

- Medical School, Institute of Biochemistry and Molecular Biology, RWTH Aachen University, Aachen, Germany
Sirtuins are homologs of the yeast silencing information regulator 2 protein, an NAD+-dependent (histone) deacetylase. In mammals seven different sirtuins, SIRT1–7, have been identified, which share a common catalytic core domain but possess distinct N- and C-terminal extensions. This core domain elicits NAD+-dependent deacetylase and in some cases also ADP-ribosyltransferase, demalonylase, and desuccinylase activities. Sirtuins have been implicated in key cellular processes, including cell survival, autophagy, apoptosis, gene transcription, DNA repair, stress response, and genome stability. In addition some sirtuins are associated with disease, including cancer and neurodegeneration. These findings suggest strongly that sirtuins are tightly controlled and potentially responsive to different signal transduction pathways. Here, we review the posttranslational regulation mechanisms of mammalian sirtuins and discuss their relevance regarding the physiological processes, with which the different sirtuins are associated. The available data suggest that the N- and C-terminal extensions are the targets of posttranslational modifications (PTM) that can affect the functions of sirtuins. Mechanistically this can be explained by the interaction of these extensions with the catalytic core domain, which appears to be controlled by PTM at least in some cases. In contrast little is known about PTM and regulation of the catalytic domain itself. Together these findings point to key regulatory roles of the N- and C-terminal extensions in controlling sirtuin functions, thus connecting these regulators to different signaling pathways.
Introduction
The first paper describing acetylation of histones at lysine residues was published almost 50 years ago (Allfrey and Mirsky, 1964). Although for many years the physiological role of lysine acetylation (Kac) was undefined, the last decade has witnessed a large increase in our knowledge about the consequences of this modification. Acetylation results in the loss of the positive charge at lysines under physiological conditions and affects the chemical appearance of proteins, resulting in altered functional properties (Figure 1). These include effects on protein–protein interaction and on the catalytic activity of enzymes among others (reviewed in Kim and Yang, 2011). Parallel to these findings enzymes were discovered that are able to transfer acetyl groups from acetyl-CoA to substrates (K-acetyltransferases, KATs) and enzymes that are able to remove acetyl groups, thus providing evidence that acetylation of lysines is a reversible posttranslational modification (PTM). Besides acetylation, lysines are the target of additional PTMs, including methylation, sumoylation, and ubiquitination, which can compete with each other for one given lysine (Figure 1). Besides histones many other proteins have been recognized to be acetylated. These appear to be distributed throughout all cellular compartments. A recent survey using mass spectrometry has identified more than 1700 acetylated proteins in mammals (Kim et al., 2006; Choudhary et al., 2009). Furthermore comparative analyses revealed that acetylation sites are significantly higher conserved than phosphorylation sites (Choudhary et al., 2011). Proteins modified by lysine acetylation control diverse cellular processes such as chromatin remodeling, protein synthesis, cell cycle, nuclear transport, actin nucleation, and mitochondrial metabolism (Choudhary et al., 2011). Therefore it’s not surprising that acetylation has been linked to different diseases, including cancer and neurodegeneration. Indeed inhibitors of deacetylases have entered clinics as therapeutic drugs (Marks, 2010). Thus these as well as many other findings demonstrate that acetylation is an important PTM, which participates in controlling key physiological processes in cells.
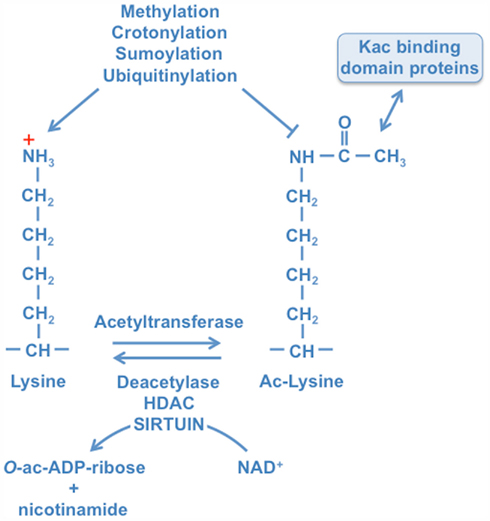
Figure 1. Lysines are targeted by multiple posttranslational modifications. Acetylation is a reversible PTM that is controlled by acetyltransferases and deacetylases. These enzymes transfer acetyl groups from acetyl-CoA to lysine residues with loss of the positive charge. Acetylated lysine residues provide docking sites for proteins that possess a Kac interaction domain, e.g., bromodomains. Lysines can also be modified by a number of additional PTMs as indicated. These modifications compete with each other, thus acetylation can potentially interfere with these other PTMs. The removal of acetyl groups is catalyzed by HDAC and sirtuin deacetylases. Sirtuins are NAD+-dependent enzymes that transfer the acetyl group onto ADP-ribose under release of nicotinamide. This results in the generation of O-acetyl-ADP-ribose, a molecule with second messenger properties.
Based on structural and functional similarities, mammalian deacetylases can be divided into four major classes. Class I HDACs are similar to yeast Rpd3 and contain HDAC1, -2, -3, and -8, whereas class II HDACs are homologs of yeast Hda1 and include HDAC4, -5, -6, -7, -9, and -10. Sirtuins which are related to yeast silent information regulator 2 (Sir2) are also termed class III HDACs, while HDAC11 forms the class IV on its own (reviewed in De Ruijter et al., 2003; Haigis and Sinclair, 2010). The sirtuins are the subject of this review. The sirtuin protein family was founded by the yeast Sir2 (Brachmann et al., 1995). The initial findings from genetic studies suggested that Sir2 controls chromatin and gene expression. In particular Sir2 was identified to participate in silencing of homothallic mating (HM) loci and telomeric chromosomal regions and to interfere with rDNA recombination (Gottlieb and Esposito, 1989; Braunstein et al., 1993). These functions have obtained wide interest because they have been linked to lifespan regulation. Indeed overexpression of Sir2 was shown to increase lifespan in yeast (Kaeberlein et al., 1999). Although similar effects were reported for Drosophila melanogaster and Caenorhabditis elegans (Tissenbaum and Guarente, 2001; Rogina and Helfand, 2004), more recent findings suggest that Sir2 does not affect longevity in these organisms (Burnett et al., 2011). While for many years the molecular base of the observations made in yeast was undefined, a break-through finding was the description of Sir2 as a nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase (Imai et al., 2000). Sir2 and Sir2-like proteins hydrolyze one NAD+ for each acetyl group removed from a substrate, with release of the nicotinamide moiety (Figure 1; Landry et al., 2000). The acetyl group is transferred to ADP-ribose to form a novel O-acetyl-ADP-ribose product (Tanner et al., 2000), which has been suggested to function as a second messenger (Tong and Denu, 2010). Very recently it was shown that SIRT5 can also remove acyl-groups from malonylated or succinylated substrate peptides, thereby forming O-malonyl-ADP-ribose or O-succinyl-ADP-ribose, respectively (Du et al., 2011; Peng et al., 2011). Additionally, distinct sirtuins (SIRT4 and SIRT6) were reported to catalyze the transfer of ADP-ribose from NAD+ to substrate proteins (Liszt et al., 2005; Ahuja et al., 2007). These observations provide evidence that some sirtuins may be able to perform more than one biochemical reaction.
Sirtuins are conserved from prokaryotes to mammals and they all share a common core domain comprising approximately 200–275 amino acids. A phylogenetic analysis of the catalytic domains allows subdividing the sirtuins into five classes, i.e., I–IV and U, with the latter only found in Gram-positive bacteria (Frye, 2000). Besides the founding member Sir2, the yeast Saccharomyces cerevisiae expresses four additional sirtuins, which are termed “homologs of sir two” (Hst1–4). Seven human sirtuins have been identified so far, which can be grouped into four of the phylogenetic classes: SIRT1, SIRT2, and SIRT3 belong to class I, SIRT4 to class II, SIRT5 to class III, and SIRT6 and SIRT7 to class IV (Frye, 2000). Of these, SIRT1 shares the highest sequence similarity with yeast Sir2 and Hst1, and SIRT2 and SIRT3 with Hst2 (North and Verdin, 2004). SIRT4 to SIRT7 are more closely related to prokaryotic sirtuins or sirtuins of D. melanogaster and C. elegans.
The mammalian sirtuins are localized in different subcellular compartments. While SIRT1, SIRT6, and SIRT7 are predominantly found in the nucleus, albeit with different subnuclear distributions, SIRT3, SIRT4, and SIRT5 are mitochondrial. SIRT2 is the only human sirtuin, which is primarily localized in the cytoplasm (Michishita et al., 2005).
The findings summarized above, including the observations on longevity in yeast, the role of NAD+ as cofactor, and the localization of the different sirtuins to distinct subcellular compartments, notably the mitochondria, suggested early on that sirtuins might have fundamental roles in metabolism. Indeed Sir2 is mediating at least in part the effects elicited by caloric restriction (reviewed in Lu and Lin, 2010). Of note is also that sirtuins in higher organisms have been suggested to contribute to longevity (reviewed in Guarente, 2011). Moreover the findings that sirtuins carry out NAD+-dependent reactions suggest an involvement of these enzymes in mammalian metabolic control and offer the possibility for modulation of their activity by small molecules. The involvement of sirtuins in many physiological processes (see also below) suggests that these enzymes themselves are most likely controlled by different signaling pathways in response to both extracellular and intracellular cues. The use of NAD+ must be controlled because of its central function in metabolic pathways, suggesting that enzymes that consume NAD+ will most likely be part of feedback control mechanisms of such pathways. In addition the enzymatic processes of deacetylation and of ADP-ribosylation need to be regulated to adjust for optimal, functionally relevant levels of substrate acetylation and ADP-ribosylation. Despite the many reasons for posttranslational control of sirtuin function, we know relatively little about such mechanisms. The available evidence suggests that the N- and C-terminal extensions relative to the catalytic core domains of the seven mammalian sirtuins are targets of PTMs, while hardly any information is available on how the catalytic domain itself is controlled (Figure 2). Here we summarize what has been learned about the regulation of mammalian sirtuins by PTMs.
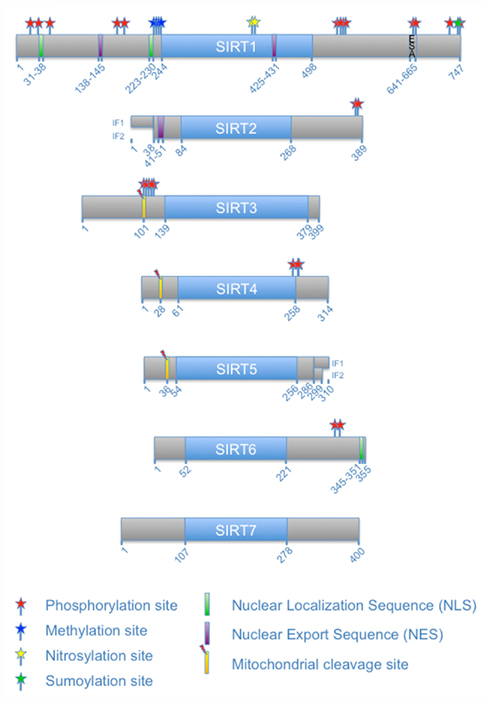
Figure 2. Schematic overview of human sirtuins and their PTMs. The seven mammalian sirtuins are schematically indicated with the blue boxes depicting the sirtuin-typic catalytic core domain. The catalytic domains are flanked by distinct N- and C-terminal extensions (gray boxes). The numbers below indicate amino acid numbers for orientation. Two isoforms (IF) are shown for SIRT2 and SIRT5, respectively. The ESA (“essential for SIRT1 activity”) sequence of SIRT1 (see below) is indicated. PTMs, nuclear localization sequences, nuclear export sequences, and proteolytic cleavage sites are indicated. The precise amino acids modified by the different PTMs are given in Table 1.
Sirtuins and Their Regulation by PTMs
All sirtuins share a common catalytic domain, which binds NAD+. In contrast to this conserved core domain, the enzymes differ in sequence and length of their C- and N-terminal extensions. These are well suited to participate in the regulation of sirtuins and indeed most PTMs that have been identified to date target these extensions (Figure 2). Several scenarios can be imagined. The extensions may communicate with the catalytic domain, thereby controlling the activity of sirtuins. For example the C-terminal extension of yeast Hst2 interacts with the NAD+-binding region, while the N-terminal region of Hst2 engages with the Kac substrate binding site, suggesting different modes of autoregulation (Zhao et al., 2003). Furthermore the N-terminal extension has been suggested to function in trimer formation, which might influence enzymatic activity. Together these findings indicate a more general role of these C- and N-terminal sequences in the regulation of sirtuin function.
SIRT1
Among the seven human sirtuins, SIRT1 shares the highest sequence homology with yeast Sir2 (Voelter-Mahlknecht and Mahlknecht, 2006). In addition SIRT1, similar to its ancestor Sir2, is primarily localized in the nucleus and involved in chromatin remodeling as it deacetylates several lysine residues of histones, including acetylated lysines 9 of histones H3 (H3K9ac), H3K14ac, H4K16ac, and H1K26ac (Vaquero et al., 2004). Moreover SIRT1 targets also non-histone proteins and its activity can be regulated by its ability to shuttle between nuclear and cytoplasmic compartments (Tanno et al., 2007; Hisahara et al., 2008). The increasing number of known SIRT1 substrates includes the transcription factor and tumor suppressor p53 as well as several other transcriptional regulators and cofactors, among them NF-κB, members of the forkhead family (FOXOs), peroxisome proliferator-activated receptors (PPAR), and p300 (reviewed in Rahman and Islam, 2011). Molecular studies revealed that SIRT1 is involved in the regulation of diverse cellular processes ranging from lipid and glucose metabolism to aging and stress response. Of particular relevance for many of these processes is the AMP-activated protein kinase (AMPK)-SIRT1 signaling axis. AMPK is activated in response to increasing amount of AMP and thus functions as an energy sensor that responds to cellular metabolic stress, including calorie restriction (reviewed in Fulco and Sartorelli, 2008). Maybe not surprising then is the finding that Sirt1 knockout mice have a high prenatal or early postnatal death rate (Cheng et al., 2003; McBurney et al., 2003).
With the identification of the tumor suppressor p53 as a SIRT1 substrate, a role of this enzyme in tumor formation was postulated (Luo et al., 2001; Vaziri et al., 2001; Langley et al., 2002). Upon deacetylation by SIRT1, the activity of p53 is reduced and thus SIRT1 appears to function as an oncoprotein (Chen et al., 2005; Kim et al., 2007; Yuan et al., 2011). However, there are also reports that describe SIRT1 as a tumor suppressor (Yi and Luo, 2010). These alternative activities are possibly the result of cell-type specific effects and/or a consequence of distinct regulation of SIRT1 that might differentially affect the activities of substrates.
SIRT1 is by far the largest human sirtuin with 747 amino acids due to its extensive N- and C-terminal extensions (Figure 1). The N-terminal extension of SIRT1 contains two functional nuclear localization sequences (NLS) and two nuclear export sequences (NES). These are responsible for the nucleo-cytoplasmic shuttling of SIRT1 (Tanno et al., 2007), which determines at least in part the enzyme’s ability to interact with distinct substrates (Hisahara et al., 2008). Furthermore the nuclear-cytoplasmic distribution of SIRT1 is regulated by signals, for example during differentiation (Tanno et al., 2007). While SIRT1 is nuclear in proliferating C2C12 myoblasts, it is cytoplasmic in differentiated cells. Moreover inhibition of PI3K prevents the nuclear localization of SIRT1 in proliferating cells, suggesting that PI3K-dependent signaling controls the shuttling. Whether the PI3K signaling cascade targets directly SIRT1 or some accessory factor or factors is not known. One kinase that might be involved in this process is JNK, although this kinase is not typically activated downstream of PI3K. JNK interacts with SIRT1 upon oxidative stress, phosphorylates SIRT1 at Ser27, Ser47, and Thr530, thereby enhancing its nuclear localization (Nasrin et al., 2009). Furthermore these phosphorylations increase the enzymatic activity of SIRT1 in a substrate-specific manner with histone H3, but not p53, becoming a better substrate. In contrast to the findings with JNK, mTOR-dependent phosphorylation of Ser47 alone results in inhibition of SIRT1 deacetylase activity (Back et al., 2011). Thus combinatorial effects of different phosphorylations appear to control SIRT1 function.
Using a mass spectrometry approach, 13 phosphorylation sites were identified in SIRT1 (Sasaki et al., 2008). Seven of these sites are located in the N-terminal region, including Ser27 and Ser47, and six in the C-terminal region, including Thr530 (Figure 2). Two of the identified sites, Thr530 and Ser540, are potential substrates of cyclin B/cyclin-dependent kinase 1 (CDK) complexes. The functional analysis suggests that these two phosphorylation sites are required for normal cell cycle progression. For example, while wild-type SIRT1 rescues the growth defect of cells lacking endogenous Sirt1, a mutant, in which Thr530 and Ser540 are substituted by alanines, is unable to rescue the knockout cells (Sasaki et al., 2008).
In addition to the sites mentioned above, four protein kinase CK2 phosphorylation sites have been identified in murine Sirt1. These are Ser154, Ser649, Ser651, and Ser683 in the N- and C-terminal extensions (Kang et al., 2009). Two of these sites have been described in human SIRT1 at the corresponding amino acids Ser659 and Ser661 (Zschoernig and Mahlknecht, 2009). It has been suggested that phosphorylation by CK2 stimulates catalytic activity of Sirt1 and its ability to interact with p53, one of its substrates (Kang et al., 2009). Whether all four CK2 sites are required for the observed effects remains to be determined. Of note is that Ser659 and Ser661 lie within a region of SIRT1 that is referred to as the ESA (essential for SIRT1 activity) motif (Figure 3). This spans a small region from amino acids 641–665 in human SIRT1 (Kang et al., 2011). The ESA interacts with the catalytic domain, activates the catalytic activity, and increases the affinity for substrates. Moreover the binding site for ESA in the catalytic domain is also the interaction site of DBC1, an endogenous SIRT1 inhibitor (Kim et al., 2008). The two CK2 phosphorylation sites flank one of the two identified key residues within ESA that are important to control catalytic activity (Figure 3). Thus it is well possible that these phosphorylation sites modulate the interaction of the C-terminal region with the catalytic domain and therefore are potentially of considerable functional relevance. One possible explanation is that the phosphorylation of the two sites within ESA regulates SIRT1 activity and substrate recognition by modulating the interaction of the ESA with the catalytic domain. This might affect the catalytic center and substrate binding through an allosteric mechanism. Additionally it might affect binding of DCB1, which is a SIRT1 inhibitor. Thus the control of SIRT1 function by its own C-terminal domain and the regulation of this interaction by CK2, although molecularly not fully explored yet, may represent an important regulatory mechanism (Figure 3).
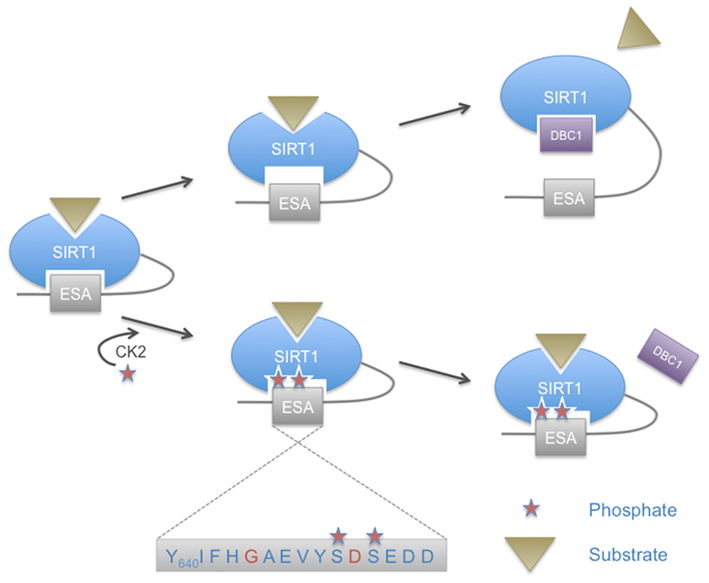
Figure 3. Control of SIRT2 catalytic activity by the ESA motif and regulation by phosphorylation. Two CK2 phosphorylation sites lie within the essential for SIRT1 activity (ESA) sequence motif found in the C-terminal extension of SIRT1. These phosphorylation sites flank one of the two key residues of the ESA motif (indicated in red). CK2-mediated phosphorylation is proposed to enhance the interaction of ESA with the catalytic core, thereby increasing SIRT1 affinity for substrates and enhancing catalytic activity. The ESA motif competes with DBC1 binding, a negative regulator of SIRT1. Enhanced interaction of ESA with the core domain of SIRT2 in response to CK2 phosphorylation would prevent binding of DBC1 and thus abrogate its inhibitory effect.
Besides phosphorylation, SIRT1 is modified by additional PTMs, including sumoylation. SUMO, a small ubiquitin-related modifier, can be attached in vitro close to the C-terminal end of SIRT1 at Lys734, which lies within a sumoylation consensus sequence (ΨKXE). This modification increases catalytic activity as measured by p53 deacetylation (Yang et al., 2007). Upon stress SIRT1 associates with the nuclear desumoylase SENP1, which reduces the catalytic activity of SIRT1 and consequently allows efficient activation of p53. How modification by SUMO stimulates the catalytic activity of SIRT1 is not known. However the recent findings that the C-terminal region is key to enhance SIRT1 activity suggests that sumoylation may participate in this regulation. A possibility is that sumoylation enhances the interaction of the ESA motif with the catalytic domain or modifies CK2 phosphorylation (Figure 3). But clearly other explanations are also possible, including the subnuclear relocalization of sumoylated SIRT1, which might affect accessibility to substrates, or allosteric effects of the sumoylation (reviewed in Wilkinson and Henley, 2010). Thus it appears that sumoylation of SIRT1 is relevant for stress control in cells.
Furthermore, SIRT1 is also targeted by methylation. The methyltransferase Set7/9 interacts with and methylates SIRT1 at Lys233, Lys235, Lys236, and Lys238 of the N-terminal extension. Although it is unclear whether methylation affects directly SIRT1 deacetylase activity, the interaction of Set7/9 with SIRT1 disrupts the binding of SIRT1 with p53. Consequently p53 acetylation and transactivating activity is enhanced (Liu et al., 2011). Despite the lack of information about direct consequences of lysine methylation, it is worth remembering that lysines can be modified by multiple PTMs and thus methylation may compete with acetylation and ubiquitination (Figure 1; reviewed in Yang and Seto, 2008).
Recently it was reported that nuclear SIRT1 is transnitrosylated by nitrosylated GAPDH (Kornberg et al., 2010). As a consequence acetylation of PGC-1α, a SIRT1 substrate, increases in cells, suggesting that SIRT1 deacetylation activity is inhibited by nitrosylation. Mutational analysis implies that two cysteines, Cys387 and Cys390, within the catalytic core of SIRT1 are targeted by nitrosylation. These cysteines are of special interest because they participate in the coordination of a structurally relevant Zn2+ ion and nitrosylation might result in protein misfolding (Kornberg et al., 2010). It will be of interest to define whether nitrosylation is a general regulatory mechanism of sirtuins.
Similar to yeast Hst2, purified endogenous SIRT1 can exist as a homotrimer (Zhao et al., 2003; Vaquero et al., 2004). Because SIRT2 was also purified as a homotrimer (Vaquero et al., 2006), this structural organization may be characteristic for sirtuins. Structural analysis of the Hst2 trimer suggests that the N-terminal region is involved in trimer formation. Whether this is also true for SIRT1 and SIRT2 remains to be determined. Because of the many PTMs that have been mapped in the N-terminal extension of SIRT1 (Table 1), it is well possible that trimer formation is regulated by signaling, an aspect that needs further exploration.
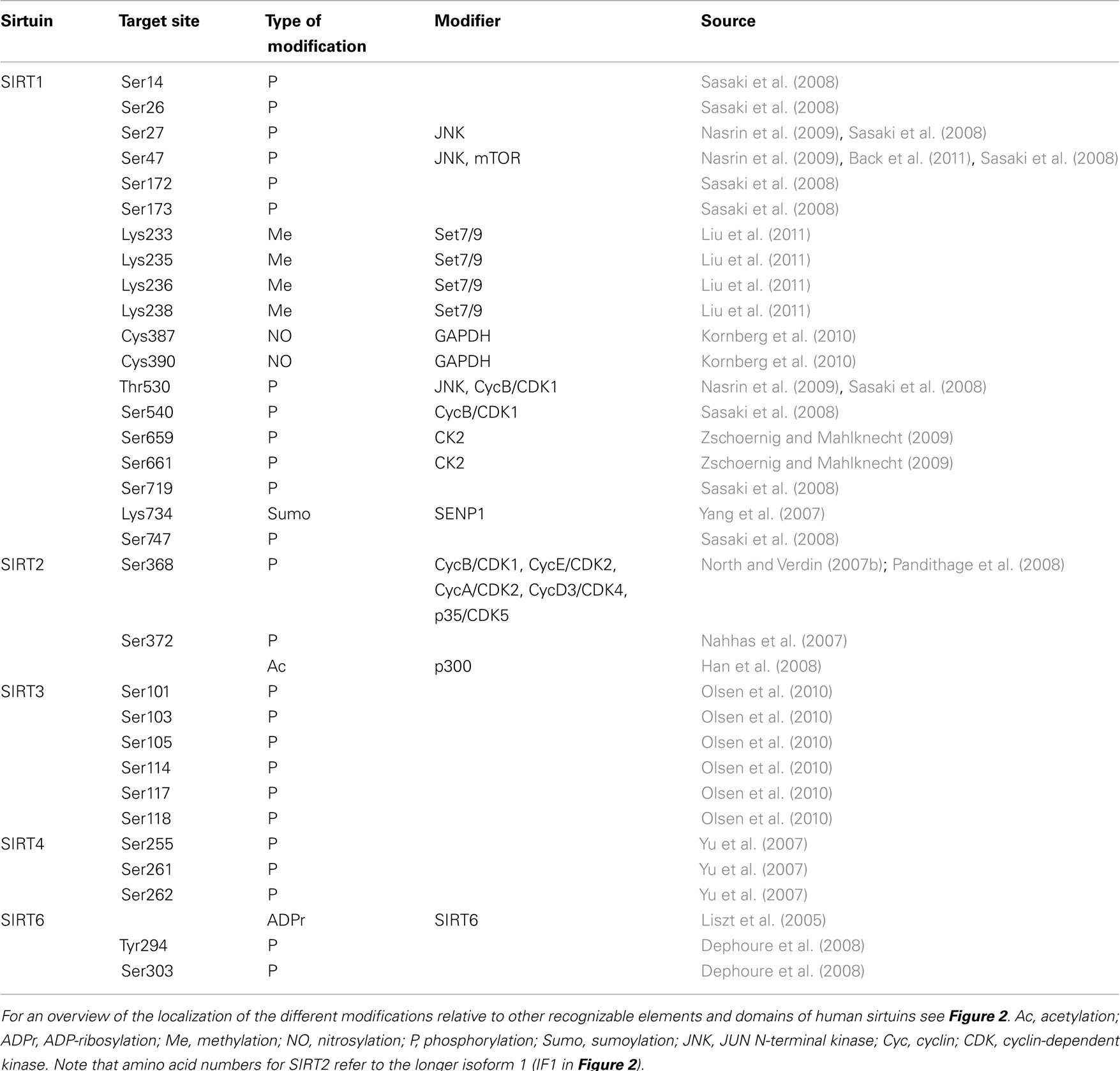
Table 1. Summary of posttranslational modification sites of human sirtuins.
SIRT2
SIRT2 is ubiquitously expressed and the only sirtuin, which is predominantly localized in the cytoplasm (Afshar and Murnane, 1999; Michishita et al., 2005; Voelter-Mahlknecht et al., 2005). SIRT2 was purified as a homotrimer out of cell extracts similar to SIRT1 and Hst2 (see above; Vaquero et al., 2006). It is not known how trimer formation is regulated and what the consequences are for SIRT2 function. Human SIRT2 is expressed in at least two isoforms. The longer SIRT2 variant 1 consists of 389 amino acids whereas variant 2 lacks the first 37 N-terminal amino acids, thus being comprised of 352 amino acids (Figure 2). The catalytic domain of SIRT2 variant 1 and 2 is located between amino acids 84–268 and 47–231, respectively (Voelter-Mahlknecht et al., 2005). The predominant cytoplasmic localization of SIRT2 is dictated by an NES in its N-terminal extension (amino acids 41–51 and 4–14 of the long and short protein variants, respectively; Wilson et al., 2006; North and Verdin, 2007a). In the cytoplasm, SIRT2 colocalizes at least in part with the microtubule network. Consistent with this finding is that Lys40 of α-tubulin is a SIRT2 substrate (North et al., 2003). Moreover, SIRT2 can also translocate into the nucleus and a predominant nuclear or chromatin-associated SIRT2 localization is detected during G2/M transition and in mitosis of the cell cycle (Vaquero et al., 2006; North and Verdin, 2007a). It is unclear how SIRT2 translocates into the nucleus because the protein lacks any obvious NLS. Nuclear substrates of SIRT2 include H4K16ac and H3K56ac, modifications that are implicated in DNA damage response and cancer (Vaquero et al., 2006; Vempati et al., 2010). Also it remains to be defined how the cell cycle-dependent nuclear localization is regulated. One possibility is that signals in late G2 control the activity of the NES. Indeed in chemically synchronized cells SIRT2 is hyperphosphorylated during the G2/M transition and in M phase, which is paralleled by a mobility shift in SDS-PAGE (Dryden et al., 2003; North and Verdin, 2007b). This correlates with the nuclear translocation of SIRT2 and suggests a regulatory role for Cyclin B/CDK1 and other mitosis-specific kinases (reviewed in Morgan, 2008). But alternative mechanisms may also be in place, such as stimulation of nuclear uptake and/or tight interaction with nuclear structures in late G2 or mitosis.
Particularly high SIRT2 protein expression in the brain is found in myelin-forming oligodendrocytes, correlating with the expression profiles of the differentiation markers CNPase (2′,3′-cyclic nucleotide 3′-phosphodiesterase) and MBP (myelin basic protein; Li et al., 2007; Southwood et al., 2007; Werner et al., 2007). Interestingly only the shorter variant 2 is present in the myelin-enriched fraction of adult mouse brain or in the cytoplasm of murine cerebellar granule cells (Suzuki and Koike, 2007; Werner et al., 2007), suggesting that the N-terminal region is involved in controlling the subcytoplasmic localization.
SIRT2 positively regulates the transcription factor sterol response element binding protein 2 (SREBP-2) thereby promoting cholesterol biosynthesis in neurons (Luthi-Carter et al., 2010). Cholesterol influences membrane thickness and fluidity and is essential for myelin membrane growth (Saher et al., 2005). However, cholesterol is also reported to have a detrimental effect in neurons and presents a risk factor in neurodegenerative diseases like Alzheimer’s (AD) and Parkinson’s diseases (PD; reviewed in Stefani and Liguri, 2009; Huang et al., 2011). Consistent with these findings, SIRT2 inhibition reduces toxicity of mutant huntingtin by decreasing sterol biosynthesis (Luthi-Carter et al., 2010). Similarly SIRT2 knockdown or SIRT2 inhibition decreases α-synuclein toxicity, a protein frequently mutated in and associated with PD (Outeiro et al., 2007). Together these findings implicate SIRT2 in the control of toxicity resulting from aggregation-prone proteins in both neurons and oligodendrocytes.
Beside SREBP-2, SIRT2 is also involved in the regulation of other transcription factors, including NF-κB (Rothgiesser et al., 2010), FOXO1 (Jing et al., 2007; Zhao et al., 2010), and FOXO3 (Wang et al., 2007). Thus, through modulating these transcriptional regulators, SIRT2 affects most likely multiple cellular processes, such as signaling, gene expression, and autophagy.
To date two phosphorylation sites have been identified in SIRT2 (Figure 2 and Table 1). They are located in the C-terminal extension in close proximity to each other at Ser368/331 and Ser372/335 (numbering according to the two translational variants). The phosphorylation of each site results in a mobility shift of the protein on SDS-PAGE, which results in a characteristic triple band pattern (Nahhas et al., 2007; Pandithage et al., 2008). Ser368/331 is part of a Cyclin/CDK consensus motif and has been demonstrated to be a substrate of Cyclin B/CDK1, Cyclin E/CDK2, Cyclin A/CDK2, Cyclin D3/CDK4, and p35/CDK5 (North and Verdin, 2007b; Pandithage et al., 2008). Consistent with these in vitro studies is the finding that Ser368/331 is phosphorylated when cells enter S phase, suggesting that this phosphorylation is not the signal for nuclear accumulation of SIRT2, which begins in late G2 (Pandithage et al., 2008). Phosphorylation of SIRT2 at Ser368/331 reduces its enzymatic activity as measured by deacetylation of core histones and of α-tubulin (Pandithage et al., 2008). Moreover SIRT2 interferes with neurite outgrowth in primary neurons, correlating with α-tubulin deacetylation, a process that is antagonized by phosphorylating Ser368/331 providing evidence that the C-terminal extension of SIRT2 controls activity in cells (Pandithage et al., 2008). So far, nothing is known about a kinase responsible for phosphorylation of Ser372/335 or about its influence on SIRT2 function.
Phosphorylation of SIRT2 by p35/CDK5 is of special interest because similar to SIRT2, CDK5 is highly expressed in the brain and its protein levels are upregulated in differentiating cells, i.e., oligodendrocytes (He et al., 2011). Moreover CDK5 is capable to interfere with SIRT2 function by phosphorylating Ser368/331 in primary neurons, as mentioned above (Pandithage et al., 2008). Furthermore CDK5 is an important cell cycle suppressor in postmitotic neurons (Cicero and Herrup, 2005). This activity requires nuclear localization of CDK5, which is mediated by an interaction with p27 (Zhang et al., 2008). Upon stress, e.g., induced by β-amyloid expression in an AD model, association between p27 and CDK5 is disrupted resulting in reduced nuclear CDK5 levels (Zhang et al., 2010). An increasing number of publications provide evidence that CDK5 also plays a role in many non-neuronal tissues (reviewed in Lalioti et al., 2010). For example CDK5 is required for the DNA damage response, suggesting that this kinase participates in stress signaling (Turner et al., 2008). Together with the above-summarized studies implicating SIRT2 in stress response in cells of the nervous system the findings suggest that the interaction with and regulation by CDK5 may be part of a stress signaling network.
Additionally SIRT2 is acetylated by the KAT p300. This acetylation, although the site of modification has not been mapped, interferes with the catalytic activity of SIRT2 (Han et al., 2008). Predictions of acetylation sites indicate that the C-terminal extension provides multiple target lysine residues (Li et al., 2006), further supporting the concept that the N- and C-terminal regions are particularly relevant to control catalytic activities of sirtuins.
SIRT3
Three sirtuins are located in mitochondria. Of these SIRT3 is the best studied. It is broadly expressed including brown but not white adipose tissue (Shi et al., 2005). Indeed, SIRT3 is required for PGC-1α-mediated differentiation of brown adipose tissue in an estrogen-related receptor α (ERRα)-dependent manner (Kong et al., 2010; Giralt et al., 2011). The transcriptional coactivator PGC-1α regulates genes involved in energy metabolism, suggesting that SIRT3 participates in this process (Shi et al., 2005). Moreover SIRT3 regulates the cellular response to oxidative stress and calorie restriction. Thus, upon cellular stress, e.g., increase in reactive oxygen species (ROS) or nutrient deprivation, human SIRT3 transcription is stimulated (Shi et al., 2005; Chen et al., 2011), and the protein translocates to the mitochondrial inner membrane (IMS; Michishita et al., 2005; Scher et al., 2007). There it deacetylates and thereby activates the enzymes isocitrate dehydrogenase 2 (Idh2) and superoxide dismutase 2 (SOD2), which are involved in reducing cellular oxidants, including oxidized glutathione (GSSG) and reactive oxygen species (ROS) (Schlicker et al., 2008; Qiu et al., 2010; Someya et al., 2010; Chen et al., 2011). Recently, it has been discovered that SIRT3 acts as a tumor suppressor. Sirt3-deficient mice show increased genomic instability as a result of enhanced superoxide levels. Mouse embryonic fibroblasts (MEFs) of such animals are transformed by a single oncoprotein, i.e., with MYC or RAS (Kim et al., 2010). In addition, SIRT3 activates several key enzymes associated with fatty-acid oxidation (3-hydroxy-3-methylglutaryl-CoA synthase/HMGCS2, Long-chain acyl-CoA dehydrogenase/LCAD, Acetyl-CoA synthetase 2/AceCS2) and the urea cycle (ornithine transcarbamoylase/OTC) (Hallows et al., 2006, 2011; Hirschey et al., 2010; Shimazu et al., 2010). Together these findings suggest that upon caloric restriction SIRT3, in addition to SIRT1, plays a key role in modulating mitochondrial activities and stimulating the use of alternative energy sources by promoting β-oxidation and amino acid catabolism.
The N-terminal extension of SIRT3 contains a mitochondrial targeting signal peptide (Figure 2). During import of SIRT3 into the mitochondrial matrix, the protein is proteolytically cleaved at position 101 and thus enzymatically activated (Schwer et al., 2002). It has been postulated that the proteolytically shortened N-terminal region and the C-terminal extension form a module that might regulate the access of substrate proteins to the active site (Schlicker et al., 2008).
Presently we know very little about the regulation of SIRT3 function. The biological significance, as summarized briefly above, would suggest strongly that SIRT3 is regulated by signaling. Indeed, six phosphorylated serine residues (out of a total of eight possible sites) between positions 101 and 118 have been identified in a high-resolution mass spectrometry-based phosphoproteome analysis (Table 1) (Olsen et al., 2010). But their biological relevance or influence on SIRT3 function has not been analyzed yet. These phosphorylation sites are close to the mitochondrial cleavage site in the N-terminal extension. Therefore it is possible that phosphorylation modulates the enzymatic activity of SIRT3 in mitochondria either by regulating the proteolytic cleavage, by influencing the interaction between the N- and C-terminal extension, or by regulating the interaction of the N-terminal region with the catalytic domain.
SIRT4
SIRT4 is an additional mitochondrial sirtuin (Michishita et al., 2005; Haigis et al., 2006). It resides as a soluble protein in the mitochondrial matrix (Ahuja et al., 2007; Nakamura et al., 2008). Similar to the other sirtuins, SIRT4 is ubiquitously expressed (Michishita et al., 2005; Haigis et al., 2006; Ahuja et al., 2007). Sirt4 knockout mice are viable and fertile and did not display apparent phenotypic abnormalities. However these mice exhibit increased insulin levels when compared to wild-type littermates (Haigis et al., 2006). This anomaly points to a function of SIRT4 in the insulin producing β-cells of the pancreatic islets. Indeed, SIRT4 negatively regulates glutamate dehydrogenase (GDH) via ADP-ribosylation. GDH is a mitochondrial enzyme, which catalyzes the conversion of glutamate to α-ketoglutarate in the tricarboxylic acid (TCA) cycle and induces insulin secretion (Haigis et al., 2006). A second possible explanation for the increased insulin levels in Sirt4-deficient mice is that IDE (insulin-degrading enzyme) interacts with SIRT4 (Ahuja et al., 2007). IDE regulates insulin levels and SIRT4 appears to be a negative regulator of this enzyme. Whether this occurs through direct interaction or by ADP-ribosylation has not been determined. It is worth pointing out that so far no deacetylase activity of SIRT4 has been identified. It remains to be determined whether this enzyme is indeed deficient of deacetylase activity or whether this is a reflection of the lack of appropriate substrates.
Similar to SIRT3, SIRT4 possesses a mitochondrial targeting signal in the N-terminal region and is proteolytically cleaved within the N-terminal extension upon entry into the mitochondrial matrix resulting in a 28 amino acids shortened protein (Ahuja et al., 2007; Figure 2). It is not known whether the proteolytic cleavage of SIRT4 influences its enzymatic activity, as was reported for SIRT3 (Schwer et al., 2002).
Three phosphorylation sites have been identified in SIRT4 at Ser255, Ser261, and Ser262 in a proteomics approach (Yu et al., 2007). These sites are unique, as far as deduced from the currently available analysis of PTMs of sirtuins, in that they are located within or in close proximity to the catalytic domain (Figure 2 and Table 1). Whether these are functionally relevant has not been determined.
SIRT5
SIRT5, the third mitochondrial sirtuin, is ubiquitously expressed (Michishita et al., 2005; Nakagawa et al., 2009). Very little is known about SIRT5 function. Sirt5 knockout mice develop inconspicuously until at least 18 months of age (Lombard et al., 2007). However, they exhibit significantly elevated blood ammonia levels compared to wild-type animals after caloric restriction or fasting, which is presumably caused by a deregulated urea cycle. In support SIRT5 can deacetylate and activate the carbamoyl phosphate synthetase 1 (CPS1), a mitochondrial enzyme of the urea cycle (Nakagawa et al., 2009). It has been suggested that elevated mitochondrial NAD+ levels in response to starvation activate SIRT5 and in turn CPS1 is stimulated and initiates the detoxification of excess ammonia under physiological conditions. In addition to its deacetylase activity SIRT5 was very recently reported to elicit also NAD+-dependent demalonylase and desuccinylase activities (Du et al., 2011; Peng et al., 2011). In line with this observation CPS1 succinylation at Lys1291 is strongly increased in Sirt5 knockout mice compared to wild-type littermates. Furthermore SIRT5 can in vitro deacetylate the mitochondrial IMS protein cytochrome c, which is involved in oxidative metabolism and apoptosis (Schlicker et al., 2008). Up to now the functional relevance of this observation has not been clarified.
SIRT5 is expressed as two distinct transcriptional variants due to alternative splicing, encoding proteins with distinct C-terminal regions (Figure 2). Both isoforms can be cleaved after the first 36 amino acids at a consensus sequence for the mitochondrial processing peptidase upon entry into the mitochondrial matrix (Michishita et al., 2005). Similar to SIRT4 no data are available about a relationship between the N-terminal truncation of SIRT5 and its enzymatic activity (Schwer et al., 2002).
Both SIRT5 isoforms display mitochondrial localization. SIRT5 can enter the IMS and the mitochondrial matrix (Schlicker et al., 2008). In contrast to the cleaved isoform two (IF2, derived from the shorter splice variant), which seems to reside exclusively in the mitochondria, cleaved IF1 is found additionally in the cytoplasm. It appears that the different C-termini of the two SIRT5 isoforms are responsible for their distinct subcellular distribution. The C-terminal extension of IF2 is rich in hydrophobic amino acids and functions as a mitochondrial membrane insertion signal (Matsushita et al., 2011). Presently no PTM of SIRT5 are described besides the proteolytic cleavage and thus nothing is known about the role of this protein in signaling processes.
SIRT6
SIRT6 is expressed in most tissues (Liszt et al., 2005; Mostoslavsky et al., 2006). It is, similar to SIRT1 and SIRT7, predominantly localized in the nucleus (Liszt et al., 2005; Michishita et al., 2005), where it associates with chromatin (Mostoslavsky et al., 2006). A nuclear localization signal was discovered between amino acids 345 and 351 in the distal region of the C-terminal extension of SIRT6 (Figure 2). This signal is necessary and sufficient for proper nuclear localization of the protein (Tennen et al., 2010). In comparison to other sirtuin knockout mice, Sirt6-deficient mice display a severe phenotype. Despite normal development for several weeks after birth, these mice die at about 1 month of age due to degenerative processes of multiple organs. These processes include loss of subcutaneous fat and metabolic defects displayed by dramatic drops of serum glucose and insulin-like growth factor 1 (IGF1) levels. Additional symptoms are lordokyphosis, colitis, and a severe lymphopenia (Mostoslavsky et al., 2006). One suggestion is that this phenotype is the consequence of a loss of Sirt6-mediated inhibition of NF-κB target gene expression (Kawahara et al., 2009, 2011). The absence of SIRT6-dependent repression of HIF1α might also account for the phenotype (Zhong et al., 2010). Under physiologic conditions SIRT6 interacts with these transcription factors, i.e., NF-κB and HIF1α, and is transported to their target gene promoters where it deacetylates H3K9ac or H3K56ac (Michishita et al., 2005; Kawahara et al., 2009; Yang et al., 2009). In both cases, the binding of the respective transcription factor to its target gene promoters is enhanced in Sirt6-deficient cells due to locally elevated acetylation levels of H3K9. Further investigations revealed that upon TNF-α signaling, SIRT6 binds to many promoters, which are highly enriched for NF-κB, SP1, STAT1/3, ELK1, E2F1, and FOXO1/4 binding motifs (Kawahara et al., 2011). Thus SIRT6 appears to have widespread activities as a regulator of transcription, in particular of genes whose products are involved in glucose and lipid metabolisms.
Moreover SIRT6 seems to be involved in DNA repair and thus in the maintenance of genomic integrity. Indeed MEFs derived from Sirt6-deficient mice are more sensitive to irradiation and display multiple chromosomal aberrations (Mostoslavsky et al., 2006). SIRT6 associates with chromatin in response to DNA damage and stabilizes the DNA-dependent protein kinase (DNA-PK) at DNA double-strand breaks (DSBs; McCord et al., 2009). Also SIRT6 deacetylates CtIP [C-terminal binding protein (CtBP) interacting protein] in response to DNA damage, which promotes the ability of CtIP to mediate DSB repair by homologous recombination (Kaidi et al., 2010). Finally SIRT6 is required for telomere maintenance (Tennen and Chua, 2011). Together these findings provide strong evidence for a role of SIRT6 in controlling genomic stability.
The conserved core domain of SIRT6 is not sufficient to deacetylate H3K9ac or H3K56ac (Tennen et al., 2010). For SIRT6 the available evidence suggests that the N-terminal region is essential for deacetylase activity. This is reminiscent of the findings with other sirtuins, which require either N- or C-terminal regions to activate catalytic function as described above.
In addition to its deacetylase activity, SIRT6 has been reported to be able to mono-ADP-ribosylate substrates. One substrate identified is PARP1/ARTD1, which is activated by SIRT6 (Mao et al., 2011). This provides an additional link to genomic stability because ARTD1 is a DNA damage sensor and upon activation synthesizes ADP-ribose polymers that are docking sites for repair enzymes (reviewed in Kleine and Luscher, 2009). SIRT6 can also auto-ADP-ribosylate but the site of modification and the functional relevance are unclear (Liszt et al., 2005). It remains to be elucidated whether the N-terminal extension is essential for the ADP-ribosylation activity of SIRT6 as it is for the deacetylase activity. Of note is that besides ADP-ribosylation, two C-terminal phosphorylation sites at Tyr294 and Ser303 have been discovered in a proteomic approach (Dephoure et al., 2008; Figure 2 and Table 1). It remains to be seen whether these phosphorylations influence SIRT6 function.
SIRT7
Out of the seven human sirtuins, SIRT7 is the least studied. It is a nuclear protein that is concentrated in the nucleoli where it interacts with components of the rDNA transcription machinery, like RNA polymerase I (Pol I) and the rDNA transcription factor UBF (Michishita et al., 2005). SIRT7 positively regulates rDNA transcription (Ford et al., 2006; Grob et al., 2009). Knockdown of SIRT7 in human cancer cell lines blocks cell proliferation and causes apoptosis. This drastic effect implies that SIRT7 is required for cancer cell viability (Ford et al., 2006). Further evidence supporting this hypothesis is provided by enhanced SIRT7 expression levels in breast carcinoma biopsies compared to normal tissue (Ashraf et al., 2006). The tumor suppressor p53 is a substrate of SIRT7 and thus this sirtuin appears to interfere with p53 function, similar to SIRT1 (Vakhrusheva et al., 2008). Sirt7 knockout mice are viable but suffer from progressive heart hypertrophy, accompanied by inflammation and decreased stress resistance, possibly a consequence of altered p53 activity. Indirect evidence suggests that SIRT7 is phosphorylated during mitosis by a CDK complex, but no sites have been mapped nor functional consequences defined (Grob et al., 2009).
Conclusion
Sirtuins have been identified as key regulators of multiple cellular processes, mainly by functioning as NAD+-dependent deacetylases but also as demalonylases, desuccinylases, and ADP-ribosyltransferases. Despite the many processes that are governed, at least in part, by sirtuins, relatively little is known about how these regulators are controlled. This is somewhat surprising because sirtuins are implicated in many signaling processes. But obviously their regulation has not been evaluated in great detail.
Sirtuins share a conserved catalytic domain, but differ in their N- and C-terminal extensions. Apparent from the available data is that the N- and C-terminal regions of the different sirtuins that extend beyond the catalytic domains mediate regulation. Multiple PTMs target these extensions, including phosphorylation, methylation, sumoylation, proteolytic cleavage, and possibly others. Most of the relevant enzymes are not known presently and thus the pathways that target sirtuins are poorly understood. Nevertheless the emerging theme suggests that the N- and C-terminal regions function as signal receivers that transmit information from signaling pathways to the catalytic domain. In addition initial findings suggest that these signals, which target the extensions of sirtuins, may also control properties of the different sirtuins beyond catalytic activity. Although the evidence is not complete, trimerization, substrate specificity, and subcellular localization are likely to be controlled by these extensions. To understand sirtuin biology more thoroughly, it will be necessary to account for all PTMs that control the N- and C-terminal extensions and to integrate and connect these findings with distinct signaling pathways to associate sirtuins with physiological processes. This will need to be combined with more definitive studies addressing the functions of the extensions both for intra- as well as intermolecular interactions (Figure 4).
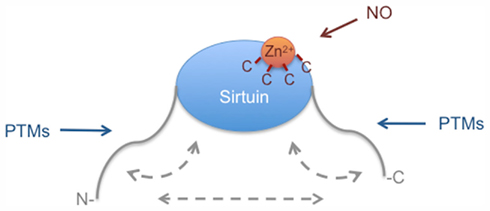
Figure 4. Summary of the regulation of the catalytic activity of Sirtuins. Most of the currently known PTMs that target sirtuins are directed to the N- and C-terminal extensions. These extensions may control sirtuin function by interacting with the catalytic core domain or with each other. Additionally the extensions may control intermolecular interactions (not indicated). Different PTMs target the N- and C-terminal extensions, thereby possibly controlling either intra- or intermolecular interactions. The regulation of Zn2+ binding by nitrosylation is potentially a common regulatory mechanism of sirtuins. For more details see the text.
While the available evidence, although still incomplete, suggests that the N- and C-terminal extensions are important to control sirtuin function at multiple levels, considerably less is known about posttranslational regulation that directly targets the catalytic domain of either of the seven sirtuins. It is presently not clear whether this reflects a true lack of direct regulation or whether we simply have not identified the relevant processes yet. Since many other enzymes are controlled by directly modulating the catalytic domain, it seems more likely that the latter explanation is correct. The findings on nitrosylation of SIRT1 indicate that the catalytic domain of at least this sirtuin is indeed regulated. Nitrosylation is potentially controlling all sirtuins because this modification targets cysteines that are important to coordinate Zn2+ binding. Sirtuins have been recognized as targets for clinical intervention. Understanding the repertoire of control mechanisms that target sirtuins will likely provide additional targets worth considering. Thus unraveling the function and regulation of sirtuins is important not only to understand basic cellular processes but also for clinical applications.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our colleagues for many helpful discussions. The work performed in the authors’ laboratory was supported by the IZKF Aachen.
References
Afshar, G., and Murnane, J. P. (1999). Characterization of a human gene with sequence homology to Saccharomyces cerevisiae SIR2. Gene 234, 161–168.
Ahuja, N., Schwer, B., Carobbio, S., Waltregny, D., North, B. J., Castronovo, V., Maechler, P., and Verdin, E. (2007). Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 282, 33583–33592.
Allfrey, V. G., and Mirsky, A. E. (1964). Structural modifications of histones and their possible role in the regulation of RNA synthesis. Science 144, 559.
Ashraf, N., Zino, S., Macintyre, A., Kingsmore, D., Payne, A. P., George, W. D., and Shiels, P. G. (2006). Altered sirtuin expression is associated with node-positive breast cancer. Br. J. Cancer 95, 1056–1061.
Back, J. H., Rezvani, H. R., Zhu, Y., Guyonnet-Duperat, V., Athar, M., Ratner, D., and Kim, A. L. (2011). Cancer cell survival following DNA damage-mediated premature senescence is regulated by mammalian target of rapamycin (mTOR)-dependent inhibition of sirtuin 1. J. Biol. Chem. 286, 19100–19108.
Brachmann, C. B., Sherman, J. M., Devine, S. E., Cameron, E. E., Pillus, L., and Boeke, J. D. (1995). The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 9, 2888–2902.
Braunstein, M., Rose, A. B., Holmes, S. G., Allis, C. D., and Broach, J. R. (1993). Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes Dev. 7, 592–604.
Burnett, C., Valentini, S., Cabreiro, F., Goss, M., Somogyvari, M., Piper, M. D., Hoddinott, M., Sutphin, G. L., Leko, V., Mcelwee, J. J., Vazquez-Manrique, R. P., Orfila, A. M., Ackerman, D., Au, C., Vinti, G., Riesen, M., Howard, K., Neri, C., Bedalov, A., Kaeberlein, M., Soti, C., Partridge, L., and Gems, D. (2011). Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 477, 482–485.
Chen, W. Y., Wang, D. H., Yen, R. C., Luo, J., Gu, W., and Baylin, S. B. (2005). Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 123, 437–448.
Chen, Y., Zhang, J., Lin, Y., Lei, Q., Guan, K. L., Zhao, S., and Xiong, Y. (2011). Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 12, 534–541.
Cheng, H.-L., Mostoslavsky, R., Saito, S. A. I., Manis, J. P., Gu, Y., Patel, P., Bronson, R., Appella, E., Alt, F. W., and Chua, K. F. (2003). Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 100, 10794–10799.
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., Olsen, J. V., and Mann, M. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840.
Choudhary, C., Weinert, B. T., Wagner, S. A., Horn, H., Henriksen, P., Liu, W. S. R., Olsen, J. V., and Jensen, L. J. (2011). Proteome-wide mapping of the Drosophila acetylome demonstrates a high degree of conservation of lysine acetylation. Sci. Signal. 4, ra48.
Cicero, S., and Herrup, K. (2005). Cyclin-dependent kinase 5 is essential for neuronal cell cycle arrest and differentiation. J. Neurosci. 25, 9658–9668.
De Ruijter, A. J. M., Van Gennip, A. H., Caron, H. N., Kemp, S., and Van Kuilenburg, A. B. P. (2003). Histone deacetylases (HDACs): characterization of the classicalHDAC family. Biochem. J. 370, 737–749.
Dephoure, N., Zhou, C., Villen, J., Beausoleil, S. A., Bakalarski, C. E., Elledge, S. J., and Gygi, S. P. (2008). A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767.
Dryden, S. C., Nahhas, F. A., Nowak, J. E., Goustin, A.-S., and Tainsky, M. A. (2003). Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell. Biol. 23, 3173–3185.
Du, J., Zhou, Y., Su, X., Yu, J. J., Khan, S., Jiang, H., Kim, J., Woo, J., Kim, J. H., Choi, B. H., He, B., Chen, W., Zhang, S., Cerione, R. A., Auwerx, J., Hao, Q., and Lin, H. (2011). Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809.
Ford, E., Voit, R., Liszt, G., Magin, C., Grummt, I., and Guarente, L. (2006). Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 20, 1075–1080.
Frye, R. A. (2000). Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 273, 793–798.
Fulco, M., and Sartorelli, V. (2008). Comparing and contrasting the roles of AMPK and SIRT1 in metabolic tissues. Cell Cycle 7, 3669–3679.
Giralt, A., Hondares, E., Villena, J. A., Ribas, F., Diaz-Delfin, J., Giralt, M., Iglesias, R., and Villarroya, F. (2011). Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the Sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J. Biol. Chem. 286, 16958–16966.
Gottlieb, S., and Esposito, R. E. (1989). A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell 56, 771–776.
Grob, A., Roussel, P., Wright, J. E., Mcstay, B., Hernandez-Verdun, D., and Sirri, V. (2009). Involvement of SIRT7 in resumption of rDNA transcription at the exit from mitosis. J. Cell. Sci. 122, 489–498.
Guarente, L. (2011). Franklin H. Epstein lecture: sirtuins, aging, and medicine. N. Engl. J. Med. 364, 2235–2244.
Haigis, M. C., Mostoslavsky, R., Haigis, K. M., Fahie, K., Christodoulou, D. C., Murphy, A. J., Valenzuela, D. M., Yancopoulos, G. D., Karow, M., Blander, G., Wolberger, C., Prolla, T. A., Weindruch, R., Alt, F. W., and Guarente, L. (2006). SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126, 941–954.
Haigis, M. C., and Sinclair, D. A. (2010). Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295.
Hallows, W. C., Lee, S., and Denu, J. M. (2006). Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. U.S.A. 103, 10230–10235.
Hallows, W. C., Yu, W., Smith, B. C., Devries, M. K., Ellinger, J. J., Someya, S., Shortreed, M. R., Prolla, T., Markley, J. L., Smith, L. M., Zhao, S., Guan, K. L., and Denu, J. M. (2011). Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol. Cell 41, 139–149.
Han, Y., Jin, Y.-H., Kim, Y.-J., Kang, B. Y., Choi, H.-J., Kim, D.-W., Yeo, C.-Y., and Lee, K.-Y. (2008). Acetylation of Sirt2 by p300 attenuates its deacetylase activity. Biochem. Biophys. Res. Commun. 375, 576–580.
He, X., Takahashi, S., Suzuki, H., Hashikawa, T., Kulkarni, A. B., Mikoshiba, K., and Ohshima, T. (2011). Hypomyelination phenotype caused by impaired differentiation of oligodendrocytes in Emx1-cre mediated Cdk5 conditional knockout mice. Neurochem. Res. 36, 1293–1303.
Hirschey, M. D., Shimazu, T., Goetzman, E., Jing, E., Schwer, B., Lombard, D. B., Grueter, C. A., Harris, C., Biddinger, S., Ilkayeva, O. R., Stevens, R. D., Li, Y., Saha, A. K., Ruderman, N. B., Bain, J. R., Newgard, C. B., Farese, R. V. Jr., Alt, F. W., Kahn, C. R., and Verdin, E. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125.
Hisahara, S., Chiba, S., Matsumoto, H., Tanno, M., Yagi, H., Shimohama, S., Sato, M., and Horio, Y. (2008). Histone deacetylase SIRT1 modulates neuronal differentiation by its nuclear translocation. Proc. Natl. Acad. Sci. U.S.A. 105, 15599–15604.
Huang, X., Auinger, P., Eberly, S., Oakes, D., Schwarzschild, M., Ascherio, A., Mailman, R., and Chen, H. (2011). Serum cholesterol and the progression of Parkinson’s disease: results from DATATOP. PLoS ONE 6, e22854. doi: 10.1371/journal.pone.0022854
Imai, S., Armstrong, C. M., Kaeberlein, M., and Guarente, L. (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800.
Jing, E., Gesta, S., and Kahn, C. R. (2007). SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 6, 105–114.
Kaeberlein, M., Mcvey, M., and Guarente, L. (1999). The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580.
Kaidi, A., Weinert, B. T., Choudhary, C., and Jackson, S. P. (2010). Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 329, 1348–1353.
Kang, H., Jung, J. W., Kim, M. K., and Chung, J. H. (2009). CK2 is the regulator of SIRT1 substrate-binding affinity, deacetylase activity and cellular response to DNA-damage. PLoS ONE 4, e6611. doi: 10.1371/journal.pone.0006611
Kang, H., Suh, J. Y., Jung, Y. S., Jung, J. W., Kim, M. K., and Chung, J. H. (2011). Peptide switch is essential for sirt1 deacetylase activity. Mol. Cell 44, 203–213.
Kawahara, T. L., Michishita, E., Adler, A. S., Damian, M., Berber, E., Lin, M., Mccord, R. A., Ongaigui, K. C., Boxer, L. D., Chang, H. Y., and Chua, K. F. (2009). SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136, 62–74.
Kawahara, T. L., Rapicavoli, N. A., Wu, A. R., Qu, K., Quake, S. R., and Chang, H. Y. (2011). Dynamic chromatin localization of sirt6 shapes stress- and aging-related transcriptional networks. PLoS Genet. 7, e1002153. doi: 10.1371/journal.pgen.1002153
Kim, E. J., Kho, J. H., Kang, M. R., and Um, S. J. (2007). Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol. Cell 28, 277–290.
Kim, G. W., and Yang, X. J. (2011). Comprehensive lysine acetylomes emerging from bacteria to humans. Trends Biochem. Sci. 36, 211–220.
Kim, H. S., Patel, K., Muldoon-Jacobs, K., Bisht, K. S., Aykin-Burns, N., Pennington, J. D., Van Der Meer, R., Nguyen, P., Savage, J., Owens, K. M., Vassilopoulos, A., Ozden, O., Park, S. H., Singh, K. K., Abdulkadir, S. A., Spitz, D. R., Deng, C. X., and Gius, D. (2010). SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17, 41–52.
Kim, J. E., Chen, J., and Lou, Z. (2008). DBC1 is a negative regulator of SIRT1. Nature 451, 583–586.
Kim, S. C., Sprung, R., Chen, Y., Xu, Y., Ball, H., Pei, J., Cheng, T., Kho, Y., Xiao, H., Xiao, L., Grishin, N. V., White, M., Yang, X.-J., and Zhao, Y. (2006). Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618.
Kong, X., Wang, R., Xue, Y., Liu, X., Zhang, H., Chen, Y., Fang, F., and Chang, Y. (2010). Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 5, e11707. doi: 10.1371/journal.pone.0011707
Kornberg, M. D., Sen, N., Hara, M. R., Juluri, K. R., Nguyen, J. V., Snowman, A. M., Law, L., Hester, L. D., and Snyder, S. H. (2010). GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 12, 1094–1100.
Lalioti, V., Pulido, D., and Sandoval, I. V. (2010). Cdk5, the multifunctional surveyor. Cell Cycle 9, 284–311.
Landry, J., Slama, J. T., and Sternglanz, R. (2000). Role of NAD(+) in the deacetylase activity of the SIR2-like proteins. Biochem. Biophys. Res. Commun. 278, 685–690.
Langley, E., Pearson, M., Faretta, M., Bauer, U. M., Frye, R. A., Minucci, S., Pelicci, P. G., and Kouzarides, T. (2002). Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 21, 2383–2396.
Li, A., Xue, Y., Jin, C., Wang, M., and Yao, X. (2006). Prediction of Nepsilon-acetylation on internal lysines implemented in Bayesian Discriminant Method. Biochem. Biophys. Res. Commun. 350, 818–824.
Li, W., Zhang, B., Tang, J., Cao, Q., Wu, Y., Wu, C., Guo, J., Ling, E.-A., and Liang, F. (2007). Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha-tubulin. J. Neurosci. 27, 2606–2616.
Liszt, G., Ford, E., Kurtev, M., and Guarente, L. (2005). Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem. 280, 21313–21320.
Liu, X., Wang, D., Zhao, Y., Tu, B., Zheng, Z., Wang, L., Wang, H., Gu, W., Roeder, R. G., and Zhu, W. G. (2011). Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proc. Natl. Acad. Sci. U.S.A. 108, 1925–1930.
Lombard, D. B., Alt, F. W., Cheng, H. L., Bunkenborg, J., Streeper, R. S., Mostoslavsky, R., Kim, J., Yancopoulos, G., Valenzuela, D., Murphy, A., Yang, Y., Chen, Y., Hirschey, M. D., Bronson, R. T., Haigis, M., Guarente, L. P., Farese, R. V. Jr., Weissman, S., Verdin, E., and Schwer, B. (2007). Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814.
Lu, S. P., and Lin, S. J. (2010). Regulation of yeast sirtuins by NAD(+) metabolism and calorie restriction. Biochim. Biophys. Acta 1804, 1567–1575.
Luo, J., Nikolaev, A. Y., Imai, S., Chen, D., Su, F., Shiloh, A., Guarente, L., and Gu, W. (2001). Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107, 137–148.
Luthi-Carter, R., Taylor, D. M., Pallos, J., Lambert, E., Amore, A., Parker, A., Moffitt, H., Smith, D. L., Runne, H., Gokce, O., Kuhn, A., Xiang, Z., Maxwell, M. M., Reeves, S. A., Bates, G. P., Neri, C., Thompson, L. M., Marsh, J. L., and Kazantsev, A. G. (2010). SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 7927–7932.
Mao, Z., Hine, C., Tian, X., Van Meter, M., Au, M., Vaidya, A., Seluanov, A., and Gorbunova, V. (2011). SIRT6 promotes DNA repair under stress by activating PARP1. Science 332, 1443–1446.
Marks, P. A. (2010). Histone deacetylase inhibitors: a chemical genetics approach to understanding cellular functions. Biochim. Biophys. Acta 1799, 717–725.
Matsushita, N., Yonashiro, R., Ogata, Y., Sugiura, A., Nagashima, S., Fukuda, T., Inatome, R., and Yanagi, S. (2011). Distinct regulation of mitochondrial localization and stability of two human Sirt5 isoforms. Genes Cells 16, 190–202.
McBurney, M. W., Yang, X., Jardine, K., Hixon, M., Boekelheide, K., Webb, J. R., Lansdorp, P. M., and Lemieux, M. (2003). The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol. Cell. Biol. 23, 38–54.
McCord, R. A., Michishita, E., Hong, T., Berber, E., Boxer, L. D., Kusumoto, R., Guan, S., Shi, X., Gozani, O., Burlingame, A. L., Bohr, V. A., and Chua, K. F. (2009). SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging 1, 109–121.
Michishita, E., Park, J. Y., Burneskis, J. M., Barrett, J. C., and Horikawa, I. (2005). Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 16, 4623–4635.
Mostoslavsky, R., Chua, K. F., Lombard, D. B., Pang, W. W., Fischer, M. R., Gellon, L., Liu, P., Mostoslavsky, G., Franco, S., Murphy, M. M., Mills, K. D., Patel, P., Hsu, J. T., Hong, A. L., Ford, E., Cheng, H. L., Kennedy, C., Nunez, N., Bronson, R., Frendewey, D., Auerbach, W., Valenzuela, D., Karow, M., Hottiger, M. O., Hursting, S., Barrett, J. C., Guarente, L., Mulligan, R., Demple, B., Yancopoulos, G. D., and Alt, F. W. (2006). Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329.
Nahhas, F., Dryden, S. C., Abrams, J., and Tainsky, M. A. (2007). Mutations in SIRT2 deacetylase which regulate enzymatic activity but not its interaction with HDAC6 and tubulin. Mol. Cell. Biochem. 303, 221–230.
Nakagawa, T., Lomb, D. J., Haigis, M. C., and Guarente, L. (2009). SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137, 560–570.
Nakamura, Y., Ogura, M., Tanaka, D., and Inagaki, N. (2008). Localization of mouse mitochondrial SIRT proteins: shift of SIRT3 to nucleus by co-expression with SIRT5. Biochem. Biophys. Res. Commun. 366, 174–179.
Nasrin, N., Kaushik, V. K., Fortier, E., Wall, D., Pearson, K. J., De Cabo, R., and Bordone, L. (2009). JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS ONE 4, e8414. doi: 10.1371/journal.pone.0008414
North, B. J., Marshall, B. L., Borra, M. T., Denu, J. M., and Verdin, E. (2003). The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11, 437–444.
North, B. J., and Verdin, E. (2004). Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 5, 224.
North, B. J., and Verdin, E. (2007a). Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE 2, e784. doi: 10.1371/journal.pone.0000784
North, B. J., and Verdin, E. (2007b). Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J. Biol. Chem. 282, 19546–19555.
Olsen, J. V., Vermeulen, M., Santamaria, A., Kumar, C., Miller, M. L., Jensen, L. J., Gnad, F., Cox, J., Jensen, T. S., Nigg, E. A., Brunak, S., and Mann, M. (2010). Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3.
Outeiro, T. F., Kontopoulos, E., Altmann, S. M., Kufareva, I., Strathearn, K. E., Amore, A. M., Volk, C. B., Maxwell, M. M., Rochet, J.-C., Mclean, P. J., Young, A. B., Abagyan, R., Feany, M. B., Hyman, B. T., and Kazantsev, A. G. (2007). Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317, 516–519.
Pandithage, R., Lilischkis, R., Harting, K., Wolf, A., Jedamzik, B., Lüscher-Firzlaff, J., Vervoorts, J., Lasonder, E., Kremmer, E., Knöll, B., and Lüscher, B. (2008). The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J. Cell Biol. 180, 915–929.
Peng, C., Lu, Z., Xie, Z., Cheng, Z., Chen, Y., Tan, M., Luo, H., Zhang, Y., He, W., Yang, K., Zwaans, B.M., Tishkoff, D., Ho, L., Lombard, D., He, T.C., Dai, J., Verdin, E., Ye, Y., and Zhao, Y. (2011). The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics 10, M111.012658.
Qiu, X., Brown, K., Hirschey, M. D., Verdin, E., and Chen, D. (2010). Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 12, 662–667.
Rahman, S., and Islam, R. (2011). Mammalian Sirt1: insights on its biological functions. Cell Commun. Signal. 9, 11.
Rogina, B., and Helfand, S. L. (2004). Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. U.S.A. 101, 15998–16003.
Rothgiesser, K. M., Erener, S., Waibel, S., Lüscher, B., and Hottiger, M. O. (2010). SIRT2 regulates NF-κB dependent gene expression through deacetylation of p65 Lys310. J. Cell. Sci. 123, 4251–4258.
Saher, G., Brugger, B., Lappe-Siefke, C., Mobius, W., Tozawa, R., Wehr, M. C., Wieland, F., Ishibashi, S., and Nave, K. A. (2005). High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 8, 468–475.
Sasaki, T., Maier, B., Koclega, K., Chruszcz, M., and Gluba, W. (2008). Phosphorylation regulates SIRT1 function. PLoS ONE. 3, e4020. doi: 10.1371/journal.pone.0004020
Scher, M. B., Vaquero, A., and Reinberg, D. (2007). SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 21, 920–928.
Schlicker, C., Gertz, M., Papatheodorou, P., Kachholz, B., Becker, C. F., and Steegborn, C. (2008). Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J. Mol. Biol. 382, 790–801.
Schwer, B., North, B. J., Frye, R. A., Ott, M., and Verdin, E. (2002). The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 158, 647–657.
Shi, T., Wang, F., Stieren, E., and Tong, Q. (2005). SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 280, 13560–13567.
Shimazu, T., Hirschey, M. D., Hua, L., Dittenhafer-Reed, K. E., Schwer, B., Lombard, D. B., Li, Y., Bunkenborg, J., Alt, F. W., Denu, J. M., Jacobson, M. P., and Verdin, E. (2010). SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 12, 654–661.
Someya, S., Yu, W., Hallows, W. C., Xu, J., Vann, J. M., Leeuwenburgh, C., Tanokura, M., Denu, J. M., and Prolla, T. A. (2010). Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812.
Southwood, C. M., Peppi, M., Dryden, S., Tainsky, M. A., and Gow, A. (2007). Microtubule deacetylases, SirT2 and HDAC6, in the nervous system. Neurochem. Res. 32, 187–195.
Stefani, M., and Liguri, G. (2009). Cholesterol in Alzheimer’s disease: unresolved questions. Curr. Alzheimer Res. 6, 15–29.
Suzuki, K., and Koike, T. (2007). Mammalian Sir2-related protein (SIRT) 2-mediated modulation of resistance to axonal degeneration in slow Wallerian degeneration mice: a crucial role of tubulin deacetylation. Neuroscience 147, 599–612.
Tanner, K. G., Landry, J., Sternglanz, R., and Denu, J. M. (2000). Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc. Natl. Acad. Sci. U.S.A. 97, 14178–14182.
Tanno, M., Sakamoto, J., Miura, T., Shimamoto, K., and Horio, Y. (2007). Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 282, 6823–6832.
Tennen, R. I., Berber, E., and Chua, K. F. (2010). Functional dissection of SIRT6: identification of domains that regulate histone deacetylase activity and chromatin localization. Mech. Ageing Dev. 131, 185–192.
Tennen, R. I., and Chua, K. F. (2011). Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem. Sci. 36, 39–46.
Tissenbaum, H. A., and Guarente, L. (2001). Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410, 227–230.
Tong, L., and Denu, J. M. (2010). Function and metabolism of sirtuin metabolite O-acetyl-ADP-ribose. Biochim. Biophys. Acta 1804, 1617–1625.
Turner, N. C., Lord, C. J., Iorns, E., Brough, R., Swift, S., Elliott, R., Rayter, S., Tutt, A. N., and Ashworth, A. (2008). A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 27, 1368–1377.
Vakhrusheva, O., Smolka, C., Gajawada, P., Kostin, S., Boettger, T., Kubin, T., Braun, T., and Bober, E. (2008). Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 102, 703–710.
Vaquero, A., Scher, M., Lee, D., Erdjument-Bromage, H., Tempst, P., and Reinberg, D. (2004). Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 16, 93–105.
Vaquero, A., Scher, M. B., Lee, D. H., Sutton, A., Cheng, H.-L., Alt, F. W., Serrano, L., Sternglanz, R., and Reinberg, D. (2006). SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 20, 1256–1261.
Vaziri, H., Dessain, S. K., Ng Eaton, E., Imai, S. I., Frye, R. A., Pandita, T. K., Guarente, L., and Weinberg, R. A. (2001). hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159.
Vempati, R. K., Jayani, R. S., Notani, D., Sengupta, A., Galande, S., and Haldar, D. (2010). p300-mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. J. Biol. Chem. 285, 28553–28564.
Voelter-Mahlknecht, S., Ho, A. D., and Mahlknecht, U. (2005). FISH-mapping and genomic organization of the NAD-dependent histone deacetylase gene, Sirtuin 2 (Sirt2). Int. J. Oncol. 27, 1187–1196.
Voelter-Mahlknecht, S., and Mahlknecht, U. (2006). Cloning, chromosomal characterization and mapping of the NAD-dependent histone deacetylases gene sirtuin 1. Int. J. Mol. Med. 17, 59–67.
Wang, F., Nguyen, M., Qin, F. X.-F., and Tong, Q. (2007). SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6, 505–514.
Werner, H. B., Kuhlmann, K., Shen, S., Uecker, M., Schardt, A., Dimova, K., Orfaniotou, F., Dhaunchak, A., Brinkmann, B. G., Möbius, W., Guarente, L., Casaccia-Bonnefil, P., Jahn, O., and Nave, K.-A. (2007). Proteolipid protein is required for transport of sirtuin 2 into CNS myelin. J. Neurosci. 27, 7717–7730.
Wilkinson, K. A., and Henley, J. M. (2010). Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 428, 133–145.
Wilson, J. M., Le, V. Q., Zimmerman, C., Marmorstein, R., and Pillus, L. (2006). Nuclear export modulates the cytoplasmic Sir2 homologue Hst2. EMBO Rep. 7, 1247–1251.
Yang, B., Zwaans, B. M. M., Eckersdorff, M., and Lombard, D. B. (2009). The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle 8, 2662–2663.
Yang, X., and Seto, E. (2008). Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell 31, 449–461.
Yang, Y., Fu, W., Chen, J., Olashaw, N., Zhang, X., Nicosia, S. V., Bhalla, K., and Bai, W. (2007). SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat. Cell Biol. 9, 1253–1262.
Yi, J., and Luo, J. (2010). SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta 1804, 1684–1689.
Yu, L. R., Zhu, Z., Chan, K. C., Issaq, H. J., Dimitrov, D. S., and Veenstra, T. D. (2007). Improved titanium dioxide enrichment of phosphopeptides from HeLa cells and high confident phosphopeptide identification by cross-validation of MS/MS and MS/MS/MS spectra. J. Proteome Res. 6, 4150–4162.
Yuan, F., Xie, Q., Wu, J., Bai, Y., Mao, B., Dong, Y., Bi, W., Ji, G., Tao, W., Wang, Y., and Yuan, Z. (2011). MST1 promotes apoptosis through regulating Sirt1-dependent p53 deacetylation. J. Biol. Chem. 286, 6940–6945.
Zhang, J., Cicero, S. A., Wang, L., Romito-Digiacomo, R. R., Yang, Y., and Herrup, K. (2008). Nuclear localization of Cdk5 is a key determinant in the postmitotic state of neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 8772–8777.
Zhang, J., Li, H., and Herrup, K. (2010). Cdk5 nuclear localization is p27-dependent in nerve cells: implications for cell cycle suppression and caspase-3 activation. J. Biol. Chem. 285, 14052–14061.
Zhao, K., Chai, X., Clements, A., and Marmorstein, R. (2003). Structure and autoregulation of the yeast Hst2 homolog of Sir2. Nat. Struct. Biol. 10, 864–871.
Zhao, Y., Yang, J., Liao, W., Liu, X., Zhang, H., Wang, S., Wang, D., Feng, J., Yu, L., and Zhu, W.-G. (2010). Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 12, 665–675.
Zhong, L., D’Urso, A., Toiber, D., Sebastian, C., Henry, R. E., Vadysirisack, D. D., Guimaraes, A., Marinelli, B., Wikstrom, J. D., Nir, T., Clish, C. B., Vaitheesvaran, B., Iliopoulos, O., Kurland, I., Dor, Y., Weissleder, R., Shirihai, O. S., Ellisen, L. W., Espinosa, J. M., and Mostoslavsky, R. (2010). The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140, 280–293.
Keywords: phosphorylation, acetylation, proteolytic cleavage, NAD+-dependent deacetylation, ADP-ribosylation, sumoylation, methylation
Citation: Flick F and Lüscher B (2012) Regulation of sirtuin function by posttranslational modifications. Front. Pharmacol. 3:29. doi: 10.3389/fphar.2012.00029
Received: 15 November 2011; Paper pending published: 09 January 2012;
Accepted: 14 February 2012; Published online: 28 February 2012.
Edited by:
Tiago F. Outeiro, University of Lisbon, PortugalReviewed by:
Armando A. Genazzani, Università del Piemonte Orientale, ItalyRonen Marmorstein, The Wistar Institute, USA
Copyright: © 2012 Flick and Lüscher. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Bernhard Lüscher, Medical School, Institute of Biochemistry and Molecular Biology, RWTH Aachen University, Pauwelsstrasse 30, 52074 Aachen, Germany. e-mail:bHVlc2NoZXJAcnd0aC1hYWNoZW4uZGU=