

- The 2nd Medical Clinic, Molecular Cardiology, Medical Center of the Johannes Gutenberg University, Mainz, Germany
Reactive oxygen species (ROS) and signaling events are involved in the pathogenesis of endothelial dysfunction and represent a major contribution to vascular regulation. Molecular signaling is highly dependent on ROS. But depending on the amount of ROS production it might have toxic or protective effects. Despite a large number of negative outcomes in large clinical trials (e.g., HOPE, HOPE-TOO), antioxidant molecules and agents are important players to influence the critical balance between production and elimination of reactive oxygen and nitrogen species. However, chronic systemic antioxidant therapy lacks clinical efficacy, probably by interfering with important physiological redox signaling pathways. Therefore, it may be a much more promising attempt to induce intrinsic antioxidant pathways in order to increase the antioxidants not systemically but at the place of oxidative stress and complications. Among others, heme oxygenase (HO) has been shown to be important for attenuating the overall production of ROS in a broad range of disease states through its ability to degrade heme and to produce carbon monoxide and biliverdin/bilirubin. With the present review we would like to highlight the important antioxidant role of the HO system and especially discuss the contribution of the biliverdin, bilirubin, and biliverdin reductase (BVR) to these beneficial effects. The BVR was reported to confer an antioxidant redox amplification cycle by which low, physiological bilirubin concentrations confer potent antioxidant protection via recycling of biliverdin from oxidized bilirubin by the BVR, linking this sink for oxidants to the NADPH pool. To date the existence and role of this antioxidant redox cycle is still under debate and we present and discuss the pros and cons as well as our own findings on this topic.
Introduction
The endothelial integrity is crucial for physiologic organ function and protects against vascular inflammation, sepsis, prothrombotic activity, and atherosclerosis (Bassenge et al., 2005). Disruption of this blood–tissue (endothelial) barrier results in endothelial dysfunction which is a hallmark of most cardiovascular complications (Cai and Harrison, 2000; Munzel et al., 2008). Reactive oxygen species (ROS) and signaling events are involved in the pathogenesis of endothelial dysfunction and represent a major contribution to vascular regulation. ROS are important regulators for cellular and metabolic conditions. Molecular signaling is highly dependent on ROS. But depending on the amount of ROS production it might have toxic or protective effects (Bachschmid et al., 2005; Daiber and Münzel, 2006; Ullrich and Kissner, 2006).
Despite a large number of negative outcomes in large clinical trials (HOPE, HOPE-TOO as well as a prospective study with vitamin C in postmenopausal women with diabetes mellitus; Yusuf et al., 2000; Muntwyler et al., 2002; Lee et al., 2004; Mann et al., 2004; Lonn et al., 2005), antioxidant molecules and agents are important players to influence the critical balance between production and elimination of reactive oxygen and nitrogen species (RONS). The latter assumption is supported by a great number of experimental animal studies (Watanabe et al., 1997; Bassenge et al., 1998; Crawford et al., 1998; Elhaimeur et al., 2002) as well as human studies with rather small numbers of patients and acute infusion of antioxidants (Heitzer et al., 1996; Gori et al., 2001) indicating that antioxidants may be highly beneficial in improving endothelial dysfunction. The main reasons for the failure of chronic oral antioxidant therapy could be as follows: vitamin E and C act as pro-oxidants (e.g., tocopheryl and ascorbyl radicals), the coronary artery disease (CAD) of included patients is already irreversible, the CAD patients are already treated with drugs displaying antioxidant properties [e.g., angiotensin-converting enzyme (ACE) inhibitors and angiotensin-receptor blockers (ARBs)], chronic antioxidant therapy inhibits intrinsic ischemic preconditioning which relies on RONS formation or oral vitamin treatment does not result in high enough concentrations of the antioxidants at the place of oxidative stress (Chen et al., 2012).
Some of these reasons would favor the acute infusion of vitamin C in accordance with respective observations. However, infusion of antioxidants cannot be applied to a large number of patients over a long period and in addition may be affected by some of the draw backs listed above under chronic administration (e.g., interference with cellular redox signaling, suppression of oxidant-triggered ischemic preconditioning). Therefore, it may be a much more promising attempt to induce intrinsic antioxidant pathways in order to increase the antioxidants not systemically but at the place of oxidative stress and complications. A number of experimental animal studies have demonstrated that overexpression of ROS sources aggravates cardiovascular complications, whereas their suppression improves these adverse effects: the genetic deletion of NADPH oxidase subunits improved myocardial infarction damage and survival of mice (Doerries et al., 2007) and prevented angiotensin-II (Landmesser et al., 2002) as well as renovascular (clip model; Jung et al., 2004) induced hypertension, oxidative stress, and endothelial dysfunction. In contrast, overexpression of NADPH oxidase subunits further aggravated these complications (Dikalova et al., 2005). Vice versa, deletion of the mitochondrial manganese superoxide dismutase (MnSOD) increased age-dependent mitochondrial oxidative stress and endothelial dysfunction (Wenzel et al., 2008a) and deficiency in glutathione peroxidase-1 potentiated atherosclerosis and vascular complications in ApoE−/− mice (Torzewski et al., 2007). The recent observation that overexpression of mitochondrial superoxide dismutase improves angiotensin-II triggered hypertension and vascular dysfunction in mice shows that the different ROS sources can directly influence each other (Dikalova et al., 2010) and that especially the cross-talk between the mitochondrial and NADPH oxidase redox axis may play an important role for various diseases (Daiber, 2010).
Among others, heme oxygenase (HO) has been shown to be important for attenuating the overall production of ROS through its ability to degrade heme and to produce carbon monoxide (CO), biliverdin/bilirubin, and the release of free iron with subsequent ferritin induction (Vitek and Schwertner, 2007; Abraham and Kappas, 2008). It should be noted that free iron itself is toxic for the cell and may contribute to cellular oxidative damage (e.g., by reduction of free iron(III) to iron(II) by superoxide and subsequent reaction of ferrous iron with hydrogen peroxide to yield hydroxyl radicals. The latter step is termed “Fenton reaction,” the overall process is the “Haber–Weiss-cycle” (Daiber and Münzel, 2006). Therefore, it is of great importance to keep the free iron levels low, which also includes that induction of HO is followed by subsequent induction of ferritin. Heme and P450 overload has been demonstrated to confer cytotoxic effects (Eipel et al., 2007), which provides a simple basis for the highly beneficial action of HO consisting of the direct detoxification of free heme. Besides the highly protective effects of heme oxygenase (HO-1) in various pathophysiological states, its down-stream targets bilirubin and biliverdin have reducing properties and are recognized as potent antioxidants (Stocker et al., 1987), but it is not fully understood how the bile pigments act in this molecular process and the role of biliverdin reductase (BVR) needs to be further explored. The localization of heme degradation enzymes (e.g., HO-1 and BVR) to plasma membrane caveolae and evidence on caveolin-1 interaction with HO-1 and modulation of its activity make the HO-1 system an attractive player in the vascular antioxidant system since also the redox-sensitive endothelial nitric oxide synthase (eNOS) as well as superoxide producing enzymes are located in the plasma membrane caveolae (Kim et al., 2004).
Heme oxygenase-1 degrades heme to biliverdin, which is then reduced by BVR to bilirubin. During the last decade BVR has attracted more attention on the background of an active antioxidant cycle providing a catalytic sink for RONS as well as cytoprotection (Baranano et al., 2002; Sedlak and Snyder, 2004). However, this concept is still under debate with a number of highly controversial reports (Maines, 2007; Pachori et al., 2007; Florczyk et al., 2008; Maghzal et al., 2009; Sedlak and Snyder, 2009; Young et al., 2009; Jansen et al., 2010). With the present review we provide an overview on our own results, the current literature and critically discuss the pros and cons of the antioxidant and cytoprotective concept underlying the action of BVR.
The Concept of Biliverdin Reductase and Oxidants Forming a Catalytic Antioxidant Cycle
In 2002 an attractive concept was put forward by Snyder and colleagues consisting of the BVR mediated reduction of biliverdin (BV) to bilirubin (BR) being the much more potent antioxidant and subsequent oxidation of BR by hydrogen peroxide back to BV forming a catalytic antioxidant cycle that is driven by NADPH, the reducing cofactor of BVR (see Figure 1; Baranano et al., 2002; Greenberg, 2002; Sedlak and Snyder, 2004). Especially lipophilic ROS such as lipid hydroperoxides and peroxyl radicals were described to feed this antioxidant cycle and later, synergistic effects of glutathione have been described (Sedlak et al., 2009). In their initial work, Baranano et al. demonstrated that BR (10 nM) increased the viability of HeLa cells in the presence of toxic concentrations of hydrogen peroxide (100 μM). The fact that 10,000-fold lower BR concentrations prevented the toxicity of H2O2 implied that there was some kind of “recycling” of BR resulting in the proposed BV/BR redox cycle. In addition, small interfering RNA (siRNA) against BVR resulted in higher ROS levels in HeLa cells and neurons, decreased the viability of neurons upon treatment with H2O2, increased caspase activity and apoptosis [envisaged by poly(ADP-ribose)polymerase (PARP) activation] in these cells. In response to the review article of Sedlak and Snyder, Antony McDonagh (1990), a pioneer on the bile pigment field, questioned the importance of the oxidative conversion of BR to BV since BR radicals are also reduced in a non-enzymatic environment by interaction with molecular oxygen – these considerations were published in a letter to the editor (McDonagh, 2004). In addition, Antony McDonagh pointed out that there is only marginal conversion of BR to BV under special conditions and generation of certain oxidants, which was supported by our observations using superoxide, peroxynitrite, hydrogen peroxide, and peroxidase systems (see chapter below). These reports stimulated us to investigate this potential sink for RONS in vitro (chemical models) and ex vivo (human endothelial cells).
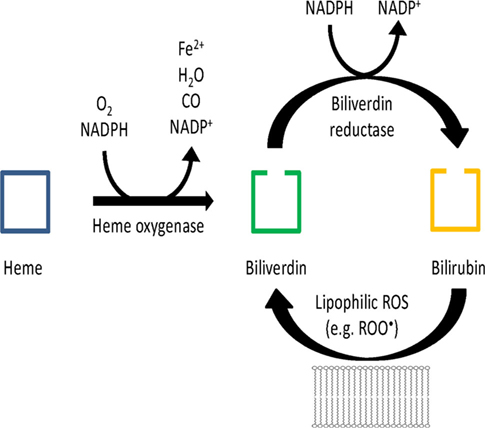
Figure 1. Hypothetical scheme of the antioxidant redox cycle of the BR/BV/BVR system. Modified from Sedlak and Snyder (2004).
Own Results
Direct Antioxidant Properties of Bilirubin versus Biliverdin in vitro
As shown by Jansen et al. (2010) bilirubin represents a superior antioxidant as compared to biliverdin when applied in high concentrations. Using cell-free systems (test tube chemistry) the direct antioxidant effects of bilirubin versus biliverdin were assessed. The peroxynitrite scavenging ability of both bile pigments was assessed by two different biochemical models: tyrosine residues in bovine serum albumin (BSA) were either nitrated by authentic peroxynitrite (ONOO−) or in situ generated ONOO− from the thermal decomposition of 3-morpholino-sydnonimine (Sin-1), a more physiological model to assess peroxynitrite-mediated oxidations. Peroxynitrite-mediated tyrosine nitration involves tyrosyl-radical-intermediates as a consequence of homolytic bond cleavage in ONO-OH yielding hydroxyl (HO• ) and nitrogen dioxide (•NO2) radicals. Therefore, inhibition of peroxynitrite-mediated BSA nitration may be regarded as ability of BR and BV to scavenge peroxynitrite-derived free radicals and/or reduction of tyrosyl-radical-intermediates. According to our results, BR is at least threefold more potent than BV in inhibiting peroxynitrite-mediated protein tyrosine nitration. The used dot blot technique excludes any interference of the BR or BV color since the compounds are removed during transfer of the sample to the membrane.
Superoxide scavenging ability of BR and BV was determined in two different systems. In the first one superoxide was constantly generated by xanthine oxidase (XO) and hypoxanthine whereas in the second one we used authentic superoxide (KO2) to exclude any inhibitory effects of the compounds on enzymatic activity (e.g., one of the compounds could be an inhibitor of XO). Since the color of BR and BV may lead to false-positive results using direct optical methods, we used HPLC-based detection of fluorescent 2-hydroxyethidium (2-HE), the specific oxidation product of dihydroethidium and superoxide. XO-derived superoxide was decreased in a concentration-dependent fashion by BR whereas BV had no inhibitory effect and even significantly increased the signal pointing toward pro-oxidative effects. It should be noted that the absolute increase in superoxide signal by BV was small (approximately 10%). Using KO2 from a saturated stock solution in dimethyl sulfoxide (DMSO), the above described differences even became more pronounced: BR decreased the formation of 2-hydroxyethidium in a concentration-dependent fashion whereas BV, this time, dramatically increased its yield up to 2.5-fold over KO2 control without BR or BV. This may be taken as evidence for redox-cycling of BV amplifying the superoxide formation rate. Again, the used HPLC technique excludes any interference of the BR or BV color since the compounds are separated from the product during their way through the column.
No Evidence for Conversion of Bilirubin to Biliverdin by Different Oxidants in vitro
Bilirubin or BV (each 50 μM) were incubated with authentic peroxynitrite, Sin-1, KO2, hydrogen peroxide alone or together with horseradish peroxidase (HRP). The latter would generate peroxidase compound I, having hydroxyl radical-like, one-electron abstracting properties. There was a pronounced loss of BR in all systems used indicating that BR reacted with any reactive species formed. In contrast, BV was converted less efficiently (not at all by superoxide), only the peroxidase/H2O2 system completely consumed BV. Special emphasis was put on the conversion of BR to BV by the different oxidants, the process that was previously described by Snyder and coworkers (Baranano et al., 2002; Sedlak and Snyder, 2004). However, we were unable to detect any significant conversion of BR to BV questioning the role of BR/BV as catalysts in the break-down of RONS. We also observed no conversion of BR to BV by mitochondrial oxidative stress but also no significant conversion of BV to BR by intrinsic mitochondrial BVR. Also here, we strictly used an HPLC technique excluding any interference of the BR or BV color since the compounds are separated during their way through the column.
(In)Direct Antioxidant Properties of Bilirubin versus Biliverdin ex vivo
Efficacy of BR and BV as antioxidants was also tested in isolated heart mitochondria (see Figure 2). Mitochondrial respiratory chain was stimulated with succinate and blocked by the complex III inhibitor antimycin A, which directs ROS toward the intermembrane space. ROS and RNS formation was detected by the luminol analog L-012 enhanced chemiluminescence (ECL) detecting intra- and extra-mitochondrial reactive species. BR sensitively decreased the L-012 signal at submicromolar concentrations being threefold more efficient as compared to BV. In contrast, suppression of the intra-mitochondrial superoxide formation, that was measured by the yield of 2-hydroxyethidium, required 1000-fold higher concentrations of BR and the difference between BR and BV was less pronounced.
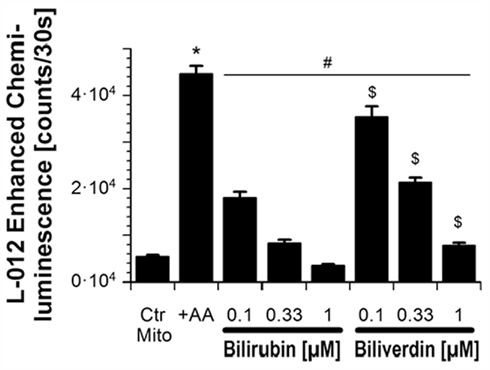
Figure 2. Effects of BR and BV on mitochondrial oxidative stress. Adopted from Jansen et al. (2010). With permission of Rightslink® by Copyright Clearance Center (Elsevier).
The inhibitory effects of BR and BV on oxidative burst in phorbol ester-stimulated neutrophils was detected by L-012 ECL indicating less pronounced differences between BR and BV, as compared to mitochondria. BR and BV decreased the signal in a concentration-dependent fashion but BR was only marginally more potent than BV. The less pronounced difference may be due to the involvement of myeloperoxidase/H2O2 in the signal intensity. The reactivity of BR and BV may be very similar for the reactive species derived from the peroxidase/H2O2 system in accordance with the almost identical break-down pattern of BR and BV caused by HRP/H2O2 (see third paragraph in the preceeding chapter). However, the superoxide-specific product 2-hydroxyethidium revealed a more pronounced antioxidative effect of BR as compared to BV, which even caused a marginal increase in the signal. Regarding the weak differences between BR and BV in the chemiluminescence assay it should be taken into account that the color of the bile pigments may have interfered with the ECL signal. Therefore, the HPLC data may be more reliable.
Antioxidant Properties of the Heme Oxygenase System and Effects of Biliverdin Reductase Silencing in Human Endothelial Cells
Knock-out models by genetic gene deletion are useful tools to investigate enzyme and protein functions. Until now, BVR knock-out mice do not exist and inhibitors are not commercially available. Therefore, silencing of the BVR gene by siRNA is often used as molecular method in most experiments. We investigated BVR silencing in human umbilical vein endothelial cells (HUVECs) and observed significant effects in resting cells without oxidative stimulus characterized by an increase in intracellular oxidative stress (measured by intracellular 2-HE formation). With lipopolysaccharide (LPS)-elicited RONS formation we established a model for cellular oxidative stress in HUVECs and endothelial cell activation, which reflects endotoxin-triggered endothelial dysfunction and may be used as a clinical model for septic shock.
Treatment of HUVECs with siBVR or siHO-1 resulted in a >90% decrease at the messenger ribonucleic acid (mRNA) level which was measured by reverse transcription-polymerase chain reaction (RT-PCR) and envisaged on agarose gels. Basal superoxide levels in these cells were determined by 2-hydroxyethidium formation, which was significantly increased in BVR-silenced cells. After characterization of the ideal conditions for cellular oxidative stress and protection by HO-1 induction these findings were applied to BVR and HO-1-silenced HUVECs. Hemin decreased the LPS-triggered oxidative stress [dihydrorhodamine 123 (DHR) fluorescence] in control cells whereas this protective effect was almost lost in BVR-deficient cells and completely absent in HO-1-depleted cells. When the BVR substrate BV was added to lipopolysaccharide-stimulated HUVECs the decrease in DHR fluorescence in control cells was more pronounced at a percentage level as compared to BVR-silenced cells (20.1 versus 15.4%).
These findings were supported by a second parameter for oxidative stress, protein tyrosine nitration. 3-nitrotyrosine staining even revealed a more pronounced effect of BVR and HO-1 silencing. In LPS-stimulated HUVECs hemin caused a decrease in 3-nitrotyrosine staining at 3.3 and 10 μM whereas nitration was significantly increased in BVR- and HO-1-deficient cells and the protective effect of hemin was completely lost. In addition, the substrate for BR synthesis by BVR decreased the level of 3-nitrotyrosine- positive proteins in a concentration-dependent fashion in control cells whereas this protective effect of BV was lost in BVR-silenced cells. The highest concentration of BV (10 μM) even tended to increase the nitration instead of preventing it, supporting the observed redox-cycling by BV (see second paragraph in the preceeding chapter). Treatment of HUVECs with siBVR or siHO-1 resulted in a >80% or complete decrease at the protein level which was measured by Western blotting.
A last set of experiments aimed to demonstrate the importance of chronic indirect effects of the HO-1/BVR product BR. LPS treatment of an immortal endothelial cell line (EA.hy 926 cells) decreased the expression of the tetrahydrobiopterin (BH4) synthase GTP-cyclohydrolase-1 (GCH-1). BV had no effect on endothelial expression of this important enzyme whereas BR as well as the CO-releasing molecule-2 (CORM-2) derived CO dramatically increased the GCH-1 protein levels. It should be noted that BH4 levels are crucial for the function of eNOS and BH4 deficiency leads to uncoupling of eNOS and subsequent formation of RONS. Therefore prevention of GCH-1 down-regulation or degradation may represent an important antioxidant pathway. Previous reports have demonstrated that oxidative stress may trigger the proteasomal degradation of GCH-1 (Xu et al., 2007, 2009). Especially for the endothelium it may be crucial to maintain the BH4 levels under conditions of increased oxidative stress to prevent eNOS uncoupling and endothelial dysfunction. Interestingly, the levels of the dihydrobiopterin (BH2) recycling enzyme dihydrofolate reductase behave complementary to those of GCH-1 in accordance with our previous results in diabetic rats (Wenzel et al., 2008b; Oelze et al., 2011).
Other Reports in Favor of an Important Role of Biliverdin Reductase for Antioxidant and Cytoprotective Effects of the Heme Oxygenase System
Previous reports suggested an efficient catalytic cycle between BV and BR (Baranano et al., 2002; Sedlak and Snyder, 2004; Liu et al., 2006) and others have observed conversion of BR to BV by peroxynitrite (Kaur et al., 2003), or peroxyl radicals (Baranano et al., 2002). In support of these previous findings and a role of BVR in cytoprotection, Young et al. (2009) demonstrated that angiotensin-II induced superoxide formation in renal tubular epithelial cells and inner medullary collecting duct (IMCD3) cells is aggravated by silencing of BVR. Superoxide formation was measured using dihydroethidine (DHE) fluorescence and lucigenin-derived chemiluminescence. BVR silencing resulted in a significantly decreased level of BVR protein (>30% of control cells) as well as diminished cellular bilirubin concentrations (50% of control cells). Co-treatment with BV could decrease the angiotensin-II induced superoxide formation in control cells, whereas it further aggravated the ROS levels in BVR-silenced cells. This is in good accordance with our observations on increased superoxide levels by biliverdin in the presence of xanthine oxidase or authentic superoxide, most probably via redox-cycling (see the preceeding chapter). However, it should be noted that Young et al. also observed BVR/BR independent antioxidant action of HO-1, since hemin co-treatment improved superoxide formation in BVR-silenced cells, indicating an important contribution of CO (and other so far not identified routes). The beneficial effect of hemin could of course also be related to increased break-down of superoxide in the presence of hemin leading to other reactive intermediates or non-toxic products that are not detected by DHE fluorescence. Of note, we have observed a more pronounced burden of oxidative stress in endothelial cells treated with HO-1 siRNA as compared to BVR siRNA suggesting on top protective pathways mediated by HO-1 activation (most probably CO formation) as compared with the BVR pathway alone (see the preceeding chapter).
Another previous report by Liu et al. (2006) demonstrated increased cellular oxidative stress in BVR-deficient cells using a model of experimental autoimmune encephalomyelitis. In detail, BVR treatment of encephalomyelitic rats improved the tissue levels of 8-isoprostane in the spinal cord but also the clinical score of the disease (50% for BVR and 66% for HO-1 of encephalomyelitic rats). In isolated cells, BVR silencing had much more pronounced effects on increase in ROS levels and decrease in cell viability as compared to HO-1 inhibition by Sn-pophyrin. The down-stream product of BVR, BR, showed a much more pronounced effect on erythrocyte hemolysis by the complement in the classical pathway and lymphocyte activity (antibody-dependent cell-mediated cytotoxic activities) as compared to the antioxidant glutathione. However, since HO-1 treatment or inhibition displayed weaker effects as compared to BVR treatment or silencing, the data of this study rather point to an effect of BVR that is independent of the antioxidant action of BR.
Panahian et al. (1999) reported on enhanced neuronal expression of BVR after permanent focal cerebral ischemia and speculated that this may be a new cellular mechanism for protecting neurons against ischemic injury and oxidative stress. Cerebral ischemia was induced by permanent middle cerebral artery occlusion and increased BVR expression obviously contributed to increased survival of neurons in the peri-ischemic region. Previous reports introduced an additional function for BVR. The protein contains a region with a leucine zipper deoxyribonucleic acid (DNA)-binding motif and binds as a homodimer to a region of the HO-1 promoter that contains two AP-1 sites (Ahmad et al., 2002). By this action BVR may prevent cellular damage not only by catalyzing the formation of the antioxidant bilirubin, but also by transcriptional activation of HO-1 triggering the efflux of potentially toxic iron from cells exposed to oxidative stress (Baranano and Snyder, 2001). The latter observation was also shared by us since BVR silencing also partially decreased HO-1 protein levels, which may be related to a role of BVR for transcriptional activation of HO-1 (Jansen and Daiber, unpublished).
Finally, it was shown by Maines and coworkers that BVR without any action of BR may change the cellular antioxidant profile by regulation of protein kinase C (PKC-βII) activity (Maines, 2007; Maines et al., 2007), which may represent an important BVR-dependent protective pathway based on PKC-ζ under inflammatory conditions [e.g., tumor necrosis factor-α (TNF-α) signaling; Lerner-Marmarosh et al., 2007]. This pathway involves insulin receptor 1/2 signaling, the insulin/growth factor signaling cascade for protein/ DNA synthesis and glucose transport down-stream of mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K). This kinase activity of BVR provides a great link between heme metabolism and cell signaling. Further down-stream in this signaling cascade, BVR may also directly interact with the transcription factors ATF-2/CREB, c-fos, c-jun preventing apoptosis. Even protective actions of BVR on arsenite toxicity were described that were completely independent and not shared by HO-1 (Miralem et al., 2005).
There is also further wide-spread evidence for beneficial effects of BVR in various disease models. In a model of oxidative damage in maternal cholestasis during pregnancy, upregulation of BVR was suggested to confer protection of the placental–fetal unit since the viability of human choriocarcinoma JAr cells was significantly increased by bilirubin in a concentration-dependent fashion (Perez et al., 2008). Another report demonstrated that the antioxidants biliverdin/bilirubin might play an important role in the protection of the nitrergic neurotransmitter against oxidative stress in murine gastric fundus (De Backer and Lefebvre, 2008). The authors demonstrated, that neurotransmission (neuronal nitric oxide synthase, nNOS-dependent relaxation in murine jejunal circular smooth muscle strips) was impaired by the superoxide generator LY83583, a process that was prevented by BR and to a smaller extend by BV but not by CO. From these observations and the colocalization of HO-2 and BVR with nNOS they concluded that the BR pathway but not the CO pathway down-stream of heme oxygenase-2 (HO-2) confer antioxidant protection of the neurotransmission process in the gastric fundus although this protective action was not observed under normal physiological conditions but only in response to oxidant challenges. In a model of cell senescence, Kim et al. (2011) found a striking decrease in BVR expression in senescent human fibroblasts (HDF cells) as compared to young HDF cells. Vice versa, senescent fibroblasts had significantly increased ROS levels in comparison with young HDF cells, a difference that was eliminated by BVR siRNA treatment suggesting a role of BVR in the young fibroblasts in the detoxification of ROS. Hydrogen peroxide caused an appreciable induction of HO-1 in the young fibroblasts (with high expression of BVR) whereas this antioxidant response was absent in the senescent HDF cells (with low BVR expression). Again, this differential stress response was eliminated when siRNA treatment against BVR was applied. As a functional read-out, the depletion of BVR isoform A could reduce the cell number by >60% in young HDFs even without additive exogenous oxidative stress, a process that was accompanied by increased expression of the expression of the senescence marker SA-β-gal. A recent study demonstrated that BV rescues the HO-2 knock-out mouse phenotype of unresolved chronic inflammation following corneal epithelial injury and the authors suggested a beneficial role of the upregulated BVR in this model (Bellner et al., 2011).
Other Reports Not in Favor of an Important Role of Biliverdin Reductase for Antioxidant and Cytoprotective Actions of the Heme Oxygenase System
According to a recent publication of Maghzal et al. (2009) revisiting the previous findings of Snyder and colleagues neither BVR overexpression nor silencing changed hydrogen peroxide toxicity in HeLa cells, an immortal human cancer cell line. In detail, Stocker and coworkers could show that lipid peroxidation-mediated oxidation of BR in chloroform, did not yield BV (see similar observations by us, preceeding chapters). Similarly, H2O2 did not oxidize albumin-bound BR to BV, and only modest yields of BV were observed upon in vitro oxidation of albumin- or ligandin-bound BR in the presence of peroxyl radicals. The viability of HeLa cells was not decreased in the presence of hydrogen peroxide after depletion of cellular BVR protein and activity in vitro using siRNA against BVR. Vice versa BVR overexpression failed to enhance protection of HeLa cells against H2O2-mediated damage, irregardless of whether BR or BV were added to the cells as substrate for the putative redox cycle. The authors also showed that overexpression of human BVR in HeLa cells did not decrease H2O2-induced ROS levels. Finally, they highlighted that HO-1 overexpression but not BVR overexpression rescued cell survival of HeLa cells in the presence of hydrogen peroxide. In a letter to the editor, Snyder and colleagues criticized the experimental setup in the work of Maghzal et al. with special emphasis on the need for providing BV by either exogenous addition or endogenous activation of the HO-1 system (Sedlak and Snyder, 2009), as done in our cell culture experiments (see preceeding chapters). In addition, these authors criticized the high hydrogen peroxide concentrations employed by Stocker and coworkers as well as the time point at which cell death and ROS measurements were detected (4–8 h) in contrast to the maximum protection observed by BVR at 24 h. In the direct reply letter to the editor, Stocker and Maghzal pointed out that previous work supporting a role for BVR in cytoprotection and antioxidant effects (e.g., Miralem et al., 2005; Maines, 2007) provided beneficial data that was largely independent of the presence of BR and the reductase activity of BVR and were therefore not in support of the antioxidant BV/BR/BVR redox cycle (Stocker and Maghzal, 2009). Even more important, these authors emphasized that BVR depletion also failed to affect the death of cells loaded with BV or BR prior to exposure to a 40-fold molar excess of H2O2 (Maghzal et al., 2009), whereas Snyder and associates reported BR to be able to defend against a 10,000-fold excess of H2O2 (Baranano et al., 2002).
In his recent study, Anthony McDonagh demonstrated that the peroxyl radical generator 2,2′-azobis(2-amidinopropane) hydrochloride (AAPH) did not result in the formation of BV from BR in air-saturated solution in the absence of serum albumin (McDonagh, 2010). He even observed consumption of BV under the same conditions arguing against an accumulation of BV under cellular conditions in the presence of peroxyl radical formation. In contrast, he described marginal BV formation from BR in the presence of serum albumin and air-saturated solution, which was even present in oxygen-free (argon flushed) solutions, whereas oxygen-saturation eliminated the conversion of BR to BV. This clearly pointed against a peroxyl-driven conversion of BR to BV since peroxyl radical generation from AAPH requires oxygen and most pronounced BR to BV conversion was observed in the absence of oxygen and required the presence of serum albumin. Therefore, Anthony McDonagh proposed that secondary protein radicals formed by the interaction of AAPH with serum albumin rather account for the conversion of BR to BV than peroxyl radicals, suggested to be the converting species by Snyder and colleagues.
Outlook and Therapeutic Implications
Antioxidant and Cytoprotective Role of BVR but Evidence against the BV/BR/BVR Redox Cycle
The data presented in our previous published work (Jansen et al., 2010) support the concept that BR is a more potent antioxidant than BV, but also provide evidence that direct and indirect antioxidant effects of the bile pigments contribute to the beneficial profile of the HO-1 pathway (e.g., GCH-1 in EA.hy cells; see Figure 3 for summary). Despite our observations that at high concentrations of the bile pigments scavenging of RONS is more (peroxynitrite or superoxide) or less (H2O2 or H2O2/peroxidase) pronounced and that BV undergoes redox-cycling in the presence of superoxide, we could not establish an important role for an antioxidant cycle based on oxidative conversion of BR to BV and reductive reversal by BVR. The antioxidant effects of BV and BR are concentration-dependent. About 10 μM BR reflect the physiological situation of endothelial cells, which are in direct contact with the blood stream. The model used by Jansen et al. for cellular oxidative stress is based on LPS-elicited RONS formation in HUVECs, which reflects endotoxin-triggered endothelial dysfunction and may be used as a cellular model for septic shock. In this respect, identification of BVR as an important constituent of the antioxidant defense system in human endothelial cells has important implications for the prevention of endothelial dysfunction in cardiovascular diseases that are associated with increased oxidative stress or chronic inflammatory conditions. HO-1 silencing had more pronounced effects on oxidative stress than BVR deletion and accordingly HO-1 dependent protection involves other (CO- or ferritin-dependent) pathways besides the formation of BR or presence of BVR. Most probably, BR (and/or BVR) and CO act synergistically in conferring antioxidant protection of endothelial cells. The antioxidant effects of CO in this synergistic action are not based on direct RONS scavenging but rather on regulation of NADPH oxidase activity, P450 activity and induction, opening of potassium channels, angiogenesis, activation of soluble guanylyl cyclase, angiogenesis, interference with MAPK signaling, and many more (Abraham and Kappas, 2008). The antioxidant effects of BR in this synergistic action are partially based on direct RONS scavenging but also on regulation of NADPH oxidase activity, neutrophil adhesion and inflammatory pathways (Abraham and Kappas, 2008) as well as control of redox sensitive protein levels (e.g., GCH-1; Jansen et al., 2010; Schuhmacher et al., 2011). Since BV, in our hands, displayed redox-cycling under certain conditions, we speculate that removal of BV may be even more important than the formation of BR – or at least both processes contribute to the antioxidant role of BVR.
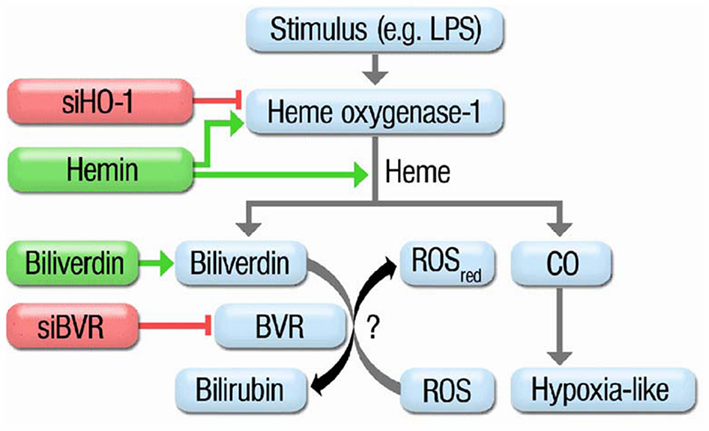
Figure 3. Scheme summarizing the protective routes in the heme oxygenase-1 system. Treatment of HUVECs with oxidative stimuli (e.g., LPS) results in induction of HO-1 which catalyzes the break-down of metallo-porphyrins to CO (hypoxia-like pathway) or biliverdin. Biliverdin is further converted by biliverdin reductase (BVR) yielding the potent antioxidant bilirubin. siRNA against HO-1 blocks induction of HO-1. Hemin is a potent inducer of HO-1 but also a substrate for it resulting in increased CO and BR formation. Biliverdin is the substrate of BVR leading to an increase in BR. siRNA against BVR impairs the conversion of BV to BR. Previous reports speculated on a catalytic cycle of BV to BR conversion by BVR and subsequent oxidation of BR to BV by ROS. Adopted from Jansen et al. (2010). With permission of Rightslink® by Copyright Clearance Center (Elsevier).
According to a recent publication of Maghzal et al. (2009) revisiting the previous findings of Snyder and colleagues neither BVR overexpression nor silencing changed hydrogen peroxide toxicity in vitro. Also these authors did not observe any significant oxidative conversion of BR to BV by hydrogen peroxide and only modest BV formation from BR in a chemical model of lipid peroxidation in chloroform or by peroxyl radicals. Whereas the latter observation is in good agreement with our present findings since we also did not observe significant conversion of BR by various oxidants or mitochondrial ROS, we disagree with the conclusion that BR and BVR have no crucial effect on antioxidant protection conferred by HO-1 under cellular conditions. We think that hydrogen peroxide is an unfortunate model to disproof a role for BVR/BR since we here show that hydrogen peroxide has only minor reactivity toward BR and BVR knock-down resulted in increased cellular oxidative damage in a septic shock model based on LPS-triggered peroxynitrite formation in HUVECs. These considerations are supported by findings of Young et al. (2009) demonstrating that angiotensin-II induced oxidative damage in renal tubular epithelial cells is aggravated by silencing of BVR. These authors also observed BVR/BR independent antioxidant action of HO-1 indicating an important contribution of CO and/or BV as well. In a letter to the editor, Snyder and colleagues criticized the experimental setup in the work of Maghzal et al. with special emphasis on the need for providing BV by either exogenous addition or endogenous activation of the HO-1 system (Sedlak and Snyder, 2009), as done in our cell culture experiments.
Therapeutic Potential of BVR in Organic Nitrate Tolerance and Other Neurological/Cardiovascular Complications
Regarding the therapeutic use of modulating BVR activity, it would be interesting whether BVR deficiency would aggravate organic nitrate induced vascular damage characterized by endothelial dysfunction and nitrate tolerance. Vice versa, it would be of clinical importance whether BVR overexpression may improve the adverse effects of chronic organic nitrate therapy. During the past years we were able to demonstrate that HO-1 induction by hemin was able to prevent nitroglycerin (GTN) induced vascular dysfunction and oxidative stress, whereas suppression of HO-1 activity by apigenin induced a tolerance-like effect in rats under pentaerithrityl tetranitrate (PETN) therapy (Wenzel et al., 2007). Previous reports demonstrated that PETN therapy is devoid of nitrate tolerance, endothelial dysfunction, and oxidative stress (Jurt et al., 2001; Gori et al., 2003; Schnorbus et al., 2010) and that this beneficial profile is at least partially based on the induction of HO-1 (Oberle et al., 1999, 2002, 2003) but also on activation of other cardio-protective genes (Pautz et al., 2009) as well as improvement of endothelial progenitor cell function (Thum et al., 2007). Recently, we have demonstrated that the beneficial profile of PETN is lost in HO-1 deficient mice and PETN therapy results in induction of nitrate tolerance as well as vascular oxidative stress (Schuhmacher et al., 2010). In a subsequent study we have identified the transcription factor Nrf2 as an important mediator of PETN-dependent HO-1 induction since cells with silenced Nrf2 did not show increased HO-1 promoter activity in response to PETN treatment anymore (Schuhmacher et al., 2011). Finally, BR caused a concentration-dependent decrease in nitroglycerin-induced mitochondrial RONS formation (Wenzel et al., 2007) suggesting that this powerful antioxidant may contribute to the prevention of adverse side-effects under chronic PETN therapy by eliminating toxic RONS species. Since our previous findings have revealed a dramatic increase in peroxynitrite (Daiber et al., 2005a,b), associated with enhanced protein tyrosine nitration (Warnholtz et al., 2002; Schuhmacher et al., 2009) and loss of function of prostacyclin synthase (Hink et al., 2003) under chronic nitroglycerin therapy, it may be speculated that the potent RONS (namely peroxynitrite) scavenging properties of bilirubin (Minetti et al., 1998; Kaur et al., 2003) contribute to the suppression of nitrate tolerance by HO-1 induction. Therefore, it would be of great clinical importance to test the effect of BVR silencing or pharmacological inhibition and vice versa investigate the effect of BVR overexpression or pharmacological induction/activation on chronic outcome of PETN therapy. This would help to clarify whether the conversion of BV to BR by BVR does significantly contribute to the beneficial profile of HO-1 induction under PETN therapy.
Recent reports propose a link between BVR and beneficial effects of statins in neurodegenerative disorders such as Alzheimer disease (Barone et al., 2012; Schneider and Simons, 2012). In detail, atorvastatin significantly increased BVR isoform A protein levels, phosphorylation and activity only in parietal cortex. Additionally, these authors found significant negative correlations between BVR isoform A and oxidative stress indices, as well as discrimination learning error scores. Finally, BVR isoform A up-regulation and post-translational modifications significantly correlated with β-secretase protein levels in the brain, suggesting a possible role for BVR isoform A in amyloid-β peptide (Aβ) formation. Preceding work has shown that nitrosative and oxidative stress-induced modifications on hippocampal BVR isoform A are an early event in the pathogenesis of Alzheimer disease (Barone et al., 2011). The Maines group reported on protection of cardiomyocytes by synergistic BVR and HO-2 upregulation as a potential strategy against cardiac dysfunction (Ding et al., 2011). In addition to these novel observations, a common BVR isoform A polymorphism was found to be associated with essential hypertension in Kazaks (Lin et al., 2011). A recent review by Maines highlights the beneficial role of BVR in insulin signaling suggesting a therapeutic potential of BVR in diabetes (Maines, 2011). The unique features and therapeutic potential of BVR were reviewed by Florczyk et al. (2008) and the same group also demonstrates that overexpression of BVR enhances resistance to chemotherapeutics (Florczyk et al., 2011).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Margot Neuser for graphical assistance. The present work was supported by generous financial support by the Johannes Gutenberg University and Medical Center Mainz (MAIFOR and Forschungsfonds grants to Andreas Daiber) and by the Margarete-Waitz-Foundation (stipend to Thomas Jansen).
Abbreviations
2-HE, 2-hydroxyethidium; AAPH, 2,2′-azobis(2-amidinopropane) hydrochloride; ACE, angiotensin-converting enzyme; ApoE−/−, apolipoprotein E deficiency (knock-out); ARB, angiotensin-receptor blocker; Aβ, amyloid-β peptide; BH2, dihydrobiopterin; BH4, tetrahydrobiopterin; BR, bilirubin; BSA, bovine serum albumin; BV, biliverdin; BVR, biliverdin reductase; CAD, coronary artery disease; CO, carbon monoxide; CORM-2, CO-releasing molecule-2; DHE, dihydroethidine (dihydroethidium); DHR, dihydrorhodamine 123; DMSO, dimethyl sulfoxide; DNA, deoxyribonucleic acid; ECL, enhanced chemiluminescence; eNOS, endothelial nitric oxide synthase (NOS-3); GCH-1, GTP-cyclohydrolase-1; GTN, glyceryl trinitrate (nitroglycerin); HDF, human fibroblast; HPLC, high performance liquid chromatography; HO, heme oxygenase; HO-1, heme oxygenase-1 (inducible isoform); HO-2, heme oxygenase-2 (constitutive isoform); HRP, horseradish peroxidase; HUVECs, human umbilical vein endothelial cells; KO2, potassium superoxide; L-012 luminol analog; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MnSOD, manganese superoxide dismutase (SOD2, mitochondrial isoform); mRNA, messenger ribonucleic acid; NO, nitric oxide (nitrogen monoxide); nNOS, neuronal nitric oxide synthase (NOS-1); PARP, poly(ADP-ribose)polymerase; PETN, pentaerithrityl tetranitrate; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; RONS, reactive oxygen and nitrogen species; ROS, reactive oxygen species; RT-PCR, reverse transcription-polymerase chain reaction; Sin-1,3-morpholino-sydnonimine (active metabolite of molsidomine); siBVR, small interfering RNA against BVR; siHO-1, small interfering RNA against HO-1; siRNA, small interfering RNA; TNF-α, tumor necrosis factor-α; XO, xanthine oxidase.
References
Abraham, N. G., and Kappas, A. (2008). Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 60, 79–127.
Ahmad, Z., Salim, M., and Maines, M. D. (2002). Human biliverdin reductase is a leucine zipper-like DNA-binding protein and functions in transcriptional activation of heme oxygenase-1 by oxidative stress. J. Biol. Chem. 277, 9226–9232.
Bachschmid, M., Schildknecht, S., and Ullrich, V. (2005). Redox regulation of vascular prostanoid synthesis by the nitric oxide-superoxide system. Biochem. Biophys. Res. Commun. 338, 536–542.
Baranano, D. E., Rao, M., Ferris, C. D., and Snyder, S. H. (2002). Biliverdin reductase: a major physiologic cytoprotectant. Proc. Natl. Acad. Sci. U.S.A. 99, 16093–16098.
Baranano, D. E., and Snyder, S. H. (2001). Neural roles for heme oxygenase: contrasts to nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 98, 10996–11002.
Barone, E., Di Domenico, F., Cenini, G., Sultana, R., Coccia, R., Preziosi, P., Perluigi, M., Mancuso, C., and Butterfield, D. A. (2011). Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer’s disease and amnestic mild cognitive impairment. J. Alzheimers Dis. 25, 623–633.
Barone, E., Mancuso, C., Di Domenico, F., Sultana, R., Murphy, M. P., Head, E., and Butterfield, D. A. (2012). Biliverdin reductase-A: a novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J. Neurochem. 120, 135–146.
Bassenge, E., Fink, N., Skatchkov, M., and Fink, B. (1998). Dietary supplement with vitamin C prevents nitrate tolerance. J. Clin. Invest. 102, 67–71.
Bassenge, E., Schneider, H. T., and Daiber, A. (2005). Oxidative stress and cardiovascular diseases. Dtsch. Med. Wochenschr. 130, 2904–2909.
Bellner, L., Wolstein, J., Patil, K. A., Dunn, M. W., and Laniado-Schwartzman, M. (2011). Biliverdin rescues the HO-2 null mouse phenotype of unresolved chronic inflammation following corneal epithelial injury. Invest. Ophthalmol. Vis. Sci. 52, 3246–3253.
Cai, H., and Harrison, D. G. (2000). Endothelial dysfunction in cardio-vascular diseases: the role of oxidant stress. Circ. Res. 87, 840–844.
Chen, A. F., Chen, D.-D., Daiber, A., Faraci, F. M., Li, H., Rembold, C. M., and Laher, I. (2012). Free radical biology of the cardiovascular system. Clin. Sci. doi: 10.1042/CS20110562
Crawford, R. S., Kirk, E. A., Rosenfeld, M. E., LeBoeuf, R. C., and Chait, A. (1998). Dietary antioxidants inhibit development of fatty streak lesions in the LDL receptor-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 18, 1506–1513.
Daiber, A. (2010). Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 1797, 897–906.
Daiber, A., Mulsch, A., Hink, U., Mollnau, H., Warnholtz, A., Oelze, M., and Munzel, T. (2005a). The oxidative stress concept of nitrate tolerance and the antioxidant properties of hydralazine. Am. J. Cardiol. 96, 25i–36i.
Daiber, A., Oelze, M., Coldewey, M., Kaiser, K., Huth, C., Schildknecht, S., Bachschmid, M., Nazirisadeh, Y., Ullrich, V., Mulsch, A., Munzel, T., and Tsilimingas, N. (2005b). Hydralazine is a powerful inhibitor of peroxynitrite formation as a possible explanation for its beneficial effects on prognosis in patients with congestive heart failure. Biochem. Biophys. Res. Commun. 338, 1865–1874.
Daiber, A., and Münzel, T. (2006). Oxidativer Stress, Redoxregulation und NO-Bioverfügbarkeit – experimentelle und klinische Aspekte. Darmstadt: Steinkopff Verlag Darmstadt.
De Backer, O., and Lefebvre, R. A. (2008). Investigation of a possible interaction between the heme oxygenase/biliverdin reductase and nitric oxide synthase pathway in murine gastric fundus and jejunum. Eur. J. Pharmacol. 590, 369–376.
Dikalova, A., Clempus, R., Lassegue, B., Cheng, G., McCoy, J., Dikalov, S., San Martin, A., Lyle, A., Weber, D. S., Weiss, D., Taylor, W. R., Schmidt, H. H., Owens, G. K., Lambeth, J. D., and Griendling, K. K. (2005). Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 112, 2668–2676.
Dikalova, A. E., Bikineyeva, A. T., Budzyn, K., Nazarewicz, R. R., McCann, L., Lewis, W., Harrison, D. G., and Dikalov, S. I. (2010). Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 107, 106–116.
Ding, B., Gibbs, P. E., Brookes, P. S., and Maines, M. D. (2011). The coordinated increased expression of biliverdin reductase and heme oxygenase-2 promotes cardiomyocyte survival: a reductase-based peptide counters beta-adrenergic receptor ligand-mediated cardiac dysfunction. FASEB J. 25, 301–313.
Doerries, C., Grote, K., Hilfiker-Kleiner, D., Luchtefeld, M., Schaefer, A., Holland, S. M., Sorrentino, S., Manes, C., Schieffer, B., Drexler, H., and Landmesser, U. (2007). Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ. Res. 100, 894–903.
Eipel, C., Eisold, M., Schuett, H., and Vollmar, B. (2007). Inhibition of heme oxygenase-1 protects against tissue injury in carbon tetrachloride exposed livers. J. Surg. Res. 139, 113–120.
Elhaimeur, F., Courderot-Masuyer, C., Nicod, L., Guyon, C., Richert, L., and Berthelot, A. (2002). Dietary vitamin C supplementation decreases blood pressure in DOCA-salt hypertensive male Sprague-Dawley rats and this is associated with increased liver oxidative stress. Mol. Cell. Biochem. 237, 77–83.
Florczyk, U., Golda, S., Zieba, A., Cisowski, J., Jozkowicz, A., and Dulak, J. (2011). Overexpression of biliverdin reductase enhances resistance to chemotherapeutics. Cancer Lett. 300, 40–47.
Florczyk, U. M., Jozkowicz, A., and Dulak, J. (2008). Biliverdin reductase: new features of an old enzyme and its potential therapeutic significance. Pharmacol. Rep. 60, 38–48.
Gori, T., Al-Hesayen, A., Jolliffe, C., and Parker, J. D. (2003). Comparison of the effects of pentaerythritol tetranitrate and nitroglycerin on endothelium-dependent vasorelaxation in male volunteers. Am. J. Cardiol. 91, 1392–1394.
Gori, T., Burstein, J. M., Ahmed, S., Miner, S. E., Al-Hesayen, A., Kelly, S., and Parker, J. D. (2001). Folic acid prevents nitroglycerin-induced nitric oxide synthase dysfunction and nitrate tolerance: a human in vivo study. Circulation 104, 1119–1123.
Heitzer, T., Just, H., and Munzel, T. (1996). Antioxidant vitamin C improves endothelial dysfunction in chronic smokers. Circulation 94, 6–9.
Hink, U., Oelze, M., Kolb, P., Bachschmid, M., Zou, M. H., Daiber, A., Mollnau, H., August, M., Baldus, S., Tsilimingas, N., Walter, U., Ullrich, V., and Munzel, T. (2003). Role for peroxynitrite in the inhibition of prostacyclin synthase in nitrate tolerance. J. Am. Coll. Cardiol. 42, 1826–1834.
Jansen, T., Hortmann, M., Oelze, M., Opitz, B., Steven, S., Schell, R., Knorr, M., Karbach, S., Schuhmacher, S., Wenzel, P., Munzel, T., and Daiber, A. (2010). Conversion of biliverdin to bilirubin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1-evidence for direct and indirect antioxidant actions of bilirubin. J. Mol. Cell. Cardiol. 49, 186–195.
Jung, O., Schreiber, J. G., Geiger, H., Pedrazzini, T., Busse, R., and Brandes, R. P. (2004). gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation 109, 1795–1801.
Jurt, U., Gori, T., Ravandi, A., Babaei, S., Zeman, P., and Parker, J. D. (2001). Differential effects of pentaerythritol tetranitrate and nitroglycerin on the development of tolerance and evidence of lipid peroxidation: a human in vivo study. J. Am. Coll. Cardiol. 38, 854–859.
Kaur, H., Hughes, M. N., Green, C. J., Naughton, P., Foresti, R., and Motterlini, R. (2003). Interaction of bilirubin and biliverdin with reactive nitrogen species. FEBS Lett. 543, 113–119.
Kim, H. P., Wang, X., Galbiati, F., Ryter, S. W., and Choi, A. M. (2004). Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J. 18, 1080–1089.
Kim, S. Y., Kang, H. T., Choi, H. R., and Park, S. C. (2011). Biliverdin reductase A in the prevention of cellular senescence against oxidative stress. Exp. Mol. Med. 43, 15–23.
Landmesser, U., Cai, H., Dikalov, S., McCann, L., Hwang, J., Jo, H., Holland, S. M., and Harrison, D. G. (2002). Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 40, 511–515.
Lee, D. H., Folsom, A. R., Harnack, L., Halliwell, B., and Jacobs, D. R. Jr. (2004). Does supplemental vitamin C increase cardiovascular disease risk in women with diabetes? Am. J. Clin. Nutr. 80, 1194–1200.
Lerner-Marmarosh, N., Miralem, T., Gibbs, P. E., and Maines, M. D. (2007). Regulation of TNF-alpha-activated PKC-zeta signaling by the human biliverdin reductase: identification of activating and inhibitory domains of the reductase. FASEB J. 21, 3949–3962.
Lin, R., Wang, X., Zhou, W., Fu, W., Wang, Y., Huang, W., and Jin, L. (2011). Association of a BLVRA common polymorphism with essential hypertension and blood pressure in Kazaks. Clin. Exp. Hypertens. 33, 294–298.
Liu, Y., Liu, J., Tetzlaff, W., Paty, D. W., and Cynader, M. S. (2006). Biliverdin reductase, a major physiologic cytoprotectant, suppresses experimental autoimmune encephalomyelitis. Free Radic. Biol. Med. 40, 960–967.
Lonn, E., Bosch, J., Yusuf, S., Sheridan, P., Pogue, J., Arnold, J. M., Ross, C., Arnold, A., Sleight, P., Probstfield, J., and Dagenais, G. R. (2005). Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA 293, 1338–1347.
Maghzal, G. J., Leck, M. C., Collinson, E., Li, C., and Stocker, R. (2009). Limited role for the bilirubin-biliverdin redox amplification cycle in the cellular antioxidant protection by biliverdin reductase. J. Biol. Chem. 284, 29251–29259.
Maines, M. D. (2007). Biliverdin reductase: PKC interaction at the cross-talk of MAPK and PI3K signaling pathways. Antioxid. Redox Signal. 9, 2187–2195.
Maines, M. D. (2011). Potential application of biliverdin reductase and its fragments to modulate insulin/IGF-1/MAPK/PI3-K signaling pathways in therapeutic settings. Curr. Drug Targets 11, 1586–1594.
Maines, M. D., Miralem, T., Lerner-Marmarosh, N., Shen, J., and Gibbs, P. E. (2007). Human biliverdin reductase, a previously unknown activator of protein kinase C betaII. J. Biol. Chem. 282, 8110–8122.
Mann, J. F., Lonn, E. M., Yi, Q., Gerstein, H. C., Hoogwerf, B. J., Pogue, J., Bosch, J., Dagenais, G. R., and Yusuf, S. (2004). Effects of vitamin E on cardiovascular outcomes in people with mild-to-moderate renal insufficiency: results of the HOPE study. Kidney Int. 65, 1375–1380.
McDonagh, A. F. (2010). The biliverdin-bilirubin antioxidant cycle of cellular protection: missing a wheel? Free Radic. Biol. Med. 49, 814–820.
Minetti, M., Mallozzi, C., Di Stasi, A. M., and Pietraforte, D. (1998). Bilirubin is an effective antioxidant of peroxynitrite-mediated protein oxidation in human blood plasma. Arch. Biochem. Biophys. 352, 165–174.
Miralem, T., Hu, Z., Torno, M. D., Lelli, K. M., and Maines, M. D. (2005). Small interference RNA-mediated gene silencing of human biliverdin reductase, but not that of heme oxygenase-1, attenuates arsenite-mediated induction of the oxygenase and increases apoptosis in 293A kidney cells. J. Biol. Chem. 280, 17084–17092.
Muntwyler, J., Hennekens, C. H., Manson, J. E., Buring, J. E., and Gaziano, J. M. (2002). Vitamin supplement use in a low-risk population of US male physicians and subsequent cardiovascular mortality. Arch. Intern. Med. 162, 1472–1476.
Munzel, T., Sinning, C., Post, F., Warnholtz, A., and Schulz, E. (2008). Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann. Med. 40, 180–196.
Oberle, S., Abate, A., Grosser, N., Hemmerle, A., Vreman, H. J., Dennery, P. A., Schneider, H. T., Stalleicken, D., and Schroder, H. (2003). Endothelial protection by pentaerithrityl trinitrate: bilirubin and carbon monoxide as possible mediators. Exp. Biol. Med. (Maywood) 228, 529–534.
Oberle, S., Abate, A., Grosser, N., Vreman, H. J., Dennery, P. A., Schneider, H. T., Stalleicken, D., and Schroder, H. (2002). Heme oxygenase-1 induction may explain the antioxidant profile of pentaerythrityl trinitrate. Biochem. Biophys. Res. Commun. 290, 1539–1544.
Oberle, S., Schwartz, P., Abate, A., and Schroder, H. (1999). The antioxidant defense protein ferritin is a novel and specific target for pentaerithrityl tetranitrate in endothelial cells. Biochem. Biophys. Res. Commun. 261, 28–34.
Oelze, M., Knorr, M., Schuhmacher, S., Heeren, T., Otto, C., Schulz, E., Reifenberg, K., Wenzel, P., Munzel, T., and Daiber, A. (2011). Vascular dysfunction in streptozotocin-induced experimental diabetes strictly depends on insulin deficiency. J. Vasc. Res. 48, 275–284.
Pachori, A. S., Smith, A., McDonald, P., Zhang, L., Dzau, V. J., and Melo, L. G. (2007). Heme-oxygenase-1-induced protection against hypoxia/reoxygenation is dependent on biliverdin reductase and its interaction with PI3K/Akt pathway. J. Mol. Cell. Cardiol. 43, 580–592.
Panahian, N., Huang, T., and Maines, M. D. (1999). Enhanced neuronal expression of the oxidoreductase – biliverdin reductase – after permanent focal cerebral ischemia. Brain Res. 850, 1–13.
Pautz, A., Rauschkolb, P., Schmidt, N., Art, J., Oelze, M., Wenzel, P., Forstermann, U., Daiber, A., and Kleinert, H. (2009). Effects of nitroglycerin or pentaerithrityl tetranitrate treatment on the gene expression in rat hearts: evidence for cardiotoxic and cardioprotective effects. Physiol. Genomics 38, 176–185.
Perez, M. J., Castano, B., Jimenez, S., Serrano, M. A., Gonzalez-Buitrago, J. M., and Marin, J. J. (2008). Role of vitamin C transporters and biliverdin reductase in the dual pro-oxidant and anti-oxidant effect of biliary compounds on the placental-fetal unit in cholestasis during pregnancy. Toxicol. Appl. Pharmacol. 232, 327–336.
Schneider, A., and Simons, M. (2012). Antioxidative strategies in cognitive impairment: a novel connection between biliverdin-reductase and statins. J. Neurochem. 120, 1–3.
Schnorbus, B., Schiewe, R., Ostad, M. A., Medler, C., Wachtlin, D., Wenzel, P., Daiber, A., Munzel, T., and Warnholtz, A. (2010). Effects of pentaerythritol tetranitrate on endothelial function in coronary artery disease: results of the PENTA study. Clin. Res. Cardiol. 99, 115–124.
Schuhmacher, S., Oelze, M., Bollmann, F., Kleinert, H., Otto, C., Heeren, T., Steven, S., Hausding, M., Knorr, M., Pautz, A., Reifenberg, K., Schulz, E., Gori, T., Wenzel, P., Munzel, T., and Daiber, A. (2011). Vascular dysfunction in experimental diabetes is improved by pentaerithrityl tetranitrate but not isosorbide-5-mononitrate therapy. Diabetes 60, 2608–2616.
Schuhmacher, S., Schulz, E., Oelze, M., Konig, A., Roegler, C., Lange, K., Sydow, L., Kawamoto, T., Wenzel, P., Munzel, T., Lehmann, J., and Daiber, A. (2009). A new class of organic nitrates: investigations on bioactivation, tolerance and cross-tolerance phenomena. Br. J. Pharmacol. 158, 510–520.
Schuhmacher, S., Wenzel, P., Schulz, E., Oelze, M., Mang, C., Kamuf, J., Gori, T., Jansen, T., Knorr, M., Karbach, S., Hortmann, M., Mathner, F., Bhatnagar, A., Forstermann, U., Li, H., Munzel, T., and Daiber, A. (2010). Pentaerythritol tetranitrate improves angiotensin II-induced vascular dysfunction via induction of heme oxygenase-1. Hypertension 55, 897–904.
Sedlak, T. W., Saleh, M., Higginson, D. S., Paul, B. D., Juluri, K. R., and Snyder, S. H. (2009). Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. U.S.A. 106, 5171–5176.
Sedlak, T. W., and Snyder, S. H. (2004). Bilirubin benefits: cellular protection by a biliverdin reductase antioxidant cycle. Pediatrics 113, 1776–1782.
Sedlak, T. W., and Snyder, S. H. (2009). Cycling the wagons for biliverdin reductase. J. Biol. Chem. 284, le11.
Stocker, R., and Maghzal, G. J. (2009). Reply to sedlak and snyder: the little bighorn of the biliverdin reductase amplification cycle. J. Biol. Chem. 284:le12.
Stocker, R., Yamamoto, Y., McDonagh, A. F., Glazer, A. N., and Ames, B. N. (1987). Bilirubin is an antioxidant of possible physiological importance. Science 235, 1043–1046.
Thum, T., Fraccarollo, D., Thum, S., Schultheiss, M., Daiber, A., Wenzel, P., Munzel, T., Ertl, G., and Bauersachs, J. (2007). Differential effects of organic nitrates on endothelial progenitor cells are determined by oxidative stress. Arterioscler. Thromb. Vasc. Biol. 27, 748–754.
Torzewski, M., Ochsenhirt, V., Kleschyov, A. L., Oelze, M., Daiber, A., Li, H., Rossmann, H., Tsimikas, S., Reifenberg, K., Cheng, F., Lehr, H. A., Blankenberg, S., Forstermann, U., Munzel, T., and Lackner, K. J. (2007). Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 27, 850–857.
Ullrich, V., and Kissner, R. (2006). Redox signaling: bioinorganic chemistry at its best. J. Inorg. Biochem. 100, 2079–2086.
Vitek, L., and Schwertner, H. A. (2007). The heme catabolic pathway and its protective effects on oxidative stress-mediated diseases. Adv. Clin. Chem. 43, 1–57.
Warnholtz, A., Mollnau, H., Heitzer, T., Kontush, A., Moller-Bertram, T., Lavall, D., Giaid, A., Beisiegel, U., Marklund, S. L., Walter, U., Meinertz, T., and Munzel, T. (2002). Adverse effects of nitroglycerin treatment on endothelial function, vascular nitrotyrosine levels and cGMP-dependent protein kinase activity in hyperlipidemic Watanabe rabbits. J. Am. Coll. Cardiol. 40, 1356–1363.
Watanabe, H., Kakihana, M., Ohtsuka, S., and Sugishita, Y. (1997). Randomized, double-blind, placebo-controlled study of supplemental vitamin E on attenuation of the development of nitrate tolerance. Circulation 96, 2545–2550.
Wenzel, P., Oelze, M., Coldewey, M., Hortmann, M., Seeling, A., Hink, U., Mollnau, H., Stalleicken, D., Weiner, H., Lehmann, J., Li, H., Forstermann, U., Munzel, T., and Daiber, A. (2007). Heme oxygenase-1: a novel key player in the development of tolerance in response to organic nitrates. Arterioscler. Thromb. Vasc. Biol. 27, 1729–1735.
Wenzel, P., Schuhmacher, S., Kienhofer, J., Muller, J., Hortmann, M., Oelze, M., Schulz, E., Treiber, N., Kawamoto, T., Scharffetter-Kochanek, K., Munzel, T., Burkle, A., Bachschmid, M. M., and Daiber, A. (2008a). Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc. Res. 80, 280–289.
Wenzel, P., Schulz, E., Oelze, M., Muller, J., Schuhmacher, S., Alhamdani, M. S., Debrezion, J., Hortmann, M., Reifenberg, K., Fleming, I., Munzel, T., and Daiber, A. (2008b). AT1-receptor blockade by telmisartan upregulates GTP-cyclohydrolase I and protects eNOS in diabetic rats. Free Radic. Biol. Med. 45, 619–626.
Xu, J., Wang, S., Wu, Y., Song, P., and Zou, M. H. (2009). Tyrosine nitration of PA700 activates the 26S proteasome to induce endothelial dysfunction in mice with angiotensin II-induced hypertension. Hypertension 54, 625–632.
Xu, J., Wu, Y., Song, P., Zhang, M., Wang, S., and Zou, M. H. (2007). Proteasome-dependent degradation of guanosine 5′-triphosphate cyclohydrolase I causes tetrahydrobiopterin deficiency in diabetes mellitus. Circulation 116, 944–953.
Young, S. C., Storm, M. V., Speed, J. S., Kelsen, S., Tiller, C. V., Vera, T., Drummond, H. A., and Stec, D. E. (2009). Inhibition of biliverdin reductase increases ANG II-dependent superoxide levels in cultured renal tubular epithelial cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1546–R1553.
Keywords: heme oxygenase, bile pigments, biliverdin reductase, oxidative stress, vascular function
Citation: Jansen T and Daiber A (2012) Direct antioxidant properties of bilirubin and biliverdin. Is there a role for biliverdin reductase? Front. Pharmacol. 3:30. doi: 10.3389/fphar.2012.00030
Received: 15 January 2012; Paper pending published: 05 February 2012;
Accepted: 14 February 2012; Published online: 16 March 2012.
Edited by:
Jaime Kapitulnik, The Hebrew University of Jerusalem, IsraelReviewed by:
Mahin D. Maines, University of Rochester School of Medicine, USARon Kohen, The Hebrew University of Jerusalem, Israel
Peter Gibbs, University of Rochester School of Medicine and Dentistry, USA
Copyright: © 2012 Jansen and Daiber. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Andreas Daiber, Universitätsmedizin der Johannes Gutenberg-Universität Mainz, II. Medizinische Klinik – Labor für Molekulare Kardiologie, Verfügungsgebäude für Forschung und Entwicklung – Raum 00349, Obere Zahlbacher Str. 63, 55101 Mainz, Germany. e-mail:YW5kcmVhcy5kYWliZXJAYmlvcmVkb3guY29t