
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 09 March 2012
Sec. Drug Metabolism and Transport
Volume 3 - 2012 | https://doi.org/10.3389/fphar.2012.00036
This article is part of the Research Topic The role of bile pigments in health and disease: effects on cell signaling, cytotoxicity and cytoprotection View all 20 articles
 Rosaria Santangelo1†
Rosaria Santangelo1† Cesare Mancuso2*† Simona Marchetti1 Enrico Di Stasio3 Giovambattista Pani4 and Giovanni Fadda1
Cesare Mancuso2*† Simona Marchetti1 Enrico Di Stasio3 Giovambattista Pani4 and Giovanni Fadda1
Bilirubin-IX-alpha (BR) is the final product of heme metabolism through the heme oxygenase/biliverdin reductase (HO/BVR) system. Previous papers reported on the microbicidal effects of the HO by-products biliverdin-IX-alpha, carbon monoxide and iron, through either direct or indirect mechanisms. In this paper the evidence of a virucidal effect of BR against human herpes simplex virus type 1 (HSV-1) and the enterovirus EV71 was provided. Bilirubin-IX-alpha, at concentrations 1–10 μM, close to those found in blood and tissues, significantly reduced HSV-1 and EV71 replication in Hep-2 and Vero cell lines, respectively. Bilirubin-IX-alpha inhibited viral infection of Hep-2 and Vero cells when given 2 h before, concomitantly and 2 h after viral infection. Furthermore, BR retained its antiviral activity even complexed with a saturating concentration of human serum-albumin. Moreover, 10 μM BR increased the formation of nitric oxide and the phosphorylation of c-Jun N-terminal kinase in Vero and Hep-2 cell lines, respectively, thus implying a role of these two pathways in the mechanism of antiviral activity of the bile pigment. In conclusion, these results support the antiviral effect of BR against HSV-1 and enterovirus in vitro, and put the basis for further basic and clinical studies to understand the real role of BR as an endogenous antiviral molecule.
The heme oxygenase/biliverdin reductase (HO/BVR) axis is the main metabolic pathway by which heme is degraded. The combined action of these enzymes converts heme into ferrous iron (FeII), carbon monoxide (CO), and biliverdin-IX-alpha (BV) which is the precursor of bilirubin-IX-alpha (BR; Maines, 1997; Mancuso and Barone, 2009). For several years, the by-products of the HO/BVR axis were considered mere waste products, but over the past two decades, a number of investigators have focused their attention on both HO/BVR and their products in an attempt to elucidate their true biological functions. Stocker et al. (1987b) described the antioxidant properties of BR, and 6 years later Verma et al. (1993) proposed a role for CO as an endogenous neuromodulator. These observations were followed by numerous papers demonstrating CO’s important role as a regulator of synaptic transmission, cardiac function, and vessel tone (Wu and Wang, 2005). Bilirubin-IX-alpha helps maintain the cell’s redox equilibrium by activating important pro-survival signaling pathways, such as those involving the proto-oncogene Akt or the mitogen-activated protein kinase family (MAPK), and by scavenging reactive oxygen and nitrogen species (ROS and RNS, respectively; Stocker et al., 1987b; Minetti et al., 1998; Kaur et al., 2003; Mancuso et al., 2006b, 2008). These findings suggest that up-regulation of the HO/BVR axis is a useful mechanism for improving cells’ responses to stress, and substances known to increase HO activity in vitro are being explored as potential drugs for the treatment of free radical-induced diseases (Mancuso and Barone, 2009). In addition, the up-regulation of HO was shown to play an important role against bacterial and viral infections. The overexpression of the inducible isoform of HO (HO-1) displayed a significant antiviral activity against enterovirus and hepatitis viruses (Zhu et al., 2010; Tung et al., 2011). With regard to the by-products of the HO system involved in this antiviral activity, BV was shown to interfere with human hepatitis C virus (HCV) and human herpes virus (HHV)-6 replication as well as to reduce the cytopathic effect of human immunodeficiency type 1 virus (HIV) in Huh or MT-4 cells (Mori et al., 1991; Nakagami et al., 1992; Lehmann et al., 2010; Zhu et al., 2010). In addition, CO inhibited the growth of enterovirus, E. coli, P. aeruginosa as well as S. aureus (Nobre et al., 2007; Tung et al., 2011; Desmard et al., 2012) and iron almost completely decreased HCV core mRNA and protein (Hou et al., 2009). Taken together these results underlined a major role for HO in antiviral and antibacterial defense. That said, it is noteworthy to mention that no data are available to date about the potential antiviral activity of BR produced by BVR. As mentioned before, BR was shown to serve as an efficient scavenger for ROS which, in turn, are currently viewed as effectors of the antimicrobial activity of the immune response (Bogdan et al., 2000; Fang, 2004; Fialkow et al., 2007), therefore a direct antiviral role for BR is unlikely. On the other hand, recent findings demonstrated alternative mechanisms for the cytoprotective activity of BR, including the stimulation of nitric oxide (NO) release and the modulation of the MAPK system in pheochromocytoma cells and primary cultures of rat cerebellar granules (Mancuso et al., 2008). NO plays an important role in the antiviral defense (Croen, 1993; Akaike and Maeda, 2000) and it is also an intermediate in the bactericidal activity of certain drugs (Timmins et al., 2004; Koide et al., 2009). Moreover, the up-regulation of specific MAPK, such as the c-Jun N-terminal kinase (JNK), was shown to increase the cytoprotective defense against viruses (Hrincius et al., 2010).
The aim of this work was to explore the antiviral activity of BR in vitro and the plausible intracellular systems involved in this protective effect. Since intracellular BR exists in the free form whereas in the bloodstream the bile pigment is bound to human serum-albumin (HSA), the antiviral activity of both BR and BR–HSA were studied. The viral species against which BR and BR–HSA were tested are the human herpes simplex type 1 virus (HSV-1), a DNA virus which belong to the Herpesviridae family, and enterovirus 71 (EV71) which belong to the Picornaviridae family (RNA viruses). Both these viruses are responsible for important acute and chronic diseases in humans.
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated. Bilirubin-IX-alpha (Frontier Scientific, Carnforth, Lancashire, UK) was dissolved in sodium hydroxide (0.1 M) at a concentration of 10 mM and further diluted in double-distilled water. Bilirubin-IX-alpha solution was freshly prepared before each experiment and protected from light. The formation of the BR–HSA complex, at saturating concentrations of the protein, was allowed by incubating BR and HSA for 10 min at 37°C in the dark at a ratio up to 1:2 (Mancuso et al., 2006a). The fluorescent cell-permeant dye 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) diacetate was purchased from Molecular Probes (Invitrogen Carlsbad, CA, USA) and the JNK inhibitor SP600125 was from Assay Design (Enzo Life Sciences International, Inc., Plymouth Meeting, PA, USA). Anti-pJNK Thr183/Tyr185, antibody was purchased from Cell Signaling, (Danvers, MA, USA) and the anti-α-tubulin rabbit monoclonal antibody was from Thermo Scientific (Rockford, IL, USA).
The human laryngeal carcinoma cell line (Hep-2) and African green monkey kidney epithelial cells line (Vero) were maintained in Minimum-Essential-Medium (MEM) additioned with 10% fetal calf serum (FCS), glutamine (200 mM), penicillin (100 U/ml) and streptomycin (100 μg/ml). Human herpes simplex virus type 1 (KOS strain) and EV71 were purchased from the ATCC (Milan, Italy) and propagated and titrated in Hep-2 and Vero cells, respectively. The virus titre for both HSV-1 and EV71 was calculated by the plaque assay (Saijets et al., 2003; Hung et al., 2010).
The day before the experiment, 1 × 105 Hep-2 or Vero cells were seeded in 24-multiwell plates at a density of 400 cells/mm2. After overnight incubation, cells were treated with HSV-1 or EV71 alone or in the presence of test substances as described in Protocols 1–4.
In order to mimic the different conditions during which BR could interfere with viral infectivity, four different protocols were designed.
Hep-2 and Vero cells were pre-incubated with BR (1–10 μM) for 2 h. At the end of incubation, the BR-containing medium was discarded, replaced with fresh medium and the cells infected with HSV-1 and EV71. After 2 h, the inocula were removed and the cells incubated in fresh culture medium for 24–48 h. These time points were selected considering the replication cycles of both HSV-1 and enterovirus which are 10–20 h.
Herpes simplex virus type 1 and EV71 were pre-incubated with BR (1–10 μM) alone or complexed with saturating HSA (20 μM) for 1 h and then transferred to Hep-2 and Vero monolayers for the infection. After 2 h, the inocula were removed and the cells incubated in fresh culture medium for 24–48 h. The effect of HSA per se on viral replication was also evaluated by incubating either HSV-1 or EV71 with 20 μM HSA and then transferring the inocula to Hep-2 and Vero cells as described above.
Hep-2 and Vero cells were exposed to BR (1–10 μM) alone or complexed with saturating HSA (20 μM) and concomitantly infected with HSV-1 and EV71 for 2 h. At the end of incubation, the inocula were removed and the cells incubated in fresh culture medium for 24–48 h. The antiviral effect of HSA per se was also evaluated by infecting Hep-2 and Vero cells with either HSV-1 or EV71 in the presence of 20 μM HSA as described above.
After 2 h of infection with HSV-1 and EV71, the inocula were removed and Hep-2 and Vero monolayers treated with BR (1–10 μM) for further 2 h. At the end of incubation the BR-containing medium was discarded and the cells incubated in fresh culture medium for 24–48 h.
In preliminary experiments, both Hep-2 and Vero cells were incubated with 10–100 μM NaOH for 2 h and then incubated with fresh medium for 24–48 h to evaluate whether or not this substance affected viral infectivity.
In selected experiments, performed following protocols 1 and 3, Hep-2 and Vero cells were pre-incubated with the NO synthase (NOS) inhibitor L-NG-monomethyl arginine citrate (L-NMMA, 1 mM) for 1 h and further for 2 h in the presence of BR (10 μM) or BR (10 μM)-HSA (20 μM).
The number of infected cells were calculated by immunofluorescence as described below.
At the end of the 24 or 48-h incubation, cells were washed with PBS to remove residual culture medium, gently scraped and counted. Twenty-five thousand infected cells were seeded on single slide (four slides for each well) and incubated with anti HSV-1 monoclonal antibody (Imagen Herpes simplex virus direct IF test, Dako Cytomation, UK) or Pan-enterovirus antibody blend (PAN ENTERO BLEND, Light Diagnostics, Temecula, CA, USA) according to the manufacturers’ instructions. Immunofluorescence is a simple and reliable method to assess viral infection and is routinely used to estimate either herpesvirus or enterovirus infectivity (Klespies et al., 1996; Saijets et al., 2003; Copeland et al., 2009; Prichard et al., 2009). Both total cells and immunofluorescence-positive cells in each sample were counted in at least five microscopy fields (120–160 total cells and ∼5–140 positive cells per field) by two independent blinded investigators and this latter expressed as percentage respect to total cells. The inter-investigator variability rate of cell count was less than 10%.
Intracellular NO was detected in Vero cells treated for 2 h with BR (1–10 μM) in 0% FCS MEM by using the fluorescent probe DAF-FM-DA as previously described (Mancuso et al., 2008).
Confluent Hep-2 and Vero cells were lysed in lysis buffer (150 mM NaCl, 50 mM Tris–HCl, pH 8, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, leupeptin, and pepstatin, 1 mM sodium orthovanadate) containing 1% Triton X-100 and 0.1% SDS; lysates were cleared by centrifugation, and equal amounts of protein (40 μg) for each sample were subjected to SDS-PAGE on 10% gel. Western blot analysis was performed as previously described (Mancuso et al., 2008) by using primary antibodies (see above) diluted 1:1000 and HRP-conjugated secondary antibodies as appropriate.
Data are expressed as mean ± SEM of (N) individual samples per group. Statistical analysis was performed using ANOVA combined with Dunnet’s or two-tailed Student’s t-tests for comparison within the same group or two groups, respectively. Differences were considered significant at P < 0.05. The maximal inhibitory activity values were calculated and confirmed by non-linear regression analysis using a Prism 4.0 software (GraphPad Software).
The first step was to titrate both HSV-1 and EV71 by the plaque assay. The titer for HSV-1 was calculated to be 1.3 × 107 plaque forming unit (PFU)/ml whereas for EV71 was 1 × 109 PFU/ml. At these titers both HSV-1 and EV71 completely detached the Hep-2 and Vero cell monolayers as early as 24 from infection and this did not allow us to carry out any experiment to prove the possible protective effect of 1–10 μM BR. For this reason, in all the experimental protocols described below, cells were infected with either HSV-1 or EV71 at the titer 1 × 105 PFU.
In preliminary experiments, the unspecific effects of NaOH, the vehicle for BR, on viral replication was studied. Sodium hydroxide in the range 10–100 μM (i.e., the concentrations in 1–10 μM BR) did not have any significant effect on HSV-1 or EV71 infectivity at 24–48 h of incubation (data not shown).
As shown in Figure 1, Hep-2 and Vero cells pre-incubated with BR (1–10 μM) for 2 h and then infected with HSV-1 and EV71, were markedly resistant to the lethal effect of both the viruses and the percentage of infected cells were significantly reduced both at 24 and 48 h after infection (Figure 1). However, in this experimental setting BR was slightly more efficacious against HSV-1 than EV71 at 48 h from infection (mean maximal inhibitory activity 95 and 83%, respectively) whereas the efficacy at 24 h was comparable. It is noteworthy to mention that BR displayed a greater potency in inhibiting both HSV-1 and EV71 infectivity at 24 h than at 48 h from the infection.
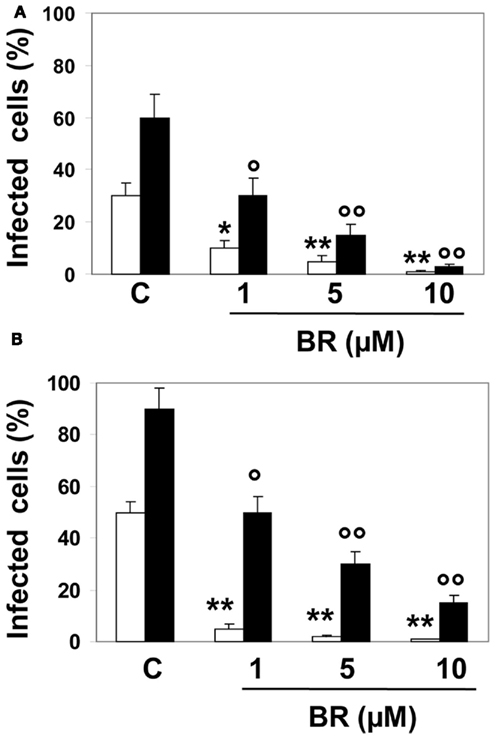
Figure 1. Effect of bilirubin (BR) on herpes simplex virus type 1 (HSV-1) and enterovirus EV71 replication. Hep-2 (A) and Vero (B) cells were pre-incubated with BR (1–10 μM) for 2 h. At the end of incubation, BR was removed and Hep-2 and Vero cells infected with HSV-1 and EV71, respectively, both at the titer 1 × 105 PFU. After 2 h, the inocula were removed and the cells incubated in culture medium for 24 (white columns) or 48 h (black columns). Data are expressed as the percentage of infected cells, mean ± SEM of six individual samples per group. *P < 0.05 and **P < 0.01 versus control (C) after 24 h from infection; °P < 0.05 and °°P < 0.01 versus control after 48 h from infection.
When HSV-1 and EV71 were pre-incubated with BR (1–10 μM) alone or complexed with HSA (20 μM) for 2 h and then transferred onto Hep-2 and Vero monolayers, the bile pigment significantly inhibited viral infectivity (Figure 2). In particular, BR alone exhibited lower efficacy against HSV-1 than EV71 at 24 h from infection (mean maximal inhibitory activity 98 versus 82%, respectively) whereas at 48 h the efficacy was comparable. Furthermore, the complexation with HSA did not alter the efficacy of BR against HSV-1 and EV71 at shorter time from infection whereas reduced the efficacy after 48 h (mean maximal inhibitory activity 54 versus 82%, respectively). Moreover, the complexation with HSA reduced the potency of BR to block HSV-1 replication after 24 h from infection.
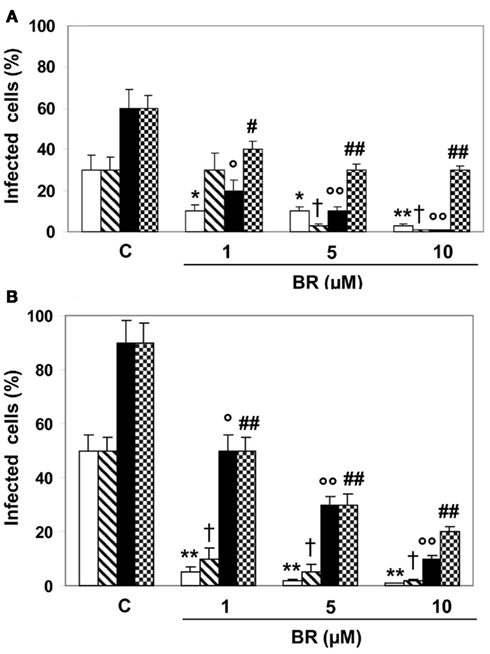
Figure 2. Direct effect of bilirubin (BR) and albumin-bound bilirubin (BR–HSA) on herpes simplex virus type 1 (HSV-1) and enterovirus EV71 and how this affects viral replication. Both HSV-1 and EV71 (1 × 105 PFU) were pre-incubated with BR (1–10 μM) alone or complexed with saturating HSA (20 μM) for 1 h and then transferred to Hep-2 (A) and Vero (B) cell monolayers for the infection. After 2 h, the inocula were removed and the cells incubated in culture medium for 24 h (white and dashed columns) or 48 h (black and dotted columns). Both dashed and dotted columns refer to HSA treatment as described in the Section “Materials and Methods.” Data are expressed as the percentage of infected cells, mean ± SEM of six individual samples per group. *P < 0.05 versus control (C) after 24 h from infection; ** or †P < 0.01 versus respective controls after 24 h from infection; ° or #P < 0.05 and °° or ##P < 0.01 versus respective controls after 48 h from infection.
In Hep-2 and Vero cells treated with BR (1–10 μM) alone or complexed with HSA (20 μM) and concomitantly infected with HSV-1 and EV71, the linear tetrapyrrole significantly modified viral infectivity (Figure 3). In details, BR alone showed a comparable efficacy against HSV-1 and EV71 at both 24 and 48 h. The complexation with HSA only partially confirmed this trend, since BR exhibited similar efficacy against HSV-1 and EV71 at 24 h, but a lower efficacy at 48 h of incubation (mean maximal inhibitory activity 62 and 82%, respectively). Similarly to Protocol 2, the complexation with HSA reduced the potency of BR to block HSV-1 replication after 24 h from infection.
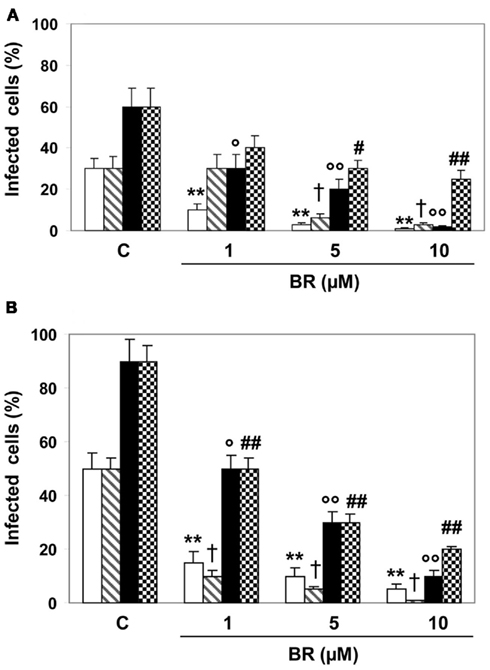
Figure 3. Effect of bilirubin (BR) and albumin-bound bilirubin (BR–HSA) given concurrently with herpes simplex virus type 1 (HSV-1) and enterovirus EV71 and how this affects viral replication. Hep-2 (A) and Vero (B) cells were exposed to BR (1–10 μM) alone or complexed with saturating HSA (20 μM) and concomitantly infected with HSV-1 and EV71 (1 × 105 PFU) for 2 h. At the end of incubation the inocula were removed and the cells incubated in culture medium for 24 (white and dashed columns) or 48 h (black and dotted columns). Both dashed and dotted columns refer to HSA treatment as described in the Section “Materials and Methods.” Data are expressed as the percentage of infected cells, mean ± SEM of six individual samples per group. *P < 0.05 versus control (C) after 24 h from infection; ** or †P < 0.01 versus respective controls after 24 h from infection; ° or #P < 0.05 and °° or ##P < 0.01 versus respective controls after 48 h from infection.
When given after viral infection, BR (1–10 μM) significantly reduced HSV-1 virulence only within the first 24 h from the infection, whereas after 48 h only the higher concentration was active (Figure 4). Conversely, BR maintained its antiviral action against EV71 at both 24 and 48 h from the infection (Figure 4).
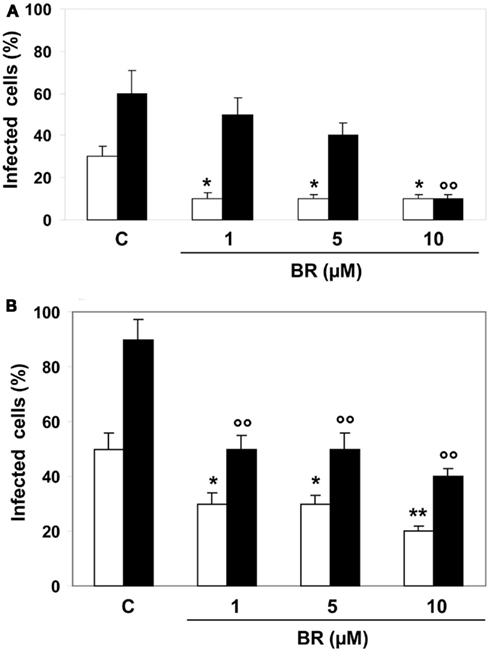
Figure 4. Effects of bilirubin (BR) on cell monolayers infected with herpes simplex virus type 1 (HSV-1) and enterovirus EV71. After 2 h of infection with HSV-1 and enterovirus (1 × 105 PFU), Hep-2 (A) and Vero (B) cell monolayers were treated with BR (1–10 μM) for further 2 h. At the end of incubation the inocula were removed and the cells incubated in culture medium for 24 (white columns) or 48 h (black columns). Data are expressed as the percentage of infected cells, mean ± SEM of six individual samples per group. *P < 0.05 and **P < 0.01 versus control (C) after 24 h from infection; °°P < 0.01 versus control after 48 h from infection.
Bilirubin-IX-alpha (1–10 μM) stimulated the phosphorylation of the member of the MAPK family JNK in Hep-2 and Vero cell lines (Figure 5A and data not shown), but a positive correlation between BR concentrations and JNK phosphorylation was not found. The JNK inhibitor SP600125 (10 μM) partially reversed the BR (10 μM)-induced block of HSV-1 and EV71 replication in Hep-2 and Vero cells at both 24 and 48 h from infection (protocols 1 and 3), but this result only approached statistical significance (data not shown).
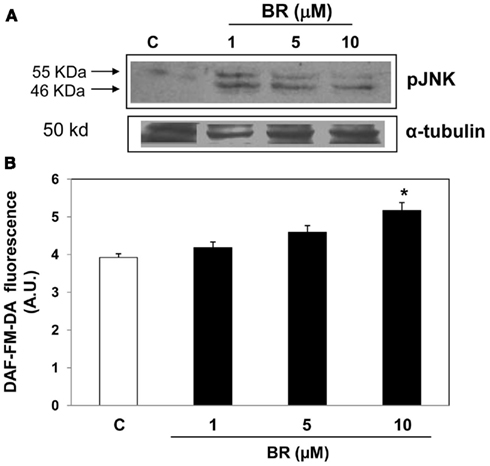
Figure 5. Effect of bilirubin (BR) on cellular expression of the c-Jun N-terminal kinase (JNK) and nitric oxide (NO) production. Hep-2 and Vero cells were pre-incubated with BR (1–10 μM) for 2 h. At the end of incubation, BR was removed, the Hep-2 cells lysed and assayed for JNK phosphorylation by Western Blot (A) as described under the Section “Materials and Methods.” α-tubulin was used as housekeeping gene to verify protein equal loading. On the contrary, Vero cells were assayed for NO production by using the fluorescent probe DAF-FM-diacetate [DAF-FM-DA, (B)] as described under the Section “Materials and Methods.” (A) Shows a representative gel. In (B), data are expressed as DAF-FM-DA fluorescence arbitrary units (A.U), mean ± SEM of six individual samples per group. *P < 0.05 versus control (C).
In addition, BR (10 μM) increased the production of NO in Vero cells in a time-dependent manner reaching statistical significance as early as 2 h after the administration (Figure 5B). The involvement of BR-dependent generation of NO in the inhibition of viral replication was confirmed in specific experiments performed by incubating Hep-2 and Vero cells with L-NMMA (1 mM) 1 h before and then in the presence of BR (10 μM, 2 h) as described in protocol 1. These experiments demonstrated that the inhibition of NOS activity reversed the BR-induced block of viral replication in HSV-1 and EV71 after 24 h and 48 h from infection (Figure 6). A similar degree of inhibition of viral replication was found when HSV-1 and Vero cells were pre-incubated with L-NMMA for 1 h and then exposed simultaneously to BR alone or complexed with HSA plus HSV-1 or EV71 according to protocol 3 (data not shown). This effect of BR was comparable at both 24 and 48 h from infection (data not shown).
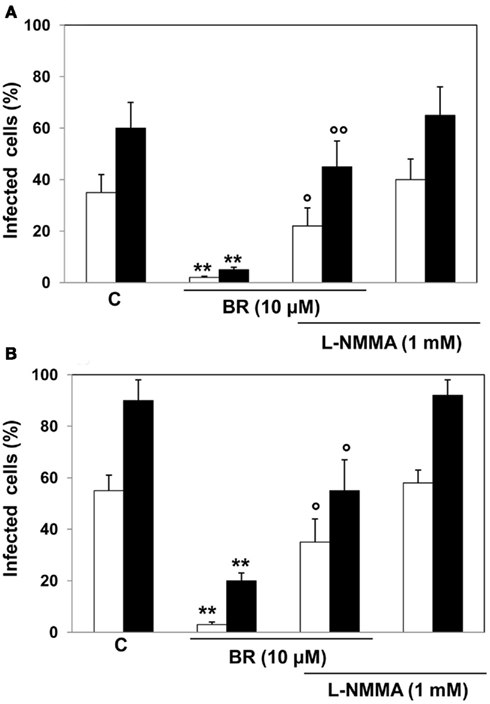
Figure 6. Effect of bilirubin (BR) plus the nitric oxide synthase inhibitor L-NG-monomethyl arginine citrate (L-NMMA) on herpes simplex virus type 1 (HSV-1) and enterovirus EV71 replication. Hep-2 (A) and Vero (B) cells were pre-incubated with 1 mM L-NMMA for 1 h and then with L-NMMA plus 10 μM BR for further 2 h. At the end of incubation, the medium containing L-NMMA plus BR was removed and Hep-2 and Vero cells infected with HSV-1 and EV71 (1 × 105 PFU), respectively. After 2 h, the inocula were removed and the cells incubated in culture medium for 24 (white columns) or 48 h (black columns). Data are expressed as the percentage of infected cells, mean ± SEM of six individual samples per group. **P < 0.01 versus respective controls (C) and °P < 0.05 and °°P < 0.01 versus 10 μM BR after 24 h (white columns) and 48 h (black columns) from viral infection.
Over the last years, several studies reported on the microbicidal activity of HO-1 and its by-products. Biliverdin-IX-alpha was shown to inhibit the replication of HCV, HHV-6, and HIV (Mori et al., 1991; Nakagami et al., 1992; Lehmann et al., 2010; Zhu et al., 2010), CO reduced the growth of E. coli, P. aeruginosa, and S. aureus (Nobre et al., 2007, 2009; Davidge et al., 2009; Desmard et al., 2012) and iron almost completely decreased HCV core mRNA and protein (Hou et al., 2009). The results shown in this paper provided original evidence about the antiviral role of BR, the formation of which requires the concerted action of both HO and BVR, thus expanding the cytoprotective role of this linear tetrapyrrole. However, in order to appreciate the novelty of our study, some considerations about the pathophysiology of the HO/BVR system are needed.
Although HO-1 is considered ubiquitous, this isoform is abundantly expressed in liver, spleen, heart, kidney, and selected brain regions and its activity increases greatly under stressful conditions (Maines, 1997). On the contrary, the constitutive HO-2 isoform prevails in neurons and testicular germ cells (Maines, 1997). Almost all organs retain BVR activity which transforms BV into BR (Maines, 1997, 2005). That said, it is plausible to wonder whether or not the antiviral activity of BV against HHV-6, HIV, or HCV (Mori et al., 1991; Nakagami et al., 1992; Lehmann et al., 2010; Zhu et al., 2010) should be attributable to its conversion into BR by the MT-4 or Huh cells used in those studies rather than a distinct effect of BV. The lack of inactivation of HHV-6 virions by BV and the consequent reduced antiviral effect lend support to this hypothesis. Another concern raised from the studies by Nakagami et al. (1992) and Mori et al. (1991) is related to the concentrations of BV used to inhibit HHV-6 and HIV replication. In these studies the antiviral effect of BV was observed at concentrations ranging from 10 to 66.7 μg/ml, equivalent to ∼17 and 113 μM, respectively, two values much higher than those expected to be produced by the HO activity in humans (Coburn, 1970). These potential biases were overcome in our study which demonstrated a direct antiviral effect for BR, the stable end product of heme metabolism, at concentrations from 1 to 10 μM which are very close to those found in human plasma (Berk et al., 1969; Bloomer et al., 1971) and about 1 order of magnitude lower than those previously used to demonstrate the antiviral effect of BV. From the analysis of the pharmacodynamic data, it emerges a different sensitivity of HSV-1 and EV71 to both free and albumin-bound BR under the various experimental conditions. A careful comparative evaluation of the mechanisms which underlie the ability of BR to inhibit HSV-1 and EV71 infectivity under the conditions which characterized Protocols 1–4, is out of the scope of this paper. However, it is important to highlight that, in this study, both BR and BR–HSA significantly inhibited the infectivity of both HSV-1 and EV71 in all the experimental settings well within the range of physiologic concentrations. A preliminary conclusion drawn from our results was that BR was much more efficacious to inhibit viral infectivity after 24 h from infection and, in particular, when the bile pigment was complexed to HSA.
The results shown in this study suggest that BR reduce viral replication by acting at several stages. The ability of BR to decrease HSV-1 and EV71 infectivity if pre-incubated with Hep-2 and Vero cells (Figure 1) resembles the pathophysiologic condition in which a virus infects cells endowed with high concentration of BR (e.g., hepatocytes, neurons). This finding is clinically relevant because nervous system and liver are both target organs for HSV-1 and enterovirus. Taking into consideration that there is not a direct clinical evidence of a reduced incidence of viral diseases in subjects affected by conditions characterized by unconjugated hyperbilirubinemia, such as Gilbert’s syndrome (Hirschfield and Alexander, 2006), we hypothesize a protective role for BR in viral diseases in analogy with the well known protective effect of the bile pigment against respiratory diseases and cardiovascular accidents (Vitek et al., 2002; Horsfall et al., 2011). With regard to the inhibition of viral infectivity by both BR and BR–HSA pre-incubated directly with virions (Figure 2), it recalls the pathophysiologic condition in which a virus comes in contact with BR or BR–HSA in the blood, as during the viremic phase, and then infects tissues. Similarly, both BR and BR–HSA reduced viral replication if given cells concomitantly with HSV-1 and EV71 (Figure 3) and this mimics the pathophysiologic condition in which a virus comes in contact with BR/BR–HSA and cells at the blood/tissue interface. However, it is well known that both herpesviruses and enteroviruses have a short viremic phase since they colonize target organs very fast and, therefore, the potential clinical importance of both these mechanisms is limited to severe diseases characterized by long-lasting veremic phase such as in immunocompromised patients (Stanberry et al., 1994). Finally, BR was efficacious in reducing viral infection even when administered to Hep-2 and Vero cells already infected by HSV-1 and EV71, respectively (Figure 4). This experimental condition reminds of the clinical situation in which jaundice is secondary to a viral infection, such as hepatitis. As a matter of fact, in viral hepatitis unconjugated BR increases when hepatic function decreases under a critical threshold and it is very often accompanied by hypoalbuminemia and hypofibrinogenemia and these alterations represent a life-threatening condition. The possible protective effect of BR at this stage is, therefore, unlikely.
Overall speaking, the mechanisms involved in the antiviral activity of BR within the cell or at the interface blood/tissue could be related either to the stimulation of intracellular pro-survival pathways, such as the MAPK system, or the production of microbicidal molecules such as NO. The BR-induced stimulation of NO release in Vero cells (Figure 5) should be considered as one of the main mechanisms involved in the antiviral effect of the linear tetrapyrrole against both HSV-1 and EV71. The importance of NO to decrease HSV-1 and EV71 infectivity was also corroborated by the evidence that the blockade of NOS, by the inhibitor L-NMMA, reverted the reduction of viral infectivity due to 10 μM BR (Figure 6). This result is not surprising and is in good agreement with previous studies which demonstrated the virucidal effect of NO (Croen, 1993; Akaike and Maeda, 2000). The JNK signaling cascade is also a crucial effector of the antiviral immune response in influenza virus (Hrincius et al., 2010), but there are no data in favor of a cytoprotective effect against HSV-1 or EV71. The lack of a positive correlation between BR concentrations and JNK phosphorylation (Figure 5A, in particular the 55-KDa isoform) could be explained keeping in mind that the bile pigment was used within the range 1–10 μM. It is interesting to recall that higher concentrations of BR (50 μM) were necessary to clearly stimulate the phosphorylation of MAPK members including JNK (Fernandes et al., 2007). In addition, the lack of any significant effect of the JNK inhibitor SP600125 (data not shown) on HSV-1 and EV71 infectivity lends support to the hypothesis that this pathway is not so efficacious to prevent viral infection as NO production. The parallel antiviral activity of BR against HSV-1 and EV71, characterized by important differences in terms of virion structure, a DNA-containing core surrounded by an envelope in HSV-1 and a RNA-containing core within a capsid without envelope in EV71, suggests that BR acts directly on the virion particles or on nucleic acids. In this light, some studies reported on a toxic effect of BR, in the presence of transition metals, on nucleic acids (Asad et al., 1999). Furthermore, the lack of direct antiviral activity of BV on HHV-6 and HSV-1 (Nakagami et al., 1992) vis-à-vis with the efficient antiviral effect of BR against HSV-1 and EV71, implies an important role of the C-10, in analogy with previous results. The absence of double bonds at the γ-bridge (which includes C-10) stabilizes the structure of BR and increases its liposolubility with respect to BV. Moreover, the C-10 can undergo redox modifications under conditions of oxidative stress (Stocker et al., 1987a; Baranano et al., 2002). According to this hypothesis, free radicals could oxidize protein-bound BR at the C-10 generating BV which, in turn, is quickly reduced back to BR by the ubiquitous BVR (Baranano et al., 2002). However, this hypothesis is still debated in the scientific community (Maghzal et al., 2009), but it cannot be excluded to explain the antiviral effect of BR toward both HSV-1 and EV71.
In conclusion, the results in this paper provide the first evidence, to our knowledge, of a virucidal effect of BR against HSV-1 and EV71. The implication of these results for the development of new antiviral drugs designed to modulate the HO/BVR system are profound and research on these subjects is ongoing in our laboratories.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Authors want to thank Mrs. Simona Galuppi, Franca Pedone, and Creola Rocchetti for the expert technical assistance. This work was supported by Fondi Ateneo to Cesare Mancuso.
Asad, S. F., Singh, S., Ahmad, A., and Hadi, S. M. (1999). Bilirubin-Cu(II) complex degrades DNA. Biochim. Biophys. Acta 1428, 201–208.
Baranano, D. E., Rao, M., Ferris, C. D., and Snyder, S. H. (2002). Biliverdin reductase: a major physiologic cytoprotectant. Proc. Natl. Acad. Sci. U.S.A. 99, 16093–16098.
Berk, P. D., Howe, R. B., Bloomer, J. R., and Berlin, N. I. (1969). Studies of bilirubin kinetics in normal adults. J. Clin. Invest. 48, 2176–2190.
Bloomer, J. R., Berk, P. D., Howe, R. B., and Berlin, N. I. (1971). Interpretation of plasma bilirubin levels based on studies with radioactive bilirubin. JAMA 218, 216–220.
Bogdan, C., Rollinghoff, M., and Diefenbach, A. (2000). Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr. Opin. Immunol. 12, 64–76.
Copeland, A. M., Newcomb, W. W., and Brown, J. C. (2009). Herpes simplex virus replication: roles of viral proteins and nucleoporins in capsid-nucleus attachment. J. Virol. 83, 1660–1668.
Croen, K. D. (1993). Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J. Clin. Invest. 91, 2446–2452.
Davidge, K. S., Sanguinetti, G., Yee, C. H., Cox, A. G., Mcleod, C. W., Monk, C. E., Mann, B. E., Motterlini, R., and Poole, R. K. (2009). Carbon monoxide-releasing antibacterial molecules target respiration and global transcriptional regulators. J. Biol. Chem. 284, 4516–4524.
Desmard, M., Foresti, R., Morin, D., Dagoussat, M., Berdeaux, A., Denamur, E., Crook, S. H., Mann, B. E., Scapens, D., Montravers, P., Boczkowski, J., and Motterlini, R. (2012). Differential antibacterial activity against Pseudomonas aeruginosa by carbon monoxide-releasing molecules. Antioxid. Redox Signal. 16, 153–163.
Fang, F. C. (2004). Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2, 820–832.
Fernandes, A., Falcao, A. S., Silva, R. F., Brito, M. A., and Brites, D. (2007). MAPKs are key players in mediating cytokine release and cell death induced by unconjugated bilirubin in cultured rat cortical astrocytes. Eur. J. Neurosci. 25, 1058–1068.
Fialkow, L., Wang, Y., and Downey, G. P. (2007). Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic. Biol. Med. 42, 153–164.
Hirschfield, G. M., and Alexander, G. J. (2006). Gilbert’s syndrome: an overview for clinical biochemists. Ann. Clin. Biochem. 43, 340–343.
Horsfall, L. J., Rait, G., Walters, K., Swallow, D. M., Pereira, S. P., Nazareth, I., and Petersen, I. (2011). Serum bilirubin and risk of respiratory disease and death. JAMA 305, 691–697.
Hou, W. H., Rossi, L., Shan, Y., Zheng, J. Y., Lambrecht, R. W., and Bonkovsky, H. L. (2009). Iron increases HMOX1 and decreases hepatitis C viral expression in HCV-expressing cells. World J. Gastroenterol. 15, 4499–4510.
Hrincius, E. R., Wixler, V., Wolff, T., Wagner, R., Ludwig, S., and Ehrhardt, C. (2010). CRK adaptor protein expression is required for efficient replication of avian influenza A viruses and controls JNK-mediated apoptotic responses. Cell. Microbiol. 12, 831–843.
Hung, H. C., Chen, T. C., Fang, M. Y., Yen, K. J., Shih, S. R., Hsu, J. T., and Tseng, C. P. (2010). Inhibition of enterovirus 71 replication and the viral 3D polymerase by aurintricarboxylic acid. J. Antimicrob. Chemother. 65, 676–683.
Kaur, H., Hughes, M. N., Green, C. J., Naughton, P., Foresti, R., and Motterlini, R. (2003). Interaction of bilirubin and biliverdin with reactive nitrogen species. FEBS Lett. 543, 113–119.
Klespies, S. L., Cebula, D. E., Kelley, C. L., Galehouse, D., and Maurer, C. C. (1996). Detection of enteroviruses from clinical specimens by spin amplification shell vial culture and monoclonal antibody assay. J. Clin. Microbiol. 34, 1465–1467.
Koide, N., Naiki, Y., Morikawa, A., Tumurkhuu, G., Dagvadorj, J., Noman, A. S., Iftekar, E. K. I., Komatsu, T., Yoshida, T., and Yokochi, T. (2009). Nystatin-induced nitric oxide production in mouse macrophage-like cell line RAW264.7. Microbiol. Immunol. 53, 295–300.
Lehmann, E., El-Tantawy, W. H., Ocker, M., Bartenschlager, R., Lohmann, V., Hashemolhosseini, S., Tiegs, G., and Sass, G. (2010). The heme oxygenase 1 product biliverdin interferes with hepatitis C virus replication by increasing antiviral interferon response. Hepatology 51, 398–404.
Maghzal, G. J., Leck, M. C., Collinson, E., Li, C., and Stocker, R. (2009). Limited role for the bilirubin-biliverdin redox amplification cycle in the cellular antioxidant protection by biliverdin reductase. J. Biol. Chem. 284, 29251–29259.
Maines, M. D. (1997). The heme oxygenase system: a regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 37, 517–554.
Maines, M. D. (2005). New insights into biliverdin reductase functions: linking heme metabolism to cell signaling. Physiology (Bethesda) 20, 382–389.
Mancuso, C., and Barone, E. (2009). The heme oxygenase/biliverdin reductase pathway in drug research and development. Curr. Drug Metab. 10, 579–594.
Mancuso, C., Bonsignore, A., Capone, C., Di Stasio, E., and Pani, G. (2006a). Albumin-bound bilirubin interacts with nitric oxide by a redox mechanism. Antioxid. Redox Signal. 8, 487–494.
Mancuso, C., Pani, G., and Calabrese, V. (2006b). Bilirubin: an endogenous scavenger of nitric oxide and reactive nitrogen species. Redox Rep. 11, 207–213.
Mancuso, C., Capone, C., Ranieri, S. C., Fusco, S., Calabrese, V., Eboli, M. L., Preziosi, P., Galeotti, T., and Pani, G. (2008). Bilirubin as an endogenous modulator of neurotrophin redox signaling. J. Neurosci. Res. 86, 2235–2249.
Minetti, M., Mallozzi, C., Di Stasi, A. M., and Pietraforte, D. (1998). Bilirubin is an effective antioxidant of peroxynitrite-mediated protein oxidation in human blood plasma. Arch. Biochem. Biophys. 352, 165–174.
Mori, H., Otake, T., Morimoto, M., Ueba, N., Kunita, N., Nakagami, T., Yamasaki, N., and Taji, S. (1991). In vitro anti-human immunodeficiency virus type 1 activity of biliverdin, a bile pigment. Jpn. J. Cancer Res. 82, 755–757.
Nakagami, T., Taji, S., Takahashi, M., and Yamanishi, K. (1992). Antiviral activity of a bile pigment, biliverdin, against human herpesvirus 6 (HHV-6) in vitro. Microbiol. Immunol. 36, 381–390.
Nobre, L. S., Al-Shahrour, F., Dopazo, J., and Saraiva, L. M. (2009). Exploring the antimicrobial action of a carbon monoxide-releasing compound through whole-genome transcription profiling of Escherichia coli. Microbiology 155, 813–824.
Nobre, L. S., Seixas, J. D., Romao, C. C., and Saraiva, L. M. (2007). Antimicrobial action of carbon monoxide-releasing compounds. Antimicrob. Agents Chemother. 51, 4303–4307.
Prichard, M. N., Quenelle, D. C., Hartline, C. B., Harden, E. A., Jefferson, G., Frederick, S. L., Daily, S. L., Whitley, R. J., Tiwari, K. N., Maddry, J. A., Secrist, J. A. III, and Kern, E. R. (2009). Inhibition of herpesvirus replication by 5-substituted 4’-thiopyrimidine nucleosides. Antimicrob. Agents Chemother. 53, 5251–5258.
Saijets, S., Ylipaasto, P., Vaarala, O., Hovi, T., and Roivainen, M. (2003). Enterovirus infection and activation of human umbilical vein endothelial cells. J. Med. Virol. 70, 430–439.
Stanberry, L. R., Floyd-Reising, S. A., Connelly, B. L., Alter, S. J., Gilchrist, M. J., Rubio, C., and Myers, M. G. (1994). Herpes simplex viremia: report of eight pediatric cases and review of the literature. Clin. Infect. Dis. 18, 401–407.
Stocker, R., Glazer, A. N., and Ames, B. N. (1987a). Antioxidant activity of albumin-bound bilirubin. Proc. Natl. Acad. Sci. U.S.A. 84, 5918–5922.
Stocker, R., Yamamoto, Y., Mcdonagh, A. F., Glazer, A. N., and Ames, B. N. (1987b). Bilirubin is an antioxidant of possible physiological importance. Science 235, 1043–1046.
Timmins, G. S., Master, S., Rusnak, F., and Deretic, V. (2004). Requirements for nitric oxide generation from isoniazid activation in vitro and inhibition of mycobacterial respiration in vivo. J. Bacteriol. 186, 5427–5431.
Tung, W. H., Hsieh, H. L., Lee, I. T., and Yang, C. M. (2011). Enterovirus 71 induces integrin beta1/EGFR-Rac1-dependent oxidative stress in SK-N-SH cells: role of HO-1/CO in viral replication(1). J. Cell. Physiol. 226, 3316–3329.
Verma, A., Hirsch, D. J., Glatt, C. E., Ronnett, G. V., and Snyder, S. H. (1993). Carbon monoxide: a putative neural messenger. Science 259, 381–384.
Vitek, L., Jirsa, M., Brodanova, M., Kalab, M., Marecek, Z., Danzig, V., Novotny, L., and Kotal, P. (2002). Gilbert syndrome and ischemic heart disease: a protective effect of elevated bilirubin levels. Atherosclerosis 160, 449–456.
Wu, L., and Wang, R. (2005). Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol. Rev. 57, 585–630.
Keywords: bilirubin, biliverdin reductase, enterovirus, heme oxygenase, herpes simplex virus, nitric oxide
Citation: Santangelo R, Mancuso C, Marchetti S, Di Stasio E, Pani G and Fadda G (2012) Bilirubin: an endogenous molecule with antiviral activity in vitro. Front. Pharmacol. 3:36. doi: 10.3389/fphar.2012.00036
Received: 09 January 2012; Paper pending published: 27 January 2012;
Accepted: 20 February 2012; Published online: 09 March 2012.
Edited by:
Mahin D. Maines, University of Rochester School of Medicine, USAReviewed by:
Paavo Honkakoski, University of Eastern Finland, FinlandCopyright: © 2012 Santangelo, Mancuso, Marchetti, Di Stasio, Pani and Fadda. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Cesare Mancuso, Institute of Pharmacology, Catholic University School of Medicine, Largo Francesco Vito, 1, 00168 Roma, Italy. e-mail:Y21hbmN1c29Acm0udW5pY2F0dC5pdA==
†Rosaria Santangelo and Cesare Mancuso have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.