 Sébastien Moniot
Sébastien Moniot


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Pharmacol. , 09 February 2012
Sec. Experimental Pharmacology and Drug Discovery
Volume 3 - 2012 | https://doi.org/10.3389/fphar.2012.00016
This article is part of the Research Topic Sirtuins in Biology and Disease View all 10 articles
Sirtuins are NAD+-dependent protein deacetylases regulating metabolism, stress responses, and aging processes. Mammalia have seven Sirtuin isoforms, Sirt1–7, which differ in their substrate specificities and subcellular localizations. The physiological functions of Sirtuins make them interesting therapeutic targets, which has stimulated extensive efforts on development of small molecule Sirtuin modulators. Yet, most Sirtuin inhibitors show limited potency and/or isoform specificity, and the mechanism of Sirtuin activation by small molecules remains obscure. Accumulating information on Sirtuin substrates, structures, and regulation mechanisms offer new opportunities for the challenging task to develop potent and specific small molecule modulators for mammalian Sirtuins for in vivo studies and therapeutic applications. We therefore recapitulate advances in structural and mechanistic studies on substrate recognition and deacetylation by Sirtuins, and in the characterization of compounds and molecular mechanisms regulating their activity. We then discuss challenges and opportunities from these findings for Sirtuin-targeted drug development efforts.
Reversible lysine (de)acetylation was long assumed to be a posttranslational protein modification mainly found in histones. It is now established to be widespread, however, with more than 6800 known acetylation sites in mammalian proteins, and thus rivaling phosphorylation in prevalence and importance (Norvell and McMahon, 2010). In fact, acetylation appears to be more ancient and acetylated lysines more conserved than phosphorylation sites, and dominant in some processes such as regulation of mitochondrial metabolism (Weinert et al., 2011). The enzymes catalyzing attachment and removal of this modification, protein acetyl transferases (PAT), and protein deacetylases (PDAC), are thus emerging as drug targets for various indications. Among the four PDAC classes (Xu et al., 2007), the Sirtuin family (class III) is unique in using NAD+ as a co-substrate, rendering Sirtuins metabolic sensors. Sirtuins were found to regulate metabolic pathways and stress responses (Bell and Guarente, 2011; Cen et al., 2011), and they contribute to some effects of calorie restriction (CR), in particular lifespan extension (Guarente and Picard, 2005). Pharmacological Sirtuin modulation has thus been identified as an attractive therapeutic approach, e.g., for supporting treatment of diabetes and prevention of malnutrition effects (Lavu et al., 2008; Haigis and Sinclair, 2010).
Mammalia have seven Sirtuin isoforms, Sirt1–7, which differ in their substrate specificities and subcellular localizations (Michan and Sinclair, 2007). Sirt1, 6, and 7 are nuclear enzymes and modify, for example, substrates contributing to chromosome stability and transcription regulation (Michan and Sinclair, 2007; Haigis and Sinclair, 2010). Sirt3, 4, and 5 are located in mitochondria – they are in fact the only deacetylases known in this organelle – and appear to regulate metabolic enzymes and stress response mechanisms (Gertz and Steegborn, 2010; Bell and Guarente, 2011). Sirt2 is mainly cytosolic and was reported to deacetylate tubulin and p300 (Black et al., 2008). Despite these examples, the relevant Sirtuin substrates are unknown for many Sirtuin-regulated processes and no or few substrate sites have been described for most mammalian isoforms. Likewise, few selective and potent inhibitors for mammalian Sirtuin isoforms are available, and their mode of action is largely unknown (Cen, 2010). Even more so, activation of Sirtuins using small molecules has been reported, but the mechanism remains enigmatic, hampering efforts to develop improved activators. We propose that Sirtuin features such as different substrates/substrate specificities and their complex catalytic mechanism offer unique opportunities for drug development, and molecular and mechanistic insights into substrate recognition, catalysis, and pharmacological modulation will be essential steps toward exploiting this potential.
All Sirtuins share a conserved catalytic core of ∼275 amino acids that is flanked by N- and C-terminal extensions. The extensions are variable in length and sequence, and they have been reported to play various roles such as ensuring a proper cellular localization, regulating the oligomerization state, and/or exerting autoregulation mechanisms (Schwer et al., 2002; Zhao et al., 2003; Tennen et al., 2010).
Whereas other PDAC families activate a water molecule for the hydrolysis reaction by using a zinc cofactor, the unique Sirtuin mechanism is based on the use of NAD+ as a co-substrate. High-resolution structures of several Sirtuins, including human Sirt2, 3, 5, and 6, in apo- or (co)substrate/inhibitor-bound forms have been reported (for a review see Sanders et al., 2010) and have helped to reveal their catalytic mechanism (Cen et al., 2011). The catalytic core adopts an oval-shaped fold composed of two globular subdomains linked by four loops, which contribute to the active site cleft between the subdomains (Figure 1A). The larger domain consists of a Rossmann-fold typical for NAD+ binding proteins and the smaller domain is formed by association of two modules inserted in the Rossmann-fold domain: a zinc-binding motif bearing the consensus sequence Cys-X2–4-Cys-X15–40-Cys-X2–4-Cys, and an α-helical region showing the highest diversity among family members. The relative orientations of the small and the Rossmann-fold domains vary in known Sirtuin structures, but these differences seem to be due to the presence or absence of substrates. Both the acetylated lysine-containing polypeptide substrates and the NAD+ co-substrate bind to the cleft at the interface of the two domains (Figure 1A). The so-called “cofactor binding loop” shows high flexibility and its conformation was shown to evolve in close relation to the catalytic events (Sanders et al., 2010), which comprise formation of a covalent 1′-O-alkylamidate intermediate between the two substrates under nicotinamide release. Subsequently, the intermediate is hydrolyzed to yield deacetylated polypeptide and 2′-O-acetyl-ADP-ribose (Cen et al., 2011). Studies on different Sirtuin substrate/substrate analog complexes indicate that the cofactor binding loop gets ordered upon substrate binding and changes to a more closed conformation upon acetyl transfer (see, e.g., Chang et al., 2002; Hoff et al., 2006), with a suggested role of this loop-relocation in expelling the first reaction product, nicotinamide. Binding of the acetyl-lysine substrates was also shown to induce a significant reorientation of the two domains relative to each other and to induce a closure of the cleft as well as the correct positioning of conserved residues for formation of the acetyl-lysine binding tunnel (Cosgrove et al., 2006). These rearrangements highlight the dynamic structure of this enzyme class. Furthermore, individual Sirtuins show specific sequence preferences due to differences in the details of their peptide binding groove shape and electrostatics (Cosgrove et al., 2006; Sanders et al., 2010). Also, they recognize in general a wide variety of substrate sequences, again indicating a high adaptability of this enzyme class to its substrates. It is thus essential to obtain structures of different isoforms and enzyme states (such as different ligand complexes) for obtaining a reliable picture of the dynamics and preferred conformations of their binding pockets for drug development.
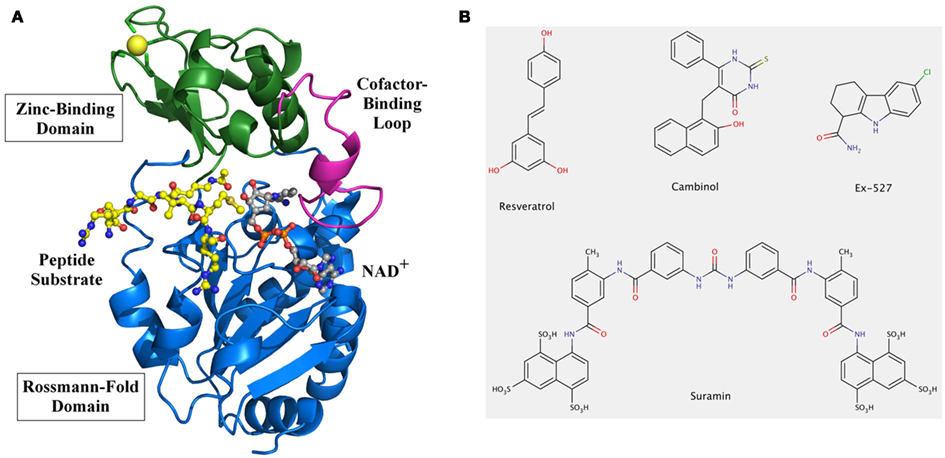
Figure 1. Overall structure and modulators of Sirtuins. (A) Structure of human Sirt3 with bound AceCS2 (acetylCoA synthetase 2) peptide and NAD+ analog. The structure of Sirt3 (PDB entry 3glr) is shown as a cartoon model with Rossmann-fold domain and Zinc-binding domain colored in blue and green, respectively. The cofactor binding loop (magenta) is in a closed conformation and binds a carba-NAD molecule (gray), which was added to the model based on a superposition with the structure of an Hst2/carba-NAD structure (1szc). The active site cleft also contains the peptide substrate AceCS2 (yellow) with the acetylated lysine directly pointing to the active site. (B) Chemical structures of known Sirtuin modulators. Compound screenings identified the polyphenol resveratrol as an activator of human Sirt1 activity against suitable substrates, and Ex527 as one of the most potent and selective Sirt1 inhibitors. The naphtol compound cambinol was obtained from structure–activity relationship studies on Sirtinol, one of the first Sirtuin inhibitors identified. Suramin, a symmetric diarylurea containing multiple anionic groups, is an inhibitor for several human Sirtuin isoforms. It is the only inhibitor, besides substrate analogs, whose complex with a Sirtuin has been described.
Sirtuins are considered attractive therapeutic targets for metabolic and aging-related diseases, which has stimulated extensive efforts for development of Sirtuin-modulating small molecules (Lavu et al., 2008; Haigis and Sinclair, 2010). Sirtuins are also special targets because they are amenable to stimulation, besides inhibition, through small molecules. Sirtuin activating compounds, initially resveratrol-related polyphenols (Figure 1B) and subsequently other compound classes (Howitz et al., 2003; Milne et al., 2007), can promote survival of human cells, extend lifespan in various species, and protect against insulin resistance (Guarente and Picard, 2005). However, their mode of action remains enigmatic and little is known about their effects on most Sirtuin isoforms. Furthermore, their physiological effects might in part or mainly rely on effects on other cellular targets, such as receptors or transport proteins (Pacholec et al., 2010). A mechanistically understood Sirtuin activator, in contrast, is isonicotinamide. Nicotinamide is the first product of the Sirtuin catalyzed reaction, released from NAD+ during formation of the alkyl imidate intermediate (see above). Rebinding of nicotinamide to the Sirtuin/intermediate complex can promote the reverse reaction to reform the substrates, and thus inhibits the deacetylation reaction (Sauve, 2010). This non-competitive mechanism is assumed to inhibit all Sirtuins, with Ki values typically reported around 0.05–0.2 mM (Sauve, 2010). Sirtuins thus appear to be affected by physiological nicotinamide concentrations, assumed to be up to 0.1 mM, and a role of nicotinamide as endogenous Sirtuin regulator is supported by in vivo studies in yeast, flies, and mammalian cells (Anderson et al., 2003; Sauve, 2010). Isonicotinamide can compete with nicotinamide for binding but cannot initiate the reverse reaction, thereby leading to apparent activation through relief of nicotinamide inhibition (Sauve et al., 2005; Cen et al., 2011). Assuming that all Sirtuins are equally inhibited by nicotinamide, isonicotinamide would be a general Sirtuin “activator.” However, data from our lab suggest that some Sirtuins show nicotinamide-insensitive deacetylase activity (Fischer et al., unpublished), indicating that nicotinamide and isonicotinamide employ isoform discriminating binding sites or modulation mechanisms. Structural and further biochemical studies on these compounds and mechanisms might enable the development of isoform selective modulators.
Various pharmacological Sirtuin inhibitors have been described, but few of them show high potencies, isoform selectivity, and favorable pharmacological properties (Cen, 2010). In fact, for most compounds effects on only few Sirtuin isoforms have been reported, and little is known about their inhibition mechanisms. For example, cambinol (Figure 1B) inhibits Sirt1 and Sirt2 with IC50 values of 50–60 μM, but has no significant effects on Sirt3 and Sirt5 (Heltweg et al., 2006). Docking studies suggest that it occupies parts of both substrate binding pockets, the one for NAD+ and the one for the polypeptide (Neugebauer et al., 2008). Such a blocking of binding site areas for both substrates was crystallographically shown for suramin (Figure 1B), a huge naphthylurea compound with antiproliferative and antiviral activity that inhibits Sirt1, Sirt2, and Sirt5 – and possibly other, not yet tested isoforms – with low micromolar potency (Schuetz et al., 2007; Trapp et al., 2007). Despite this lack of specificity, the crystal structure of a Sirt5/suramin complex (Schuetz et al., 2007) allows insights into the binding details helpful for drug development efforts, and it was used to rationalize structure–activity relationships for suramin derivatives with improved potency (Trapp et al., 2007). However, the Sirt5/suramin complex is the only published crystal structure of a Sirtuin complex with an inhibitor other than peptide or NAD+ derivatives co-crystallized for mechanistic insights, and even kinetic data to identify potential competition with one of the Sirtuin substrates is lacking in most cases (Cen, 2010). Thus, to better understand how available compounds interact with Sirtuins and how improved compounds can be obtained, mechanistic data and structural information on their complexes with Sirtuins are of paramount importance.
The large body of biochemical and structural work on Sirtuins has provided us with exciting insights in how Sirtuins recognize their substrates and how they catalyze lysine deacetylation (Sanders et al., 2010; Sauve, 2010). The differences between Sirtuin isoforms in details of structure, physiological targets, and regulators should enable identification of highly specific inhibitors, and possibly also activators. An obvious requirement toward this goal is progress in the identification of Sirtuin substrates, so that the proper Sirtuin isoform(s) can be targeted for modulating a specific cell function. Furthermore, physiological Sirtuin substrates are needed for meaningful modulation tests, as can be learned from the studies on Sirtuin activation by resveratrol, which showed that effects can be substrate-specific and thus that non-physiological substrates can lead to artificial results (Kaeberlein et al., 2005; Cen et al., 2011). These findings have led to heated discussions on the general possibility of Sirtuin activation against physiological substrates (Cen et al., 2011), but rather should stimulate studies on the molecular reasons for seemingly contradicting observations, which promise outstanding opportunities for drug development. Understanding the substrate-specific resveratrol effects offers the exiting possibility to develop modulators not only specific for one Sirtuin isoform, but maybe even affecting only deacetylation of one or few of the substrates of this isoform.
A general challenge for understanding Sirtuin interactions and mechanisms lies in their complexity, with two substrates, one of them a polypeptide that can vary in sequence and the second one releasing the product nicotinamide, which also acts as a non-competitive inhibitor. Some Sirtuins have even been proposed to catalyze physiologically other reactions than deacetylation, such as hydrolytic release of other organic acids or ADP-ribosylation (Haigis et al., 2006; Zhu et al., 2012), but it remains to be seen whether these activities are their dominant physiological functions. However, the sequence variability of the polypeptide substrate poses a challenge, because it means that many Sirtuin/substrate complexes with small differences in conformation exist, whereas only few structures of Sirtuin complexes with physiological substrate sequences have been solved. Thus, exploiting the peptide binding pockets for inhibition so far only yielded peptide mimetics and other lead compounds not yet suitable as drugs (Huhtiniemi et al., 2011; Schlicker et al., 2011), and further studies on peptide binding site differences and dynamics will be required for efficient compound improvement. A major obstacle for fully understanding Sirtuin/ligand interactions is the limited number of Sirtuin complex structures, and that the available structures were solved with several different Sirtuins, packed through various crystal lattice interactions. Comparing such complexes often does not allow assigning differences in conformation to the different Sirtuin or to the different ligand, or even the different crystal packing. Despite these limitations, using the available structural snapshots for rational drug discovery and for rationalizing results from screening has revealed a number of promising Sirtuin inhibitors (Cen, 2010). Further mechanistic studies on these compounds promise to boost Sirtuin inhibitor development by enabling more sophisticated rational approaches. An especially interesting compound should be Ex527 (Figure 1B), a potent Sirt1 inhibitor (IC50 ∼0.1 μM) with two orders of magnitude lower potency against Sirt2 and Sirt3 (Napper et al., 2005), and no effect on Sirt5 and class I/II HDACs (Solomon et al., 2006). Kinetic data suggest that it binds after nicotinamide release and prevents product release, indicating that an interaction with a reaction intermediate or product complex mediates specificity for Sirtuins. Structural insights on Ex527 inhibition will thus support development of Sirtuin-specific compounds and should reveal the molecular basis of its isoform selectivity, enabling development of similar compounds specific for other isoforms.
Molecular studies on enzyme inhibition have revealed many attractive drug development approaches, such as selective zinc-binding groups for carboanhydrase inhibition (Schlicker et al., 2008b) or non-competitive chelators for adenylyl cyclase inhibition (Steegborn et al., 2005; Schlicker et al., 2008b), and such studies on Sirtuin modulators promise similar progress for these exiting drug targets. Mechanistic studies on Sirtuins beyond the basics of substrate recognition promise additional opportunities for drug development. Many Sirtuins have regulatory regions outside the catalytic domain (Michan and Sinclair, 2007; Schlicker et al., 2008a; Tennen et al., 2010), yet structural studies so far mainly focused on the catalytic cores (Sanders et al., 2010). Also, posttranslational modifications regulating Sirtuins are just emerging (Guo et al., 2010), and their mode of action is largely unknown. Thus, although the accumulated knowledge on Sirtuins already offers approaches for their modulation, further mechanistic studies on Sirtuin modulators and on physiological regulation mechanisms promise exciting insights into Sirtuin function and in Sirtuin features exploitable for highly specific intervention with drugs.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Work on Sirtuins in the authors’ lab is partially supported by Deutsche Forschungsgemeinschaft grant STE1701/5 (to Clemens Steegborn).
Anderson, R. M., Bitterman, K. J., Wood, J. G., Medvedik, O., and Sinclair, D. A. (2003). Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423, 181–185.
Bell, E. L., and Guarente, L. (2011). The SirT3 divining rod points to oxidative stress. Mol. Cell 42, 561–568.
Black, J. C., Mosley, A., Kitada, T., Washburn, M., and Carey, M. (2008). The SIRT2 deacetylase regulates autoacetylation of p300. Mol. Cell 32, 449–455.
Cen, Y. (2010). Sirtuins inhibitors: the approach to affinity and selectivity. Biochim. Biophys. Acta 1804, 1635–1644.
Cen, Y., Youn, D. Y., and Sauve, A. A. (2011). Advances in characterization of human sirtuin isoforms: chemistries, targets and therapeutic applications. Curr. Med. Chem. 18, 1919–1935.
Chang, J. H., Kim, H. C., Hwang, K. Y., Lee, J. W., Jackson, S. P., Bell, S. D., and Cho, Y. (2002). Structural basis for the NAD-dependent deacetylase mechanism of Sir2. J. Biol. Chem. 277, 34489–34498.
Cosgrove, M. S., Bever, K., Avalos, J. L., Muhammad, S., Zhang, X., and Wolberger, C. (2006). The structural basis of sirtuin substrate affinity. Biochemistry 45, 7511–7521.
Gertz, M., and Steegborn, C. (2010). Function and regulation of the mitochondrial Sirtuin isoform Sirt5 in Mammalia. Biochim. Biophys. Acta 1804, 1658–1665.
Guo, X., Williams, J. G., Schug, T. T., and Li, X. (2010). DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem. 285, 13223–13232.
Haigis, M. C., Mostoslavsky, R., Haigis, K. M., Fahie, K., Christodoulou, D. C., Murphy, A. J., Valenzuela, D. M., Yancopoulos, G. D., Karow, M., Blander, G., Wolberger, C., Prolla, T. A., Weindruch, R., Alt, F. W., and Guarente, L. (2006). SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126, 941–954.
Haigis, M. C., and Sinclair, D. A. (2010). Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295.
Heltweg, B., Gatbonton, T., Schuler, A. D., Posakony, J., Li, H., Goehle, S., Kollipara, R., Depinho, R. A., Gu, Y., Simon, J. A., and Bedalov, A. (2006). Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 66, 4368–4377.
Hoff, K. G., Avalos, J. L., Sens, K., and Wolberger, C. (2006). Insights into the sirtuin mechanism from ternary complexes containing NAD+ and acetylated peptide. Structure 14, 1231–1240.
Howitz, K. T., Bitterman, K. J., Cohen, H. Y., Lamming, D. W., Lavu, S., Wood, J. G., Zipkin, R. E., Chung, P., Kisielewski, A., Zhang, L. L., Scherer, B., and Sinclair, D. A. (2003). Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196.
Huhtiniemi, T., Salo, H. S., Suuronen, T., Poso, A., Salminen, A., Leppanen, J., Jarho, E., and Lahtela-Kakkonen, M. (2011). Structure-based design of pseudopeptidic inhibitors for SIRT1 and SIRT2. J. Med. Chem. 54, 6456–6468.
Kaeberlein, M., Mcdonagh, T., Heltweg, B., Hixon, J., Westman, E. A., Caldwell, S. D., Napper, A., Curtis, R., Distefano, P. S., Fields, S., Bedalov, A., and Kennedy, B. K. (2005). Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 280, 17038–17045.
Lavu, S., Boss, O., Elliott, P. J., and Lambert, P. D. (2008). Sirtuins – novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discov. 7, 841–853.
Michan, S., and Sinclair, D. (2007). Sirtuins in mammals: insights into their biological function. Biochem. J. 404, 1–13.
Milne, J. C., Lambert, P. D., Schenk, S., Carney, D. P., Smith, J. J., Gagne, D. J., Jin, L., Boss, O., Perni, R. B., Vu, C. B., Bemis, J. E., Xie, R., Disch, J. S., Ng, P. Y., Nunes, J. J., Lynch, A. V., Yang, H., Galonek, H., Israelian, K., Choy, W., Iffland, A., Lavu, S., Medvedik, O., Sinclair, D. A., Olefsky, J. M., Jirousek, M. R., Elliott, P. J., and Westphal, C. H. (2007). Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450, 712–716.
Napper, A. D., Hixon, J., Mcdonagh, T., Keavey, K., Pons, J. F., Barker, J., Yau, W. T., Amouzegh, P., Flegg, A., Hamelin, E., Thomas, R. J., Kates, M., Jones, S., Navia, M. A., Saunders, J. O., Distefano, P. S., and Curtis, R. (2005). Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J. Med. Chem. 48, 8045–8054.
Neugebauer, R. C., Uchiechowska, U., Meier, R., Hruby, H., Valkov, V., Verdin, E., Sippl, W., and Jung, M. (2008). Structure-activity studies on splitomicin derivatives as sirtuin inhibitors and computational prediction of binding mode. J. Med. Chem. 51, 1203–1213.
Pacholec, M., Bleasdale, J. E., Chrunyk, B., Cunningham, D., Flynn, D., Garofalo, R. S., Griffith, D., Griffor, M., Loulakis, P., Pabst, B., Qiu, X., Stockman, B., Thanabal, V., Varghese, A., Ward, J., Withka, J., and Ahn, K. (2010). SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 285, 8340–8351.
Sanders, B. D., Jackson, B., and Marmorstein, R. (2010). Structural basis for sirtuin function: what we know and what we don’t. Biochim. Biophys. Acta 1804, 1604–1616.
Sauve, A. A., Moir, R. D., Schramm, V. L., and Willis, I. M. (2005). Chemical activation of Sir2-dependent silencing by relief of nicotinamide inhibition. Mol. Cell 17, 595–601.
Schlicker, C., Boanca, G., Lakshminarasimhan, M., and Steegborn, C. (2011). Structure-based development of novel sirtuin inhibitors. Aging 3, 852–872.
Schlicker, C., Gertz, M., Papatheodorou, P., Kachholz, B., Becker, C. F., and Steegborn, C. (2008a). Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J. Mol. Biol. 382, 790–801.
Schlicker, C., Rauch, A., Hess, K. C., Kachholz, B., Levin, L. R., Buck, J., and Steegborn, C. (2008b). Structure-based development of novel adenylyl cyclase inhibitors. J. Med. Chem. 51, 4456–4464.
Schuetz, A., Min, J., Antoshenko, T., Wang, C. L., Allali-Hassani, A., Dong, A., Loppnau, P., Vedadi, M., Bochkarev, A., Sternglanz, R., and Plotnikov, A. N. (2007). Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure 15, 377–389.
Schwer, B., North, B. J., Frye, R. A., Ott, M., and Verdin, E. (2002). The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 158, 647–657.
Solomon, J. M., Pasupuleti, R., Xu, L., Mcdonagh, T., Curtis, R., Distefano, P. S., and Huber, L. J. (2006). Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell. Biol. 26, 28–38.
Steegborn, C., Litvin, T. N., Hess, K. C., Capper, A. B., Taussig, R., Buck, J., Levin, L. R., and Wu, H. (2005). A novel mechanism for adenylyl cyclase inhibition from the crystal structure of its complex with catechol estrogen. J. Biol. Chem. 280, 31754–31759.
Tennen, R. I., Berber, E., and Chua, K. F. (2010). Functional dissection of SIRT6: identification of domains that regulate histone deacetylase activity and chromatin localization. Mech. Ageing Dev. 131, 185–192.
Trapp, J., Meier, R., Hongwiset, D., Kassack, M. U., Sippl, W., and Jung, M. (2007). Structure-activity studies on suramin analogues as inhibitors of NAD+-dependent histone deacetylases (sirtuins). ChemMedChem 2, 1419–1431.
Weinert, B. T., Wagner, S. A., Horn, H., Henriksen, P., Liu, W. R., Olsen, J. V., Jensen, L. J., and Choudhary, C. (2011). Proteome-wide mapping of the Drosophila acetylome demonstrates a high degree of conservation of lysine acetylation. Sci. Signal. 4, ra48.
Xu, W. S., Parmigiani, R. B., and Marks, P. A. (2007). Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26, 5541–5552.
Zhao, K., Chai, X., Clements, A., and Marmorstein, R. (2003). Structure and autoregulation of the yeast Hst2 homolog of Sir2. Nat. Struct. Biol. 10, 864–871.
Keywords: Sirtuin, structure, mechanism, inhibitor, activator
Citation: Moniot S, Weyand M and Steegborn C (2012) Structures, substrates, and regulators of mammalian Sirtuins – opportunities and challenges for drug development. Front. Pharmacol. 3:16. doi: 10.3389/fphar.2012.00016
Received: 16 November 2011; Accepted: 25 January 2012;
Published online: 09 February 2012.
Edited by:
Aleksey G. Kazantsev, Harvard Medical School and Massachusetts General Hospital, USAReviewed by:
Joel Tyndall, University of Otago, New ZealandCopyright: © 2012 Moniot, Weyand and Steegborn. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Clemens Steegborn, Department of Biochemistry, University of Bayreuth, Universitätsstraß e 30, 95447 Bayreuth, Germany. e-mail:Y2xlbWVucy5zdGVlZ2Jvcm5AdW5pLWJheXJldXRoLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.