, M. D., Mark, J. A., and Ewing, J. F. (1993). Heme oxygenase, a likely regulator of cGMP production in the brain: induction in vivo of HO-1 compensates for depression in NO Synthase Activity. Mol. Cell. Neurosci. 4, 396–405.
Maines, M. D., Mayer, R. D., Erturk, E., Huang, T. J., and Disantagnese, A. (1999). The oxidoreductase, biliverdin reductase, is induced in human renal carcinoma–pH and cofactor-specific increase in activity. J. Urol. 162, 1467–1472.
Mancuso, C. (2004). Heme oxygenase and its products in the nervous system. Antioxid. Redox Signal. 6, 878–887.
Mariani, E., Polidori, M. C., Cherubini, A., and Mecocci, P. (2005). Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 827, 65–75.
Maritim, A. C., Sanders, R. A., and Watkins, J. B. III. (2003). Diabetes, oxidative stress, and antioxidants: a review. J. Biochem. Mol. Toxicol. 17, 24–38.
Markesbery, W. R., and Lovell, M. A. (2007). Damage to lipids, proteins, DNA, and RNA in mild cognitive impairment. Arch. Neurol. 64, 954–956.
Marnett, L. J. (1999). Chemistry and biology of DNA damage by malondialdehyde. IARC Sci. Publ. 17–27.
McCarty, M. F. (2007). “Iatrogenic Gilbert syndrome” – a strategy for reducing vascular and cancer risk by increasing plasma unconjugated bilirubin. Med. Hypotheses 69, 974–994.
McDonagh, A. F. (2010). The biliverdin-bilirubin antioxidant cycle of cellular protection: missing a wheel? Free Radic Biol. Med. 49, 814–820.
Miller, R. L., James-Kracke, M., Sun, G. Y., and Sun, A. Y. (2009). Oxidative and inflammatory pathways in Parkinson’s disease. Neurochem. Res. 34, 55–65.
Mireles, L. C., Lum, M. A., and Dennery, P. A. (1999). Antioxidant and cytotoxic effects of bilirubin on neonatal erythrocytes. Pediatr. Res. 45, 355–362.
Miro, O., Casademont, J., Casals, E., Perea, M., Urbano-Marquez, A., Rustin, P., and Cardellach, F. (2000). Aging is associated with increased lipid peroxidation in human hearts, but not with mitochondrial respiratory chain enzyme defects. Cardiovasc. Res. 47, 624–631.
Mizuno, Y. (1990). The role of oxygen free radicals in the aging of the brain. Nihon Ronen Igakkai Zasshi 27, 182–187.
Molavi, B., and Mehta, J. L. (2004). Oxidative stress in cardiovascular disease: molecular basis of its deleterious effects, its detection, and therapeutic considerations. Curr. Opin. Cardiol. 19, 488–493.
Muralikrishna Adibhatla, R., and Hatcher, J. F. (2006). Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic. Biol. Med. 40, 376–387.
Nishikawa, Y., Stepp, D. W., Merkus, D., Jones, D., and Chilian, W. M. (2004). In vivo role of heme oxygenase in ischemic coronary vasodilation. Am. J. Physiol. Heart Circ. Physiol. 286, H2296–H2304.
Noda, A., Ning, Y., Venable, S. F., Pereira-Smith, O. M., and Smith, J. R. (1994). Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp. Cell Res. 211, 90–98.
Ohrui, T., Higuchi, M., Kanda, A., Matsui, T., Sato, E., and Sasaki, H. (2001). A patient with exacerbation of idiopathic pulmonary fibrosis which was resolved probably due to the coexisting hyperbilirubinemia? Tohoku J. Exp. Med. 193, 245–249.
Oren, D. A., Desan, P. H., Boutros, N., Anand, A., and Charney, D. S. (2002). Effects of light on low nocturnal bilirubin in winter depression: a preliminary report. Biol. Psychiatry 51, 422–425.
Pansarasa, O., Bertorelli, L., Vecchiet, J., Felzani, G., and Marzatico, F. (1999). Age-dependent changes of antioxidant activities and markers of free radical damage in human skeletal muscle. Free Radic. Biol. Med. 27, 617–622.
Park, S. C. (2011). Nuclear barrier hypothesis of aging as mechanism for trade-off growth to survival. Adv. Exp. Med. Biol. 720, 3–13.
Plastaras, J. P., Riggins, J. N., Otteneder, M., and Marnett, L. J. (2000). Reactivity and mutagenicity of endogenous DNA oxopropenylating agents: base propenals, malondialdehyde, and N(epsilon)-oxopropenyllysine. Chem. Res. Toxicol. 13, 1235–1242.
Poon, H. F., Calabrese, V., Scapagnini, G., and Butterfield, D. A. (2004). Free radicals and brain aging. Clin. Geriatr. Med. 20, 329–359.
Pratico, D., Uryu, K., Leight, S., Trojanoswki, J. Q., and Lee, V. M. (2001). Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 21, 4183–4187.
Qin, J., Goswami, R., Balabanov, R., and Dawson, G. (2007). Oxidized phosphatidylcholine is a marker for neuroinflammation in multiple sclerosis brain. J. Neurosci. Res. 85, 977–984.
Ravandi, A., Boekholdt, S. M., Mallat, Z., Talmud, P. J., Kastelein, J. J., Wareham, N. J., Miller, E. R., Benessiano, J., Tedgui, A., Witztum, J. L., Khaw, K. T., and Tsimikas, S. (2011). Relationship of IgG and IgM autoantibodies and immune complexes to oxidized LDL with markers of oxidation and inflammation and cardiovascular events: results from the EPIC-Norfolk study. J. Lipid Res. 52, 1829–1836.
Recuero, M., Munoz, T., Aldudo, J., Subias, M., Bullido, M. J., and Valdivieso, F. (2010). A free radical-generating system regulates APP metabolism/processing. FEBS Lett. 584, 4611–4618.
Riggins, J. N., and Marnett, L. J. (2001). Mutagenicity of the malondialdehyde oligomerization products 2-(3′-oxo-1′-propenyl)-malondialdehyde and 2,4-dihydroxymethylene-3-(2,2-dimethoxyethyl)glutaraldehyde in Salmonella. Mutat. Res. 497, 153–157.
Robles, S. J., and Adami, G. R. (1998). Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 16, 1113–1123.
Rosenthal, P., Pincus, M., and Fink, D. (1984). Sex- and age-related differences in bilirubin concentrations in serum. Clin. Chem. 30, 1380–1382.
Salzman, R., Pacal, L., Tomandl, J., Kankova, K., Tothova, E., Gal, B., Kostrica, R., and Salzman, P. (2009). Elevated malondialdehyde correlates with the extent of primary tumor and predicts poor prognosis of oropharyngeal cancer. Anticancer Res. 29, 4227–4231.
Schluchter, W. M., and Glazer, A. N. (1997). Characterization of cyanobacterial biliverdin reductase. Conversion of biliverdin to bilirubin is important for normal phycobiliprotein biosynthesis. J. Biol. Chem. 272, 13562–13569.
Schwertner, H. A., Jackson, W. G., and Tolan, G. (1994). Association of low serum concentration of bilirubin with increased risk of coronary artery disease. Clin. Chem. 40, 18–23.
Sedlak, T. W., Saleh, M., Higginson, D. S., Paul, B. D., Juluri, K. R., and Snyder, S. H. (2009). Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. U.S.A. 106, 5171–5176.
Sedlak, T. W., and Snyder, S. H. (2004). Bilirubin benefits: cellular protection by a biliverdin reductase antioxidant cycle. Pediatrics 113, 1776–1782.
Seki, T., Curiel, D. T., Naruse, M., and Takano, K. (2000). HO (heme oxygenase)/CO (carbon monoxide) system. Nihon Rinsho 58(Suppl. 1), 188–191.
Serrano, M., Lin, A. W., Mccurrach, M. E., Beach, D., and Lowe, S. W. (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602.
Simpson, E. P., Henry, Y. K., Henkel, J. S., Smith, R. G., and Appel, S. H. (2004). Increased lipid peroxidation in sera of ALS patients: a potential biomarker of disease burden. Neurology 62, 1758–1765.
Sohal, R. S., and Weindruch, R. (1996). Oxidative stress, caloric restriction, and aging. Science 273, 59–63.
Stein, G. H., Beeson, M., and Gordon, L. (1990). Failure to phosphorylate the retinoblastoma gene product in senescent human fibroblasts. Science 249, 666–669.
Stocker, R., and Ames, B. N. (1987). Potential role of conjugated bilirubin and copper in the metabolism of lipid peroxides in bile. Proc. Natl. Acad. Sci. U.S.A. 84, 8130–8134.
Stocker, R., Mcdonagh, A. F., Glazer, A. N., and Ames, B. N. (1990). Antioxidant activities of bile pigments: biliverdin and bilirubin. Meth. Enzymol. 186, 301–309.
Stoyanovsky, D., Murphy, T., Anno, P. R., Kim, Y. M., and Salama, G. (1997). Nitric oxide activates skeletal and cardiac ryanodine receptors. Cell Calcium 21, 19–29.
Takahashi, M., Dore, S., Ferris, C. D., Tomita, T., Sawa, A., Wolosker, H., Borchelt, D. R., Iwatsubo, T., Kim, S. H., Thinakaran, G., Sisodia, S. S., and Snyder, S. H. (2000). Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer’s disease. Neuron 28, 461–473.
Tudor, C., Lerner-Marmarosh, N., Engelborghs, Y., Gibbs, P. E., and Maines, M. D. (2008). Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem. J. 413, 405–416.
Valko, M., Leibfritz, D., Moncol, J., Cronin, M. T., Mazur, M., and Telser, J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84.
Van Hoydonck, P. G., Temme, E. H., and Schouten, E. G. (2001). Serum bilirubin concentration in a Belgian population: the association with smoking status and type of cigarettes. Int. J. Epidemiol. 30, 1465–1472.
Vitek, L., Jirsa, M., Brodanova, M., Kalab, M., Marecek, Z., Danzig, V., Novotny, L., and Kotal, P. (2002). Gilbert syndrome and ischemic heart disease: a protective effect of elevated bilirubin levels. Atherosclerosis 160, 449–456.
Ward, W. F., Qi, W., Van Remmen, H., Zackert, W. E., Roberts, L. J. 2nd, and Richardson, A. (2005). Effects of age and caloric restriction on lipid peroxidation: measurement of oxidative stress by F2-isoprostane levels. J. Gerontol. A Biol. Sci. Med. Sci. 60, 847–851.
Wegiel, B., Baty, C. J., Gallo, D., Csizmadia, E., Scott, J. R., Akhavan, A., Chin, B. Y., Kaczmarek, E., Alam, J., Bach, F. H., Zuckerbraun, B. S., and Otterbein, L. E. (2009). Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. J. Biol. Chem. 284, 21369–21378.
Wu, B., Liu, X., and Shen, J. (2008). Old biliverdin reductase: links to insulin resistance and may be a novel therapeutic target. Med. Hypotheses 71, 73–76.
Wu, T. W., Fung, K. P., Wu, J., Yang, C. C., and Weisel, R. D. (1996). Antioxidation of human low density lipoprotein by unconjugated and conjugated bilirubins. Biochem. Pharmacol. 51, 859–862.
Yuan, X. M., and Li, W. (2003). The iron hypothesis of atherosclerosis and its clinical impact. Ann. Med. 35, 578–591.
Zanetti, M., Gortan Cappellari, G., Burekovic, I., Barazzoni, R., Stebel, M., and Guarnieri, G. (2010). Caloric restriction improves endothelial dysfunction during vascular aging: effects on nitric oxide synthase isoforms and oxidative stress in rat aorta. Exp. Gerontol. 45, 848–855.
Zhang, L., Liu, W., Tanswell, A. K., and Luo, X. (2003). The effects of bilirubin on evoked potentials and long-term potentiation in rat hippocampus in vivo. Pediatr. Res. 53, 939–944.
Zhu, J., Woods, D., Mcmahon, M., and Bishop, J. M. (1998). Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 12, 2997–3007.
Zhu, X., Lee, H. G., Perry, G., and Smith, M. A. (2007). Alzheimer disease, the two-hit hypothesis: an update. Biochim. Biophys. Acta 1772, 494–502.
Keywords: bilirubin, biliverdin reductase, heme oxygenase, aging, cellular senescence, reactive oxygen species, antioxidant, lipid peroxidation
Citation: Kim SY and Park SC (2012) Physiological antioxidative network of the bilirubin system in aging and age-related diseases. Front. Pharmacol. 3:45. doi: 10.3389/fphar.2012.00045
Received: 09 January 2012; Accepted: 27 February 2012;
Published online: 14 March 2012.
Edited by:
Jaime Kapitulnik, The Hebrew University of Jerusalem, IsraelReviewed by:
Mahin D. Maines, University of Rochester School of Medicine, USACopyright: © 2012 Kim and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Sang Chul Park, Lee Gil Ya Cancer and Diabetes Institute, Gachon University, 7-45 Songdo dong, Yeonsu gu, Incheon 406-849, South Korea. e-mail:Ymx1ZW9jZWFuQGdhY2hvbi5hYy5rcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.