



95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 13 March 2012
Sec. Drug Metabolism and Transport
Volume 3 - 2012 | https://doi.org/10.3389/fphar.2012.00031
This article is part of the Research Topic The role of bile pigments in health and disease: effects on cell signaling, cytotoxicity and cytoprotection View all 20 articles
The expanse of human biliverdin reductase (hBVR) functions in the cells is arguably unmatched by any single protein. hBVR is a Ser/Thr/Tyr-kinase, a scaffold protein, a transcription factor, and an intracellular transporter of gene regulators. hBVR is an upstream activator of the insulin/IGF-1 signaling pathway and of protein kinase C (PKC) kinases in the two major arms of the pathway. In addition, it is the sole means for generating the antioxidant bilirubin-IXα. hBVR is essential for activation of ERK1/2 kinases by upstream MAPKK-MEK and by PKCδ, as well as the nuclear import and export of ERK1/2. Small fragments of hBVR are potent activators and inhibitors of the ERK kinases and PKCs: as such, they suggest the potential application of BVR-based technology in therapeutic settings. Presently, we have reviewed the function of hBVR in cell signaling with an emphasis on regulation of PKCδ activity.
Biliverdin reductase (BVR), purified to homogeneity from rat liver, was characterized as an enzyme that was capable of reducing biliverdin-IXα to bilirubin-IXα and was shown to exhibit a unique, dual cofactor/pH optimum profile (Kutty and Maines, 1981). In the course of elucidating the underlying mechanism for this unprecedented activity profile, an extensive array of functions of the human BVR (hBVR) protein was uncovered (Kapitulnik and Maines, 2009; Gibbs et al., 2012). Examination of the sequence has revealed the presence of three domains, as illustrated in Figure 1A. The N-terminal domain contains the catalytic site, while the extreme C-terminus is cysteine-rich and binds metal ions, notably Zn2+. The remainder of the molecule, as discussed below, harbors numerous regulatory motifs. An early observation made with the rat enzyme was that BVR was extensively modified posttranslationally (Huang et al., 1989), and it was subsequently demonstrated that this was a consequence of phosphorylation (Salim et al., 2001). The reductase activity of BVR required that the protein be phosphorylated. Rat or human BVR purified after expression in E. coli was found to be phosphorylated. Because E. coli has minimal Ser/Thr- or Tyr-kinase activity, the likely source of post-translational modification was autophosphorylation, indicating that BVR is a protein kinase. This kinase activity was characterized further using the human enzyme, and it was shown that BVR is a rare, soluble dual-specificity (Ser/Thr/Tyr) kinase (Lerner-Marmarosh et al., 2005). In its capacity as a Tyr-kinase, hBVR was shown to phosphorylate the insulin receptor substrate, effectively mimicking the insulin receptor kinase (IRK; Lerner-Marmarosh et al., 2005). This phosphorylation event initiates the down-regulation of glucose uptake (Tanti et al., 1994). hBVR itself was a substrate for tyrosine phosphorylation by IRK.
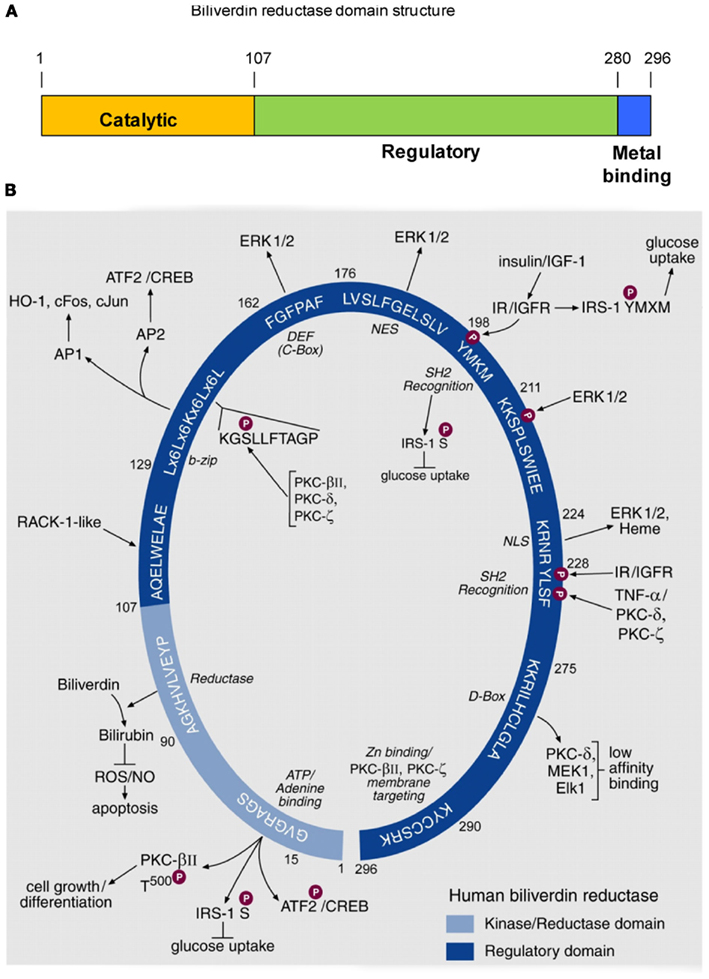
Figure 1. Domain structure of hBVR, and its functional specific sequence motifs. (A) Three domains of hBVR. The N-terminal domain includes the sequences required for catalytic activity. The regulatory domain comprises residues beyond position 107, and there is some overlap with the C-terminal metal-binding sequence. (B) Schematic presentation of consensus sequences of hBVR for which functions have been ascribed. The numbers indicated for each consensus sequences are those of the hBVR primary structure. The N-terminal segment of 99 residues is the catalytic domain of hBVR; it houses a sequence of four valines followed by the consensus for the ATP/adenine ring-binding site. The kinase activity of hBVR is responsible for its autophosphorylation (Salim et al., 2001; Lerner-Marmarosh et al., 2005). hBVR is a kinase for serine phosphorylation of IRS-1, the phosphorylation of which halts glucose uptake (Tanti et al., 1994). hBVR is also a likely kinase for T500 in the activating loop of PKCβII (Maines et al., 2007); the PKC is a key component of cell growth and differentiation. The reductase domain catalyzes reduction of biliverdin to bilirubin, a component of cellular defense mechanisms protecting against ROS (Sedlak et al., 2009) and apoptosis (Miralem et al., 2005). The sequences starting at aa 107 and 211 closely resemble sites in the primary sequence of repeats V (QAMLWDLNE) and VI (SIKIWDLE) of the Receptor for Activated C-Kinase-1 (RACK1). RACK1 is a 36-kDa protein that is similar in size to hBVR (Ron et al., 1994). Activation of PKCs, including the β, δ and ε isoforms (Ron et al., 1994), is associated with conformational change that exposes their RACK-binding sites. We predict that the presence of RACK1-like sequences in hBVR may allow its binding to PKCs. The binding would not require kinase activity of hBVR. The bZip motif binds to 7 and 8 bp AP-1 and AP-2 sites. Stress response genes are activated by AP-1, and cAMP-responsive genes are regulated by AP-2 regulatory elements. hBVR regulates expression of stress-responsive HO-1, c-Fos, c-Jun, and ATF2/CREB (Kravets et al., 2004; Miralem et al., 2005; Maines et al., 2007). Within this sequence is a motif that strongly resembles a conserved protein kinase motif (Hanks and Hunter, 1995). The high affinity ERK binding site, known either as C-Box or DEF (Jacobs et al., 1999), is the site of interaction of ERK1/2 and hBVR, positioning ERK in proximity to its kinase (Lerner-Marmarosh et al., 2008). Nuclear localization of hBVR is also critical for transport of the transcriptional regulators ERK1/2 and heme into the nucleus (Lerner-Marmarosh et al., 2008; Tudor et al., 2008). Reentry of ERK into the cytoplasm requires the intact hBVR NES (Lerner-Marmarosh et al., 2008). hBVR is directly phosphorylated by IRK upon activation by insulin or IGF-1 (Lerner-Marmarosh et al., 2005). The tyrosine in the SH2 recognition motif of hBVR, as with other SH2 recognition motif-containing proteins, is predicted to form a platform for formation of signaling complexes (Pawson and Scott, 1997). hBVR is phosphorylated by ERK, and MotifScan predicts serine in the SP sequence as the phosphorylation target site of ERK1/2. A second SH2 recognition motif follows the nuclear localization signal and is involved in activation of PKCζ by TNF-α (Lerner-Marmarosh et al., 2007). The low affinity D-Box-like sequence is the binding site for kinases and substrates in the MAPK signaling cascade. The C-terminal six residues are the Zn-binding domain of hBVR (Maines et al., 1996). Based on the reported role of Zn for plasma membrane translocation of PKCs and nuclear translocation of NF-κB (Kabu et al., 2006) we predict that the function of hBVR in translocation of PKCs β and ζ to the cell membrane may involve its associated Zn. Notably, hBVR under resting conditions is found in the cytoplasm and membrane caveolae (Kim et al., 2004). The C-terminal lysine 296 is critical for hBVR’s catalytic activity (unpublished); although it lies in a disordered region of the BVR molecule (Whitby et al., 2002), this does not preclude a catalytic function. The figure is adapted from a previously published version (Gibbs et al., 2012), and description is taken directly from the same publication.
The extensive array of motifs in hBVR is illustrated further in Figure 1B. Each of the sequences shown in the figure was proposed as a functional site in the molecule, and has subsequently been demonstrated to be active. Sequence motifs in the C-terminal regulatory domain of BVR are, to a great extent, involved in interaction with other proteins. As an example, hBVR was found to bind AP-1 sites in DNA by means of its bZip motif (Ahmad et al., 2002). The reaction of hBVR with DNA required two binding sequences in the DNA target, suggesting that hBVR binds as a dimer; site-directed mutagenesis of residues in the leucine zipper segment of the bZip domain prevented DNA binding, presumably by preventing hBVR dimerization. The bZip domain probably also allows heterodimer formation with other bZip transcription factors (Ahmad et al., 2002; Kravets et al., 2004). Two motifs, the nuclear localization (NLS) and nuclear export (NES) sequences presumably allow hBVR to interact with components of the nuclear pore. Critically for signaling activity, hBVR also interacts with several protein kinases; this interaction aids in the activation of these kinases, as well as playing a role in their translocation to the site of activity. The experiments used to characterize these sequences relied on site-directed mutagenesis and siRNA studies to disrupt each function (Ahmad et al., 2002; Lerner-Marmarosh et al., 2007, 2008; Maines, 2007; Tudor et al., 2008; Miralem et al., 2010; Gibbs et al., 2012). More recent studies have focused on the use of peptides based on the hBVR sequence as a means of disrupting protein: protein interactions, and/or modulating the enzyme activities of hBVR or its binding partners. These experiments will be discussed below. We have examined in some detail the role played by hBVR in modulating the activity of five different kinases: three members of the protein kinase C family (PKCβII, PKCζ, and PKCδ, the extracellular signal responsive kinase 2 (ERK2) and the highly atypical but clinically relevant Goodpasture antigen binding protein (GPBP). The function of BVR differs somewhat for each of these kinases.
Studies in our laboratory have indicated that hBVR activates members of the three protein kinase C (PKC) classes, and is in turn, activated by the PKCs. It is apparent, however, that the mechanism by which activation occurs differs according to the class of PKC associating with hBVR. A brief description of the structure and activation of the PKCs is presented, followed by discussion of the function of hBVR in the regulation of each class.
The PKC family of signaling proteins functions at the intersection of a wide variety of signal transduction pathways, and the kinases are therefore considered as key regulators of responses to extracellular stimuli. The PKC family consists of three subgroups, i.e., the conventional, novel, and atypical PKCs (cPKC, nPKC, aPKC, respectively), which are characterized by differences in protein structure, mechanism of activation and function.
The cPKCs, nPKCs, and aPKCs have homologous catalytic kinase domains that encompass the C-terminal half of the molecules (Figure 2; Steinberg, 2008; Newton, 2010). The primary structure of this domain is well conserved between the proteins, and it includes essential sequence motifs that are common to all members as well as being required for maturation of the kinase. These motifs include the activation loop, which contains a threonine residue that generally must be phosphorylated for catalytic activity; in the case of PKCδ, phosphorylation of this site is observed, but it is not essential. Phosphorylation of the activation loop threonine is otherwise the essential first step in maturation of the newly synthesized protein. All members have both a turn motif and a C-terminal hydrophobic motif; each motif contains serine or threonine targets for autophosphorylation, a requirement for full maturation of the enzyme. One exception is noted for the atypical kinases, which have glutamic acid, an amino acid known to mimic phosphoSer/Thr, at the hydrophobic motif phosphoacceptor site. Phosphate incorporated during the maturation process remains an integral part of the protein, with little or no turnover.
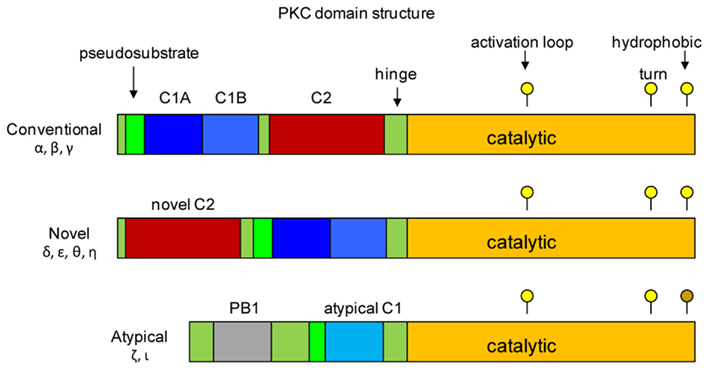
Figure 2. Structural domains of protein kinase C. The three classes of PKCs are shown. Members of each class have a catalytic domain that encompasses the C-terminus of the protein (shown in orange). Positions of residues in the catalytic domain that are phosphorylated during activation of the kinases are shown as yellow circles above each map. The threonine residue in the activation loop is the first to be phosphorylated, which allows phosphorylation of the threonine/serine in the turn motif and the serine/threonine in the hydrophobic motif, resulting in full activity of the kinase. There is no phosphorylation target in the atypical kinase hydrophobic motif; negative charge is supplied by the glutamic acid residue (indicated by the brown circle). The regulatory domains are located in the N-terminal half, and consist of C1 (blue), C2 (red) and pseudosubstrate domains (green). The positions of the C1 and C2 domains of conventional PKCs are reversed in the novel PKCs. Atypical PKCs lack a C2 domain, and have only a partial C1 domain. Redrawn from Steinberg (2008), Newton (2010).
The PKC regulatory domains are located in the N-terminal half of the molecule, and it is these domains that differentiate the different classes from each other, and enable the PKCs to mount specific responses to different stimuli. Each PKC protein contains one or two conserved regulatory domains (C1, C2) and a pseudosubstrate sequence which is located N-terminal to the C1 domain. In the resting state, the pseudosubstrate is bound by the active site of the catalytic subunit, effectively preventing substrate binding. The C2 domain is found only in the conventional and novel PKCs – in the conventional PKCs, the C2 domain binds Ca2+, which is critical for their activation. Ca2+ binding initiates a conformational change in the kinase exposing lipid biding sites in the C1 domain. The novel PKCs lack the Ca2+-binding site, and are therefore Ca2+-independent; however, they too must undergo a conformational change in response to stimuli that results in exposure of the C1 domain. The C1 region of cPKCs and nPKCs contains two sequences (C1A, C1B) that share a pattern of conserved His and Cys residues that coordinate Zn2+. These regions serve as binding sites for membrane lipids, notably phosphatidyl serine and diacylglycerol (DAG), which is a product of phospholipase C activity. Interaction with the plasma membrane is a critical step in the activation of the conventional and novel PKCs; for the cPKCs the C1 domain is inserted into the membrane to bind DAG. In resting cells the lipid binding sites of the cPKC C1 domain are masked by the C2 domain – binding of Ca2+ triggers a conformational change that unblocks the C1 domain. The atypical kinases have only a single C1-type sequence that binds phosphatidylinositoltrisphosphate or ceramide, but not DAG.
The conventional PKCβII phosphorylates hBVR in vitro, under conditions that are unfavorable to hBVR kinase activity (and hence autophosphorylation). The presence of hBVR in kinase reactions increased the Vmax of the PKC without affecting the Km for its substrate. However, both wild-type and kinase-inactive PKC could serve as substrates for the hBVR kinase activity. Peptides, based on the three motifs of PKCβII that are required for maturation of the kinase, were used as hBVR kinase substrates and the data suggested that the hBVR phosphorylation target may be in the PKCβII activation loop (Maines et al., 2007). As noted above, phosphorylation at that site is the first step in maturation of PKCβII. Peptides that included the PKCβII turn and hydrophobic motif phosphorylation sites were not substrates for the hBVR kinase activity. These experiments do not preclude the possibility that hBVR phosphorylation of other sites in PKCβII could provide an additional activation mechanism.
Protein kinase C-βII and hBVR were found to co-immunoprecipitate from extracts of cells in which both proteins were over-expressed and stimulated with phorbol myristate acetate (PMA). Confocal microscopy of PMA-stimulated cells over-expressing PKCβII and hBVR indicated that both proteins colocalized to the cell membrane. Based on site-directed mutagenesis of its sequence, two regions of hBVR were required for the protein–protein interaction, notably the C-terminal cysteine-rich metal-binding sequence (Figure 1B) and an intact ATP binding site. hBVR was shown to be a Zn2+-binding protein (Maines et al., 1996), and the hBVR metal-binding sequence closely resembles the Zn2+-binding sequences in the PKC C1 domain. It is possible that hBVR may be binding to the C1 domain via a mechanism that involves the cysteine residues of both motifs and/or Zn2+ chelation. The hBVR Gly17 residue in the nucleotide binding site was essential for activation of PKCβII, as was the sequence of four consecutive valine residues (Val11–14) located immediately upstream. The valines are unlikely to interact directly with PKCβII as they are buried within the molecule (Kikuchi et al., 2001; Whitby et al., 2002), but their mutation to alanine may alter or destabilize the BVR secondary structure so as to prevent PKCβII binding.
Activation of PKCβII by hBVR therefore occurs by two mechanisms – phosphorylation as a consequence of the hBVR kinase activity, and protein: protein interaction.
Human biliverdin reductase activates the atypical PKCζ, by a mechanism that clearly differs from that observed with PKCβII. PKCζ was able to use hBVR as a substrate in vitro but the PKC was not a substrate for the hBVR kinase activity (Lerner-Marmarosh et al., 2007). However, the presence of hBVR in PKCζ-catalyzed reactions stimulated the activity of the PKC toward its substrates and in autophosphorylation; in such reactions, stimulation of activity by kinase-inactive hBVR was at least as effective as that of the wild-type protein. Mutant hBVR proteins were used as substrates for in vitro kinase reactions to identify candidate phosphorylation sites. Mutation of either hBVR Ser149 in the S/T kinase motif (Figure 1) or Ser230 in one of the SH2-binding motifs to Ala yielded substrates that were phosphorylated to a significantly lesser extent than the wild-type protein. A protein having both of these mutations was a very poor substrate, incorporating less than 20% of the phosphate taken up by the wild-type protein. These data indicate that PKCζ phosphorylates hBVR at Ser149 and Ser230.
In response to stimulation of cells with tumor necrosis factor-α (TNF-α), hBVR, and PKCζ formed a complex in the cell, as indicated by their co-immunoprecipitation and by confocal microscopy (Lerner-Marmarosh et al., 2007). Enhancement of PKCζ activity in the cell was observed if hBVR was over-expressed; conversely, ablation of hBVR from cells with siRNA significantly inhibited the activation of PKCζ in response to TNF-α. A scrambled version of the siRNA was ineffective in inhibiting PKCζ. The inhibition of the TNF-α response by siRNA was as effective as that seen by introducing a competitive inhibitor peptide containing the PKCζ pseudosubstrate sequence. The data indicated dependence of PKCζ activation, and hence downstream signaling events, on hBVR. It is probable that binding of hBVR to PKCζ either initiates a conformational change to activate the protein, or binds to and preferentially stabilizes the active PKCζ conformation.
Protein kinase C-δ is a member of the novel class of PKCs and, as with the other PKCs described above, is both activated by the presence of hBVR in kinase reactions and utilizes hBVR as a substrate (Gibbs et al., 2012). PKCδ was not a substrate for hBVR kinase activity, however, as enhanced PKC activity was observed with kinase-inactive hBVR mutant. A constitutively active mutant of PKCδ, lacking part of the pseudosubstrate sequence (Zhao et al., 1998) was also activated by hBVR (Lerner-Marmarosh et al., in preparation). The increased activity of PKCδ is dependent on formation of a complex with hBVR, and as described above for the other PKCs, a complex that included both hBVR and PKCδ could be immunoprecipitated from extracts of IGF-1-treated cells (Gibbs et al., 2012). More direct evidence for association of the proteins was provided by an in vivo assay, using FRET-FLIM and IGF-1-treated cells. To examine whether BVR–PKCδ interaction extended to other stimuli that activate hBVR and PKCδ, cells were instead treated with PMA. Cells transfected with PKCδ alone showed a fluorescence lifetime of 2.65 ± 0.09 ns (Figures 3A,B). However, when the cells are co-transfected with pDsRed2-hBVR, the PKCδ fluorescence lifetime was reduced to 2.31 ± 0.13 ns; the diminished lifetime is indicative of close juxtaposition of the hBVR and PKCδ proteins in the FRET detection system. In PMA-treated cells, the fluorescence lifetime is 2.39 ± 0.15 ns, again indicating association of the proteins in the cell. Based on these observations, it is likely that the binding between hBVR and the PKC triggers a conformational change in the latter that facilitates its activation.
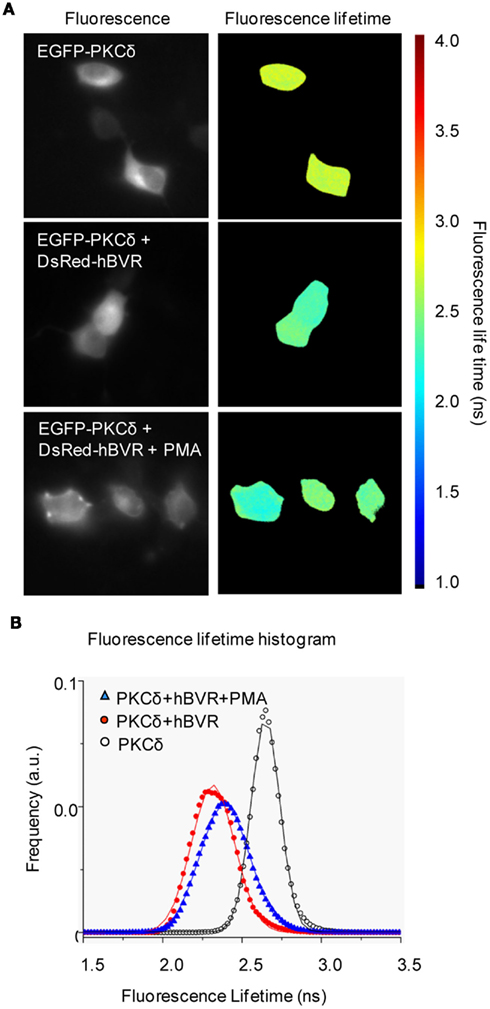
Figure 3. FRET detection of association of BVR and PKCδ in living cells. (A) FRET-FLIM images. Representative fluorescence intensity and FLIM images of HeLa cells transfected with EGFP-PKCδ, EGFP-PKCδ + DsRed2-hBVR and EGFP-PKCδ + DsRed2-hBVR treated with PMA. The scale bar ranges from 1 to 4 ns. Frequency-domain FLIM (Schlachter et al., 2009) experiments were performed using a Zeiss Axiovert 200M inverted wide-field microscope and a Lambert Instruments Fluorescence lifetime Attachment (LIFA; Lambert Instruments, Roden, The Netherlands). FLIM images were taken in the presence or absence of 100 nM PMA added before image collection. Fluorescence lifetimes were calculated from several regions and included data from multiple cells. (B) Fluorescence lifetime data are represented as histograms for each of EGFP-PKCδ (○), EGFP-PKCδ + DsRed2-hBVR (●) and EGFP-PKCδ + DsRed2-hBVR + PMA (▲) transfected cells. Curves represent FLIM data recorded from ~25 cells per condition; frequency of events is shown in arbitrary units (a.u.). Errors reported are one-half of the distribution width.
The ERK proteins are of fundamental importance in the regulation of cell proliferation and differentiation, and of the stress responses (Jackson and Foster, 2004; Boutros et al., 2008; Kim and Choi, 2010). ERK1/2 is known to activate some 50 transcription factors, including Elk and NF-κB (Ranganathan et al., 2006; Yazicioglu et al., 2007). hBVR was shown to be both an activator of extracellular receptor kinase 1/2 (ERK1/2) kinase activity, and a substrate for the kinase in vitro (Lerner-Marmarosh et al., 2008); this activation did not require hBVR kinase activity, as the kinase-inactive Val11–14, Gly17 mutant was able to activate ERK1/2. In cells stimulated with IGF-1, the ERK proteins associate with hBVR in co-immunoprecipitation experiments. A central role for hBVR in ERK-mediated signal transduction was proposed based on hBVR serving as a scaffold in the formation of a series of complexes that included ERK proteins. These included a complex containing the MAPK kinase MEK1/2 (Lerner-Marmarosh et al., 2008), the upstream activator of ERK1/2. Association of hBVR and ERK1/2 enabled translocation of ERK1/2 into and out of the nucleus, a function dependent on the hBVR NLS and NES motifs. Mutation of the NLS resulted in reduced transport of ERK2 into the nucleus, whereas expression of the NES mutant in cells resulted in significant nuclear accumulation of ERK2. Whereas the Gab1 protein is a known carrier for import to the nucleus of ERK1/2, it does not function to export the kinase (Osawa et al., 2004). On the other hand, hBVR functions as a bidirectional carrier protein for translocation of ERK1/2 into and out of the nucleus and, to date, it is the only protein known to fulfill this latter function. In the nucleus, hBVR brings ERK1/2 into proximity with the transcription factor Elk, enabling phosphorylation and activation of Elk. Ablation of hBVR with siRNA resulted in significantly reduced induction of ERK1/2 activation in response to IGF-1 treatment, and an attenuated response of Elk-dependent gene expression in response to IGF-1. The sites in hBVR required to form the complexes with ERK1/2 were determined by expressing mutant hBVR proteins in cells. Mutations in the 162FGFPAF sequence, that resembles the high affinity C-Box binding motif found in ERK1/2 associated proteins (Jacobs et al., 1999) and in the low affinity 275KKRILHCLGL D-Box-like sequence (Minden and Karin, 1997) prevented formation of the complex, and attenuated ERK activation and signaling in response to IGF-1.
Protein kinase C-δ is known to function in the ERK1/2 signaling pathway (Gorelik et al., 2007), and is believed to directly activate the ERK proteins. Examination of immunoprecipitates obtained from cells over-expressing both hBVR and PKCδ and stimulated with either IGF-1 or PMA revealed the presence of co-immunoprecipitated ERK2 (Gibbs et al., 2012). Formation of the ternary complex was required for downstream nuclear signaling mediated by ERK2. The activation of PKCδ by BVR in the absence of its kinase activity suggests that BVR may act as a scaffold to stabilize the active conformation of the PKC, while at the same time bringing the kinase into close association with its substrate. Site-directed mutagenesis implicated the hBVR C- and D-Boxes in the formation of the ternary complex. The C-Box was found to be critical for hBVR/ERK2 association, and the D-Box for hBVR/PKCδ interaction. These findings underscore the critical scaffolding function of hBVR as does the observation that siRNA-mediated reduction of hBVR protein in the cell resulted in a severe attenuation of PKCδ- and ERK2-dependent signaling, as determined from activation of Elk-dependent transcription. The siRNA-mediated inhibition was rescued by introduction of a plasmid to express hBVR into siRNA-treated cells. NF-κB is also activated in response to ERK2 (Ranganathan et al., 2006; Yazicioglu et al., 2007), and a transcription factor for expression of the HO-1 and iNOS genes. As with Elk, activation of the transcription factor, and expression of a downstream iNOS target, was shown to be blocked by siRNA treatment to ablate any of hBVR, PKCδ, ERK1/2, or MEK1/2. Restoration of protein expression by transfection with expression plasmids restored downstream NF-κB-dependent gene expression.
As described above, hBVR binds to ERK1/2 and to its upstream activating kinases MEK1/2 (Lerner-Marmarosh et al., 2008) and PKCδ (Gibbs et al., 2012). It was thought to be unlikely that two entirely distinct complexes, containing hBVR ERK2 and an upstream kinase would be formed in response to the same extracellular stimulus with IGF-1. Examination of the complex obtained by immunoprecipitation with anti-PKCδ antibodies revealed the presence of MEK1, in addition to the expected hBVR and ERK2 (Gibbs et al., 2012), suggesting the assembly of an elaborate, hBVR-anchored signal transduction complex. Because this function of hBVR is independent of its kinase activity, the role of hBVR in the complex is clearly that of a scaffold or bridge, and formation of the complex results in activation of both PKCδ and of ERK. Because unrestrained activation of the signaling complex is likely to be detrimental to the cell, it is noteworthy that the protein phosphatase, PP2A, that targets PKCδ (Srivastava et al., 2002), was also detected in the complex. This implies that the complex carries within it a means of self-regulation. PKCδ activity is commonly considered in the context of oxidant-dependent, mitochondrial-linked apoptosis (Buder-Hoffmann et al., 2009). Additionally, however, the enzyme is associated with functions that include cell cycle progression and proliferation, differentiation and tumorigenesis (Steinberg, 2008; Newton, 2010). Since hBVR has been observed to provide cytoprotective functions during oxidative stress (Miralem et al., 2005), it is possible that the association of hBVR, PKCδ and ERK1/2 in a ternary complex may provide a mechanism to protect the PKC from cleavage to its constitutively active, pro-apoptotic form, thereby favoring those functions involved in cell survival.
Our studies with protein kinases had led to our proposing two hypotheses. The first predicted that BVR would affect signaling via Toll-like receptors (TLRs) and NF-κB (Maines, 2005), and this would also involve the PKCs and MAPKs, including ERK. The second prediction was that, by virtue of its interaction with the insulin receptor (Lerner-Marmarosh et al., 2005) and role in activation of PKCβII (Maines et al., 2007), BVR would be an integral component of the PI3K/Akt signaling pathway (Maines, 2007).
It has been reported that biliverdin is a protective agent in preventing damage due to ischemia and reperfusion (I/R) in cardiac and renal transplantation (Nakao et al., 2005). It has also been reported that the cytoprotective effects of Heme Oxygenase-1 (HO-1) in I/R injury, both in rat cardiomyoblasts (H9c2 cells) and in the intact mouse heart are mediated by rat and mouse BVR (rBVR, mBVR), respectively, and involve the PI3K/Akt signaling pathway (Pachori et al., 2007). Specifically, activation of PI3K and its downstream effector Akt, as measured by phosphorylation, was increased in the presence of elevated HO-1. Inhibition of PI3K or knockdown of rBVR with siRNA attenuated the HO-1-induced phosphorylation of Akt. As described above for the PKCs, rBVR was found to co-immunoprecipitate with the phosphorylated p85 subunit of PI3K. In mouse macrophages, activation of the PI3K/Akt pathway was shown to be initiated by conversion of biliverdin to bilirubin by membrane-bound mBVR, which led to tyrosine phosphorylation of the mBVR C-terminal region (Wegiel et al., 2009), and allowed subsequent binding of mBVR to p85, activating PI3K and then Akt. The cytoprotective effect of biliverdin was negated by siRNA-induced ablation of mBVR.
Biliverdin was also observed to enable tolerance of cardiac allografts (Yamashita et al., 2004), by a means of inhibiting the activation of the proinflammatory transcription factors, nuclear factor of activated T cells (NFAT) and nuclear factor κB (NF-κB). These transcription factors both function in production of inflammatory cytokines, and stimulation of T cell proliferation. We also examined the role of biliverdin in modulating the activity of NF-κB (Gibbs and Maines, 2007), and observed an inhibition of transcription factor activation in response to stimulation of the cells with TNF-α. This inhibition could be overcome by over-expression of BVR, resulting in activation of NF-κB. Examination of NF-κB binding to its target sequence in vitro indicated that biliverdin was not an inhibitor of NF-κB binding to DNA, suggesting that biliverdin functions by quenching an upstream signaling event (Gibbs and Maines, 2007). Biliverdin was also effective in down-regulating the activity of the human BVR promoter (Gibbs et al., 2010).
Other studies have indicated that the endothelial nitric oxide synthase (eNOS) was phosphorylated by the calcium/calmodulin-dependent kinase in response to endotoxin, and that this was mediated by biliverdin binding to mBVR. This results in S-nitrosylation of mBVR, followed by its translocation to the nucleus and binding to AP-1 sites in the Toll-like receptor 4 (TLR4) promoter. This binding down-regulated TLR4 expression and subsequent resolution of the inflammatory response (Wegiel et al., 2011). The in vivo protection against acute liver damage was found to be dependent on the availability of NO. Moreover, macrophages from eNOS knockout mice displayed neither nitrosylation of BVR nor its translocation to the nucleus. Further, the expression of TLR4 was not inhibited in these cells. Recent studies also have revealed that biliverdin is a potent inhibitor of PKCδ in vitro. The mechanism of inhibition is presently unclear, although it does not appear to involve covalent modification of the protein (Lerner-Marmarosh et al., in preparation). These observations revealed a second mechanism by which BVR might activate PKCδ; in addition to the scaffolding function described above, BVR catalyzes the metabolism of a potent kinase inhibitor.
Goodpasture syndrome (GPS) is characterized by an autoimmune attack against the C-terminal non-collagenous-1 (NC1) domain of the α3 chain of the type IV collagen of basement membrane [α3(IV)NC1; Goodpasture antigen, GPA], resulting in deposits of autoantibodies along alveolar and glomerular basement membranes, which leads to hemorrhage in the lungs and a rapidly progressive glomerulonephritis (Saus, 1998; Hudson et al., 2003). The NC1 domain initiates the assembly of two individual triple helical protomers into a quaternary structure known as the hexamer; correct folding of the protein in the hexamer conceals the epitope for the autoantibody. The mechanism for its immunological exposure remains unknown; however GPBP targets the α3(IV)NC1 domain and regulates basement membrane collagen organization (Quinones et al., 1992; Raya et al., 1999; Granero et al., 2005; Revert et al., 2007); GPBP phosphorylates GPA at its N-terminus (Raya et al., 1999). Alterations in protein phosphorylation may lead to the autoimmune response by adversely modifying protein processing, resulting in epitope peptide presentation (Litersky and Johnson, 1992; Brown et al., 1995).
The association of GPBP with hBVR suggested by the yeast two-hybrid experiment was confirmed in the cell by co-immunoprecipitation and GST pull-down experiments (Miralem et al., 2010). hBVR down-regulated the TNF-α-stimulated kinase activity of GPBP without decreasing the level of the protein. In the same study, hBVR was found to stimulate GPBP gene expression by TNF-α-activated NF-κB. Ablation of hBVR with siRNA prevented GPBP gene expression, and led to a decrease in the cellular level of GPBP mRNA. Using a series of constructs expressing truncated forms of hBVR enabled the interacting domain to be mapped to the C281X10CC motif in the hBVR C-terminal 24 residues (Figure 2). These findings suggest a role for hBVR in auto-immunity, since hBVR may modulate the activity of a key protein in presentation of an autoantigen.
Several sequence motifs within hBVR (Figure 1) were identified as possible protein–protein interaction sites: the cysteine-containing 275KKRILHC and 290KYCCSRK in the C-terminal α-helix, and the two SH2-binding motifs (198YMKM and 228YLSF). As noted above, 162FGFPAF closely resembles the core of the high affinity C-Box for binding of ERK (Jacobs et al., 1999). The KKRILHC sequence resembles the core of the D-Box (KKRILHCLGL), a lower affinity binding site for MAPKs (Minden and Karin, 1997). Three peptides were initially examined for their effect on hBVR activity. The peptide KYCCSRK activated hBVR kinase activity, whereas KKRILHC and KRNRYLSF were both inhibitors (Lerner-Marmarosh et al., 2007).
The same peptides were also found to modulate the activity of PKCζ, with their effects on the PKC being similar to those observed with hBVR activity (Lerner-Marmarosh et al., 2007) – the BVR-activating peptide also activated PKCζ and allowed its translocation to the plasma membrane in response to TNF-α. Since the C-terminal lysine of the peptide was required for the PKC activation, it was proposed that the peptide enhances presentation of ATP to the kinase. The BVR inhibitor peptide KKRILHC clearly inactivated PKC activity, and also prevented transport to the membrane. The C-terminal cysteine residue of this peptide was found to be essential for kinase inactivation, and it is therefore possible that the peptide interacted with, and adversely affected the function of, the atypical C1 domain of PKCζ. Such binding might also be expected to inhibit the proposed hBVR association with the C1 domain. KRNRYLSF did not affect kinase activity of PKCζ in vitro, but did prevent translocation to the membrane in the intact cell. It is probable that this peptide prevented formation of the hBVR/PKCζ complex in response to TNF-α. Since translocation to the membrane is an integral part of PKC activation (Steinberg, 2008; Newton, 2010), it is clear that in the cell this peptide is also an inhibitor of the PKC.
The observation that, in response to IGF-1 or PMA, hBVR forms an essential complex for activation of PKCδ and ERK, which minimally includes PKCδ, MEK, and ERK1/2 (Lerner-Marmarosh et al., 2008; Gibbs et al., 2012) gave further scope to the use of BVR peptides as a means of disrupting intracellular signaling. As noted, mutation of either the C- or D-Box sequences of hBVR indicated that the interaction with ERK is dependent on the C-Box sequence, whereas the D-Box is required for interaction with PKCδ (Lerner-Marmarosh et al., 2008; Gibbs et al., 2012). Introduction into cells of peptides that include the C-Box core (FGFPAFSG) or the D-Box (KKRILHCLGLA) prevent complex formation in response to treatment with IGF-1, and thus prevent activation of ERK1/2 (Lerner-Marmarosh et al., 2008); The D-Box peptide is also sufficient to prevent association between PKCδ and ERK2, and blocks activation of PKCδ (Gibbs et al., 2012). Because the hBVR/PKCδ/ERK2 complex is critical for downstream signaling by ERK2, the finding that treatment with peptides disrupts complex formation in the cell and leads to inhibition of ERK-dependent activation of Elk-mediated gene expression offers a novel approach to regulation of ERK signaling.
Peptides were also examined for their ability to modulate hBVR-dependent down-regulation of GPBP. The peptide KKRILHC, which inhibits hBVR and PKCζ activity, was found to be as effective as the intact 296 residue hBVR protein in inhibiting GPBP kinase activity in the cell. Two other peptides that were examined, KYCCSRK and KRNRYLSF, both of which affect the activities of hBVR and PKCζ, but showed no effect on GPBP. The specificity of the KKRILHC in inhibiting GPBP suggests that it might offer a new line of attack in prevention of auto-immunity.
Altered activity of PKCδ has been implicated in a variety of disorders, including breast and lung cancer (Clark et al., 2003; McCracken et al., 2003), and in resistance to chemotherapeutic drugs. PKCδ is upregulated by estrogen (Cutler et al., 1994), and development of resistance to drugs such as tamoxifen that are used in treatment of hormone-responsive breast tumors is directly regulated by PKCδ. Resistance is enhanced by over-expression of the kinase (Nabha et al., 2005).
A major factor in the development of Parkinson’s disease is PKCδ-mediated death of dopaminergic neurons (Anantharam et al., 2002). In this context, it is noteworthy that an early event in apoptosis is cleavage of PKCδ by caspase-3 that results in the separation of the catalytic and regulatory domains (Emoto et al., 1995). Nuclear transport of the constitutively active catalytic domain accelerates apoptosis by inhibition of DNA repair functions and phosphorylation of lamin-B, resulting in a loss of nuclear integrity (Bharti et al., 1998; Cross et al., 2000; Yoshida et al., 2003). Conversely, PKCδ may be cytoprotective in retinopathy associated with type-2 diabetes. On balance, it is apparent that PKCδ regulation is critical to health, and that over- or under-expression is likely to be detrimental. Restoration of normal levels of activity of this kinase is, therefore, likely to be importance in controlling disease.
The finding that kinases upstream of ERK1/2 are dependent on the hBVR scaffolding function defines hBVR as an essential partner in ERK1/2 signaling. hBVR therefore regulates one of the three major MAPK signal transduction pathways, a carefully orchestrated series of events that initiates at the cell membrane and ultimately results in gene activation. The significance of these findings is underscored by the essential role of ERK and PKCδ in a wide spectrum of cellular functions, including ERK1/2-dependent regulation of transcription factors and proteins involved in the cellular stress response. ERK kinases have major roles in regulation of cell growth, differentiation and division; uncontrolled ERK1/2 activation is a frequent event in cancer. Such dysregulation is frequently a consequence of mutation of upstream kinases and signaling molecules in the pathway. The search for inhibitors of the ERK1/2 pathway is a major thrust of drug development research (Chappell et al., 2011). Therefore, hBVR-based peptides targeting the PKCδ/ERK and MEK/ERK pathways may well be an effective approach for therapeutic intervention in drug resistant cancers that are linked to dysregulated signaling pathways. We envision that hBVR-based peptides that inhibit ERK1/2 activation, whether by disrupting the hBVR-scaffolded complexes, or that inhibit upstream kinases, would be antiproliferative if administered over the long term.
Examination of the role of hBVR in activation of kinases, such as PKCs and MEK1/2, that function upstream of ERK1/2 has led to development of hBVR-based peptides that have introduced a new dimension to the regulation of ERK signaling. These peptides hold the potential for development of novel therapeutic agents to regulate the ERK signaling pathway, and hence in ameliorating human disease.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supported by NIH grants ES04066 and ES12187 (to Mahin D. Maines).
Ahmad, Z., Salim, M., and Maines, M. D. (2002). Human biliverdin reductase is a leucine zipper-like DNA-binding protein and functions in transcriptional activation of heme oxygenase-1 by oxidative stress. J. Biol. Chem. 277, 9226–9232.
Anantharam, V., Kitazawa, M., Wagner, J., Kaul, S., and Kanthasamy, A. G. (2002). Caspase-3-dependent proteolytic cleavage of protein kinase C delta is essential for oxidative stress-mediated dopaminergic cell death after exposure to methylcyclopentadienyl manganese tricarbonyl. J. Neurosci. 22, 1738–1751.
Bharti, A., Kraeft, S. K., Gounder, M., Pandey, P., Jin, S., Yuan, Z. M., Lees-Miller, S. P., Weichselbaum, R., Weaver, D., Chen, L. B., Kufe, D., and Kharbanda, S. (1998). Inactivation of DNA-dependent protein kinase by protein kinase C delta: implications for apoptosis. Mol. Cell. Biol. 18, 6719–6728.
Boutros, T., Chevet, E., and Metrakos, P. (2008). Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol. Rev. 60, 261–310.
Brown, K., Gerstberger, S., Carlson, L., Franzoso, G., and Siebenlist, U. (1995). Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 267, 1485–1488.
Buder-Hoffmann, S. A., Shukla, A., Barrett, T. F., Macpherson, M. B., Lounsbury, K. M., and Mossman, B. T. (2009). A protein kinase C delta-dependent protein kinase D pathway modulates ERK1/2 and JNK1/2 phosphorylation and Bim-associated apoptosis by asbestos. Am. J. Pathol. 174, 449–459.
Chappell, W. H., Steelman, L. S., Long, J. M., Kempf, R. C., Abrams, S. L., Franklin, R. A., Basecke, J., Stivala, F., Donia, M., Fagone, P., Malaponte, G., Mazzarino, M. C., Nicoletti, F., Libra, M., Maksimovic-Ivanic, D., Mijatovic, S., Montalto, G., Cervello, M., Laidler, P., Milella, M., Tafuri, A., Bonati, A., Evangelisti, C., Cocco, L., Martelli, A. M., and Mccubrey, J. A. (2011). Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget 2, 135–164.
Clark, A. S., West, K. A., Blumberg, P. M., and Dennis, P. A. (2003). Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKC delta promotes cellular survival and chemotherapeutic resistance. Cancer Res. 63, 780–786.
Cross, T., Griffiths, G., Deacon, E., Sallis, R., Gough, M., Watters, D., and Lord, J. M. (2000). PKC-delta is an apoptotic lamin kinase. Oncogene 19, 2331–2337.
Cutler, R. E. Jr., Maizels, E. T., and Hunzicker-Dunn, M. (1994). Delta protein kinase-C in the rat ovary: estrogen regulation and localization. Endocrinology 135, 1669–1678.
Emoto, Y., Manome, Y., Meinhardt, G., Kisaki, H., Kharbanda, S., Robertson, M., Ghayur, T., Wong, W. W., Kamen, R., Weichselbaum, R., and Kufe, D. (1995). Proteolytic activation of protein kinase C delta by an ICE-like protease in apoptotic cells. EMBO J. 14, 6148–6156.
Gibbs, P. E., and Maines, M. D. (2007). Biliverdin inhibits activation of NF-kappaB: reversal of inhibition by human biliverdin reductase. Int. J. Cancer 121, 2567–2574.
Gibbs, P. E., Miralem, T., Lerner-Marmarosh, N., Tudor, C., and Maines, M. D. (2012). Formation of ternary complex of human biliverdin reductase-protein kinase C delta-ERK2 protein is essential for ERK2-mediated activation of Elk1 protein, nuclear factor-kappaB, and inducible nitric-oxidase synthase (iNOS). J. Biol. Chem. 287, 1066–1079.
Gibbs, P. E., Miralem, T., and Maines, M. D. (2010). Characterization of the human biliverdin reductase gene structure and regulatory elements: promoter activity is enhanced by hypoxia and suppressed by TNF-{alpha}-activated NF-{kappa}B. FASEB J. 24, 3239–3254.
Gorelik, G., Fang, J. Y., Wu, A., Sawalha, A. H., and Richardson, B. (2007). Impaired T cell protein kinase C delta activation decreases ERK pathway signaling in idiopathic and hydralazine-induced lupus. J. Immunol. 179, 5553–5563.
Granero, F., Revert, F., Revert-Ros, F., Lainez, S., Martinez-Martinez, P., and Saus, J. (2005). A human-specific TNF-responsive promoter for Goodpasture antigen-binding protein. FEBS J. 272, 5291–5305.
Hanks, S. K., and Hunter, T. (1995). Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 9, 576–596.
Huang, T. J., Trakshel, G. M., and Maines, M. D. (1989). Detection of 10 variants of biliverdin reductase in rat liver by two-dimensional gel electrophoresis. J. Biol. Chem. 264, 7844–7849.
Hudson, B. G., Tryggvason, K., Sundaramoorthy, M., and Neilson, E. G. (2003). Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N. Engl. J. Med. 348, 2543–2556.
Jackson, D. N., and Foster, D. A. (2004). The enigmatic protein kinase C delta: complex roles in cell proliferation and survival. FASEB J. 18, 627–636.
Jacobs, D., Glossip, D., Xing, H., Muslin, A. J., and Kornfeld, K. (1999). Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 13, 163–175.
Kabu, K., Yamasaki, S., Kamimura, D., Ito, Y., Hasegawa, A., Sato, E., Kitamura, H., Nishida, K., and Hirano, T. (2006). Zinc is required for Fc epsilon RI-mediated mast cell activation. J. Immunol. 177, 1296–1305.
Kapitulnik, J., and Maines, M. D. (2009). Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol. Sci. 30, 129–137.
Kikuchi, A., Park, S. Y., Miyatake, H., Sun, D., Sato, M., Yoshida, T., and Shiro, Y. (2001). Crystal structure of rat biliverdin reductase. Nat. Struct. Biol. 8, 221–225.
Kim, E. K., and Choi, E. J. (2010). Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405.
Kim, H. P., Wang, X., Galbiati, F., Ryter, S. W., and Choi, A. M. (2004). Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J. 18, 1080–1089.
Kravets, A., Hu, Z., Miralem, T., Torno, M. D., and Maines, M. D. (2004). Biliverdin reductase, a novel regulator for induction of activating transcription factor-2 and heme oxygenase-1. J. Biol. Chem. 279, 19916–19923.
Kutty, R. K., and Maines, M. D. (1981). Purification and characterization of biliverdin reductase from rat liver. J. Biol. Chem. 256, 3956–3962.
Lerner-Marmarosh, N., Miralem, T., Gibbs, P. E., and Maines, M. D. (2007). Regulation of TNF-alpha-activated PKC-zeta signaling by the human biliverdin reductase: identification of activating and inhibitory domains of the reductase. FASEB J. 21, 3949–3962.
Lerner-Marmarosh, N., Miralem, T., Gibbs, P. E., and Maines, M. D. (2008). Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc. Natl. Acad. Sci. U.S.A. 105, 6870–6875.
Lerner-Marmarosh, N., Shen, J., Torno, M. D., Kravets, A., Hu, Z., and Maines, M. D. (2005). Human biliverdin reductase: a member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 7109–7114.
Litersky, J. M., and Johnson, G. V. (1992). Phosphorylation by cAMP-dependent protein kinase inhibits the degradation of tau by calpain. J. Biol. Chem. 267, 1563–1568.
Maines, M. D. (2005). New insights into biliverdin reductase functions: linking heme metabolism to cell signaling. Physiology (Bethesda) 20, 382–389.
Maines, M. D. (2007). Biliverdin reductase: PKC interaction at the cross-talk of MAPK and PI3K signaling pathways. Antioxid. Redox Signal. 9, 2187–2195.
Maines, M. D., Miralem, T., Lerner-Marmarosh, N., Shen, J., and Gibbs, P. E. (2007). Human biliverdin reductase, a previously unknown activator of protein kinase C betaII. J. Biol. Chem. 282, 8110–8122.
Maines, M. D., Polevoda, B. V., Huang, T. J., and Mccoubrey, W. K. Jr. (1996). Human biliverdin IX alpha reductase is a zinc-metalloprotein. Characterization of purified and Escherichia coli expressed enzymes. Eur. J. Biochem. 235, 372–381.
McCracken, M. A., Miraglia, L. J., Mckay, R. A., and Strobl, J. S. (2003). Protein kinase C delta is a prosurvival factor in human breast tumor cell lines. Mol. Cancer Ther. 2, 273–281.
Minden, A., and Karin, M. (1997). Regulation and function of the JNK subgroup of MAP kinases. Biochim. Biophys. Acta 1333, F85–F104.
Miralem, T., Gibbs, P. E., Revert, F., Saus, J., and Maines, M. D. (2010). Human biliverdin reductase suppresses Goodpasture antigen-binding protein (GPBP) kinase activity: the reductase regulates tumor necrosis factor-alpha-NF-kappaB-dependent GPBP expression. J. Biol. Chem. 285, 12551–12558.
Miralem, T., Hu, Z., Torno, M. D., Lelli, K. M., and Maines, M. D. (2005). Small interference RNA-mediated gene silencing of human biliverdin reductase, but not that of heme oxygenase-1, attenuates arsenite-mediated induction of the oxygenase and increases apoptosis in 293A kidney cells. J. Biol. Chem. 280, 17084–17092.
Nabha, S. M., Glaros, S., Hong, M., Lykkesfeldt, A. E., Schiff, R., Osborne, K., and Reddy, K. B. (2005). Upregulation of PKC-delta contributes to antiestrogen resistance in mammary tumor cells. Oncogene 24, 3166–3176.
Nakao, A., Neto, J. S., Kanno, S., Stolz, D. B., Kimizuka, K., Liu, F., Bach, F. H., Billiar, T. R., Choi, A. M., Otterbein, L. E., and Murase, N. (2005). Protection against ischemia/reperfusion injury in cardiac and renal transplantation with carbon monoxide, biliverdin and both. Am. J. Transplant. 5, 282–291.
Newton, A. C. (2010). Protein kinase C: poised to signal. Am. J. Physiol. Endocrinol. Metab. 298, E395–E402.
Osawa, M., Itoh, S., Ohta, S., Huang, Q., Berk, B. C., Marmarosh, N. L., Che, W., Ding, B., Yan, C., and Abe, J. (2004). ERK1/2 associates with the c-Met-binding domain of growth factor receptor-bound protein 2 (Grb2)-associated binder-1 (Gab1): role in ERK1/2 and early growth response factor-1 (Egr-1) nuclear accumulation. J. Biol. Chem. 279, 29691–29699.
Pachori, A. S., Smith, A., Mcdonald, P., Zhang, L., Dzau, V. J., and Melo, L. G. (2007). Heme-oxygenase-1-induced protection against hypoxia/reoxygenation is dependent on biliverdin reductase and its interaction with PI3K/Akt pathway. J. Mol. Cell. Cardiol. 43, 580–592.
Pawson, T., and Scott, J. D. (1997). Signaling through scaffold, anchoring, and adaptor proteins. Science 278, 2075–2080.
Quinones, S., Bernal, D., Garcia-Sogo, M., Elena, S. F., and Saus, J. (1992). Exon/intron structure of the human alpha 3(IV) gene encompassing the Goodpasture antigen (alpha 3(IV)NC1). Identification of a potentially antigenic region at the triple helix/NC1 domain junction. J. Biol. Chem. 267, 19780–19784.
Ranganathan, A., Yazicioglu, M. N., and Cobb, M. H. (2006). The nuclear localization of ERK2 occurs by mechanisms both independent of and dependent on energy. J. Biol. Chem. 281, 15645–15652.
Raya, A., Revert, F., Navarro, S., and Saus, J. (1999). Characterization of a novel type of serine/threonine kinase that specifically phosphorylates the human goodpasture antigen. J. Biol. Chem. 274, 12642–12649.
Revert, F., Merino, R., Monteagudo, C., Macias, J., Peydro, A., Alcacer, J., Muniesa, P., Marquina, R., Blanco, M., Iglesias, M., Revert-Ros, F., Merino, J., and Saus, J. (2007). Increased Goodpasture antigen-binding protein expression induces type IV collagen disorganization and deposit of immunoglobulin A in glomerular basement membrane. Am. J. Pathol. 171, 1419–1430.
Ron, D., Chen, C. H., Caldwell, J., Jamieson, L., Orr, E., and Mochly-Rosen, D. (1994). Cloning of an intracellular receptor for protein kinase C: a homolog of the beta subunit of G proteins. Proc. Natl. Acad. Sci. U.S.A. 91, 839–843.
Salim, M., Brown-Kipphut, B. A., and Maines, M. D. (2001). Human biliverdin reductase is autophosphorylated, and phosphorylation is required for bilirubin formation. J. Biol. Chem. 276, 10929–10934.
Schlachter, S., Elder, A. D., Esposito, A., Kaminski, G. S., Frank, J. H., Van Geest, L. K., and Kaminski, C. F. (2009). mhFLIM: resolution of heterogeneous fluorescence decays in widefield lifetime microscopy. Opt. Express 17, 1557–1570.
Sedlak, T. W., Saleh, M., Higginson, D. S., Paul, B. D., Juluri, K. R., and Snyder, S. H. (2009). Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. U.S.A. 106, 5171–5176.
Srivastava, J., Goris, J., Dilworth, S. M., and Parker, P. J. (2002). Dephosphorylation of PKC delta by protein phosphatase 2Ac and its inhibition by nucleotides. FEBS Lett. 516, 265–269.
Steinberg, S. F. (2008). Structural basis of protein kinase C isoform function. Physiol. Rev. 88, 1341–1378.
Tanti, J. F., Gremeaux, T., Van Obberghen, E., and Le Marchand-Brustel, Y. (1994). Serine/threonine phosphorylation of insulin receptor substrate 1 modulates insulin receptor signaling. J. Biol. Chem. 269, 6051–6057.
Tudor, C., Lerner-Marmarosh, N., Engelborghs, Y., Gibbs, P. E., and Maines, M. D. (2008). Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem. J. 413, 405–416.
Wegiel, B., Baty, C. J., Gallo, D., Csizmadia, E., Scott, J. R., Akhavan, A., Chin, B. Y., Kaczmarek, E., Alam, J., Bach, F. H., Zuckerbraun, B. S., and Otterbein, L. E. (2009). Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. J. Biol. Chem. 284, 21369–21378.
Wegiel, B., Gallo, D., Csizmadia, E., Roger, T., Kaczmarek, E., Harris, C., Zuckerbraun, B. S., and Otterbein, L. E. (2011). Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc. Natl. Acad. Sci. U.S.A. 108, 18849–18854.
Whitby, F. G., Phillips, J. D., Hill, C. P., Mccoubrey, W., and Maines, M. D. (2002). Crystal structure of a biliverdin IX alpha reductase enzyme-cofactor complex. J. Mol. Biol. 319, 1199–1210.
Yamashita, K., Mcdaid, J., Ollinger, R., Tsui, T. Y., Berberat, P. O., Usheva, A., Csizmadia, E., Smith, R. N., Soares, M. P., and Bach, F. H. (2004). Biliverdin, a natural product of heme catabolism, induces tolerance to cardiac allografts. FASEB J. 18, 765–767.
Yazicioglu, M. N., Goad, D. L., Ranganathan, A., Whitehurst, A. W., Goldsmith, E. J., and Cobb, M. H. (2007). Mutations in ERK2 binding sites affect nuclear entry. J. Biol. Chem. 282, 28759–28767.
Yoshida, K., Wang, H. G., Miki, Y., and Kufe, D. (2003). Protein kinase C delta is responsible for constitutive and DNA damage-induced phosphorylation of Rad9. EMBO J. 22, 1431–1441.
Keywords: biliverdin reductase, protein kinase C, signaling pathways, peptides, biliverdin
Citation: Gibbs PEM, Tudor C and Maines MD (2012) Biliverdin reductase: more than a namesake – the reductase, its peptide fragments, and biliverdin regulate activity of the three classes of protein kinase C. Front. Pharmacol. 3:31. doi: 10.3389/fphar.2012.00031
Received: 05 February 2012; Accepted: 16 February 2012;
Published online: 13 March 2012.
Edited by:
Jaime Kapitulnik, The Hebrew University of Jerusalem, IsraelReviewed by:
Jaime Kapitulnik, The Hebrew University of Jerusalem, IsraelCopyright: © 2012 Gibbs, Tudor and Maines. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Mahin. D. Maines, Department of Biochemistry and Biophysics, University of Rochester School of Medicine and Dentistry, 601 Elmwood Avenue, Rochester, NY 14624, USA. e-mail:bWFoaW5fbWFpbmVzQHVybWMucm9jaGVzdGVyLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.