
- Department of Pharmaceutical Biosciences, School of Pharmacy, University of Oslo, Oslo, Norway
Through the last decades it has become increasingly evident that disease-states involving cytokines affect the pharmacokinetics of drugs through regulation of expression and activity of drug metabolizing enzymes, and more recently also drug transporters. The clinical implication is however difficult to predict, since these effects are dependent on the degree of inflammation and may be changed when the diseases are treated. This article will give an overview of the present understanding of the effects of cytokines on cytochrome P450 enzymes and drug transporters, and highlight the importance of considering these issues in regard to increasing use of the relatively new class of drugs, namely therapeutic proteins.
Introduction
Differences in drug response among individuals are a great challenge in order to optimize drug dosage regimen for a patient. The reason for this variability is multifactorial and include genetic, environmental, and disease related factors, which may affect both pharmacodynamics and pharmacokinetics. Understanding the factors contributing to inter-individual variability in drug response is of crucial importance both in the development of new drugs and in optimization of the use of drugs already on the market.
Factors contributing to determining the pharmacokinetic profile of a drug include drug metabolizing enzymes and drug transporting proteins. The most important drug metabolizing enzymes are the phase I enzymes belonging to the cytochrome P450 (CYP) enzyme family which metabolize many structurally different xenobiotics (drug, chemicals), as well as endobiotics (steroids, fatty acids, prostaglandins; Gonzalez, 1990). There are several CYP subfamilies, of which CYP1, CYP2, and CYP3 are mainly involved in drug metabolism, and in humans 50% of the overall elimination of commonly used drugs is performed by these subfamilies (Wilkinson, 2005). Liver, the principal organ of drug elimination, is the organ with the highest abundance of CYP enzymes, while the small intestinal mucosa has been described to be the most important extra hepatic site of biotransformation (Lin and Lu, 2001). Inter-individual variability in the expression and activity of CYP enzymes is recognized as significant contributor to variation in drug response. CYP3A4 is the most prominent CYP enzyme, mainly because it is highly expressed in organs involved in drug disposition, such as liver, gastrointestinal tract, and kidney (Shimada et al., 1994; Paine et al., 2006) and because of the broad substrate specificity. The expression of this isoenzyme displays a 30- to 60-fold variability in human liver and intestine biopsies (Thummel et al., 1994; Paine et al., 1997). The reason for inter-individual variability in the expression and activity of CYP enzymes is multifactorial, but may to some degree be explained by genetic, environmental, and disease related factors.
The most studied drug transporter is the transmembrane efflux transporter P-glycoprotein (P-gp), a human ABC-transporter encoded by the ABCB1 gene (Higgins, 1992). P-gp was discovered in 1976 (Juliano and Ling, 1976) as an important multi-drug resistance (MDR) mechanism in cancer treatment. It is expressed and distributed in the luminal surface membrane of the enterocytes in the small intestine, renal proximal tubular cells, the bile canalicular membrane of hepatocytes, the capillary endothelial cells in the blood–brain barrier (BBB) and in different cell types involved in the immune response (Thiebaut et al., 1987; Sugawara et al., 1988; Cordon-Cardo et al., 1989; Klimecki et al., 1994). Based on its localization, the function of P-gp is suspected to be protection of the cells against various toxicants, among these therapeutically active drugs. As P-gp is abundant in the intestinal epithelium, one important function is to restrict oral bioavailability of drugs, and since the substrate specificity of P-gp is to a great deal overlapping with that of CYP3A4, the general view is that P-gp and CYP3A4 work together in restricting the intestinal bioavailability of drugs (Benet and Cummins, 2001).
Through the last decade, there has been an increasing awareness on drug transporters other than P-gp and their role in bioavailability, elimination, and tissue distribution of drugs. These include other ABC transporters such as multidrug-resistance associated proteins (MRPs), the SLC transporters [e.g., organic anion transporting polypeptides (OATPs), organic anion transporters (OATs), and organic cation transporters (OCTs)]. Similar to drug metabolizing enzymes, there is also a considerable variability in the expression and activity of drug transporters. This variability is only to a minor extent explained by genetic polymorphism and other causes, such as environmental influence (e.g., drug interactions) and disease-state also play a role.
Immunological response and the release of cytokines is part of the pathophysiology of various diseases like autoimmune diseases, infections, brain injuries, and cancer. It has been known for several decades that cytokines regulate the expression and activity of drug metabolizing enzymes and thus, may affect the pharmacokinetics of drugs. More recently it has become evident that this applies to drug transporters as well. This article will give an overview of the present understanding of the effects of cytokines on CYP enzymes and transporters involved in drug pharmacokinetics, and also point out the importance of considering these issues in regard to the increasing use of the relatively new class of drugs, namely therapeutic proteins and their involvement in drug–drug interactions.
Immunological Response and CYP Metabolism
Several clinical studies have reported alterations in drug pharmacokinetics in patients with inflammations, infectious diseases, and cancer as well as in critically ill patients (Aitken et al., 2006; Morgan et al., 2008; Morgan, 2009). Already in 1978 acute virus infections in asthmatic children were shown to significantly increase the terminal half-life of theophylline (Chang et al., 1978). Also during an influenza B out break asthmatic children developed a sudden decrease in theophylline clearance and were hospitalized with toxicity problems (Kraemer et al., 1982). Already in 1976 it was shown that agents causing inflammation and infection depressed hepatic CYP enzymes in rats (Renton and Mannering, 1976a,b) and thus, the decreased theophylline clearance could be explained by a down-regulation of the CYP enzyme responsible for the metabolism of theophylline (CYP1A2). Several viruses, e.g., Herpes simplex, adenovirus, and HIV, have since then been identified to depress CYP metabolism and reduce drug clearance (Anolik et al., 1982; Forsyth et al., 1982; Lee et al., 1993). Also acute hepatitis virus A infection has been shown to decrease the excretion of 7-hydroxycoumarin in children and adults, indicating a depressed CYP2A6 activity during virus infection (Pasanen et al., 1997). CYP2D6 and CYP3A4 activities have been reported to be significantly lower in patients with chronic hepatitis C compared to in healthy volunteers (Becquemont et al., 2002). Interestingly, HIV patients genotyped as CYP2D6 extensive metabolizers (EM) expressed a shift toward a poor metabolizer (PM) CYP2D6 phenotype which correlated with disease activity (O’Neil et al., 2000). Also bacterial infections cause impaired drug clearance in humans. Administration of low doses of bacterial lipopolysaccharide (LPS) to healthy volunteers has been reported to cause reduced clearance of theophylline, antipyrine, and hexobarbitone (Shedlofsky et al., 1994, 1997). In rats CYP-mediated drug metabolism is suppressed during polymicrobial sepsis, particularly in the late phase (Lee and Lee, 2005).
Several studies have reported decreased theophylline and aminopyrine clearance following influenza virus and bacillus Calmette–Guerin (BCG) vaccination in healthy volunteers (Renton et al., 1980; Kramer and McClain, 1981; Gray et al., 1983). The effect was shown to be largest in individuals with high theophylline clearance before vaccination (Meredith et al., 1985), probably those with high CYP1A2 activity. On the other hand, influenza immunization did not significantly change CYP3A4 or CYP2E1 activities, as measured by the erythromycin breath test (ERMBT) and chlorzoxazone clearance (Kim and Wilkinson, 1996; Hayney et al., 2001). However, an inverse correlation between interferon-γ (IFN-γ) production and changes in ERMBT has been reported after administration of influenza vaccine to healthy volunteers (Hayney and Muller, 2003). In this respect it is interesting to note that in vitro studies with hepatocytes cultured with IFN-γ showed a decreased CYP3A4 expression and activity (Donato et al., 1997). The observed discrepancies in effect of vaccines might be due to different purity of vaccines, variable vaccination protocols or differences in response on the various CYP enzymes.
Additionally altered pharmacokinetics is observed in patients with inflammatory diseases and cancer. The largest effect of inflammatory disease on the pharmacokinetics of drugs has been reported for patients with rheumatoid arthritis, which showed a three and fourfold higher systemic exposure of verapamil and simvastatin compared to healthy volunteers (Mayo et al., 2000; Zhang et al., 2009). Also in patients with advanced cancer, all genotyped as EM of CYP2C19, a reduction in omeprazole metabolism was observed, and all patients had a slower metabolic CYP2C19 phenotype compared to healthy volunteers (Williams et al., 2000). Similarly decreased CYP3A4-dependent CsA metabolism has been reported in bone marrow transplanted patients, and interestingly an association between high interleukin 6 (IL-6) plasma concentrations and increased CsA concentrations were found (Chen et al., 1994). Later, Frye et al. (2002) studied the relationship between plasma concentrations of IL-6 and tumor necrosis factor alpha (TNF-α) and CYP enzyme activities in patients with congestive heart failure. IL-6 and TNF-α concentrations were negatively correlated to the activities of CYP1A2 and CYP2C19, investigated by use of caffeine and mephenytoin as probe substrates. There was no significant relationship between the cytokine level and CYP2D6 and CYP2E1 activities in these patients (Frye et al., 2002). In this respect it is interesting to note that increased adverse events and discontinuing treatment of the CYP2C19 substrate imipramine has been reported in heart failure patients (Glassman et al., 1983).
To summarize, depression of metabolic capacity through CYP enzymes seems to be a common feature of a variety of diseases involving an immune response with the release of cytokines. The different CYP enzymes are to a variable degree affected, and increases in drug exposure from less than 50 to up to 400% have been observed. The potential effects of cytokines on the pharmacokinetics of a large number of drugs accounts for increased awareness in treating patients with diseases involving an immune response. Also, there are indications of differential effects, with larger effects on patients with initially high clearance through the enzyme in question. Thus, depression of CYP activity is a considerable factor contributing to inter-individual variability in drug exposure.
Immunological Response and Drug Transport
Similar to the drug metabolizing enzymes, a variety of diseases have also been shown to influence on the expression of drug transporters. P-gp is in this area by far the most extensively studied drug transporter. For example, several studies have shown intestinal P-gp to be inversely correlated with inflammatory disease activity. In a study by Ufer et al. (2009), P-pg mRNA and protein expression were decreased in patients with ulcerative colitis compared to healthy volunteers and P-gp mRNA was inversely correlated with disease activity. Also, while expression of breast cancer resistance protein (BCRP) and P-gp in inflamed mucosa is reduced in patients with ulcerative colitis, expression of these transporters is comparable in unaffected mucosa from ulcerative colitis patients and healthy volunteers (Gutmann et al., 2008). Accordingly, a post mortem study of seropositive HIV patients showed that P-gp in brain microvascular endothelial cells was decreased compared to HIV-negative controls (Langford et al., 2004). However, this picture is more complicated as different parts of the brain were differently affected; i.e., in contrast to the aforementioned decrease of P-gp in endothelial cells, P-gp immunoreactivity was increased in astroglial cells in AIDS patients with HIV encephalitis compared to HIV encephalitis-negative patients and seronegative controls (Langford et al., 2004).
Many in vitro studies have examined the effect of inflammatory mediators on expression and activity of P-gp in the brain (Bauer et al., 2005; Miller et al., 2008; Roberts and Goralski, 2008). Several studies show a difference in effect after short-term versus long-term exposure to inflammatory mediators; whereas P-gp activity is initially depressed, long-term exposure to inflammatory mediators seems to upregulate P-gp expression and activity (Hartz et al., 2006; Bauer et al., 2007). Not surprisingly, the magnitude and direction of changes in drug transport activity is dependent on both the specific cytokine and model examined (as reviewed by Roberts and Goralski, 2008). This is exemplified by the diverging results of two separate rat models. Seelbach et al. (2007) reported that P-gp expression in brain microvessels increased 3 h after induction of inflammatory pain. These results were accompanied by in situ brain perfusion studies and antinociceptive studies that showed decreased brain uptake and decreased analgesia of morphine, a P-gp substrate (Seelbach et al., 2007). In contrast, Goralski et al. (2003) showed that LPS-induced CNS inflammation decreased P-gp expression and activity. In this study radioactive labeled digoxin was increased both in the brain and liver following intracranial ventricle administration of LPS in male rats (Goralski et al., 2003). Accordingly, P-gp mRNA in the brain and mRNA of both P-gp and OATP1B1 in liver were reduced. The diverging results of in vitro and animal models call for more in vivo studies to explore the effect of inflammatory disease on drug transport in patients. So far, altered pharmacokinetics of drugs relative to disease activity has been observed by Roberts et al. (2009). This group showed that patients with acute inflammatory brain injury obtained increased levels of the morphine metabolites morphine-3-glucuoronide and morphine-6-glucuronide with increasing IL-6, while no linkage between the P-gp substrate morphine and CSF IL-6 was observed. These data suggests an inhibition or down-regulation of drug efflux transporters specific to these metabolites other than P-gp in the BBB, possibly OATPs, as postulated by the authors (Roberts et al., 2009). Taken together, data on the effect of inflammatory mediators/disease on drug exposure in the brain are not conclusive and more in vivo studies are needed to explore this issue.
There is increasing evidence for differential ability of cytokines to influence on the regulation of expression and activity of drug transporters in immune cells compared to other tissue (Liptrott and Owen, 2011). While most studies suggest a depression or down-regulation of P-gp upon an inflammatory response, a recent study showed that P-gp expression on lymphocytes in patients with systemic lupus erythematosus (SLE) correlated positively with disease activity (Tsujimura and Tanaka, 2011). Up-regulation of P-gp in peripheral blood mononuclear cells has previously been shown for a variety of diseases, by far most studied in malignant diseases, where it causes the problem of MDR (Kantharidis et al., 2000; Shtil, 2002), but also in rheumatoid arthritis (Suzuki et al., 2010), HIV (Langford et al., 2004), and in solid organ transplantation (Donnenberg et al., 2001). This observation is supported by in vitro studies where P-gp in lymphocytes is induced by various stimuli such as IL-2 (Tsujimura et al., 2004; Liptrott et al., 2009). Up-regulation of P-gp and other drug transporters resulting is a problem in the use of drugs which are P-gp substrates and have their site of action within the immune cells, where an up-regulation of P-gp leads to reduced levels of drugs at their site of action. This applies to drugs such as antiviral agents used in HIV, immunosuppressants used in autoimmune diseases and solid organ transplantation, among others.
In vitro studies in human hepatocytes also suggest a role for proinflammatory cytokines in the regulation of a wide range of drug transporters other than P-gp, such as OATPs, MRPs, OATs, and OCTs (Le Vee et al., 2008, 2011; Vee et al., 2009). However, in vivo data is lacking, and more studies are needed to explore the role of immune response in the regulation of drug transporters and its effect on the pharmacokinetics of drugs.
Mechanisms of CYP and Transporter Regulation by Cytokines
In response to infections and inflammatory diseases, cytokines like interferons (IFNs), interleukins (IL-1 and IL-6), and TNFα are produced and released from monocytes, macrophages, and stromal cells. The mechanisms by which they affect drug metabolism and transport is not fully understood, but in brief cytokines bind to receptors on the cell surface in target organs and activate intracellular signal systems regulating gene transcription of enzymes and transporters. Such receptors include Toll-like receptors (TLRs), which are presented on the surface of Kupffer cells in the liver and are involved in mediating inflammatory response. In patients with sepsis, TLR2 and TLR4 expression has been found to be significantly up-regulated in several organs (Cinel and Opal, 2009). It has been shown that CYP enzyme expression was regulated by a TLR4-dependent mechanism in a LPS-induced inflammation model (Ghose et al., 2008). Several animal studies have shown that individual CYP enzymes and transporters are down-regulated by cytokines at the level of gene transcription with decreases in mRNA and protein expression (Renton, 2004, 2005; Aitken et al., 2006; Morgan et al., 2008; Roberts and Goralski, 2008; Miller, 2010). The major mechanistic explanation involve the transcription factors pregnane X receptor (PXR) and constitutive androstane receptor (CAR), which both are involved in the expression of genes associated with drug metabolism and transport (Chang and Waxman, 2006).
For CYP3A4 and P-gp transcriptional activation is mediated by PXR and NF-κB (Bentires-Alj et al., 2003; Gu et al., 2006; Kojima et al., 2007). Moreover cytokines have been shown to induce the production of NF-κB, which directly disrupt binding of the PXR–retinoid X receptor (RXR) complex to its response element, leading to suppression of CYP3A4 expression (Gu et al., 2006). Recently suppression of CYP3A4 by IL-6 was shown to occur after the decrease of PXR in human hepatocytes (Yang et al., 2010). Several additional transcription factors may be responsible for regulation of P-gp expression. Heat-shock transcription factor 1 (HSF-1) and stimulatory protein 1 (SP-1) both have binding sites within the ABCB1 promoter (Rohlff and Glazer, 1998; Vilaboa et al., 2000), and in tumor cells P-gp expression is regulated by Y-box protein (YB-1; Ohga et al., 1998).
Studies in human hepatocytes indicate that the effect of various cytokines is gene-specific. While IL-1 down-regulated CYP2C8 and CYP3A4 mRNA expression by 75 and 95%, respectively, there was no effect on CYP2C9 or CYP2C19 (Aitken and Morgan, 2007). IL-6, on the other hand, caused a decrease in CYP2C8, CYP2C9, CYP2C19, and CYP3A4 mRNA expression. Recently IL-6 was also shown to suppress the activities of CYP3A4 and CYP1A2 in human primary hepatocytes, while anti-IL-6 monoclonal antibody partially blocked this suppression (Dickmann et al., 2011). The effects of various cytokines on individual CYP isoenzyme expression and activity investigated in vitro are summarized in Table 1. With respect to P-gp, in vitro studies in human hepatoma cells and human colon carcinoma (Caco-2) cells as well as in vivo studies in mice have shown that IL-6 and IL-2 down-regulate its expression (Piquette-Miller et al., 1998; Hartmann et al., 2001; Belliard et al., 2002; Hosten et al., 2008). On the other hand induction of P-gp by TNF-α or IL-2 has been shown in mice (Hartmann et al., 2001), human lymphocytes (Liptrott et al., 2009), and rat brain capillaries (Bauer et al., 2007). Thus, for P-gp there seems to be organ- and cytokine-specific effects. The response of cytokines on CYP protein expression correlated generally well with the effect on mRNA expression (Aitken and Morgan, 2007), while P-gp expression was strongly decreased with no change in mRNA expression in patients with inflammatory gastrointestinal disorders (Blokzijl et al., 2007).
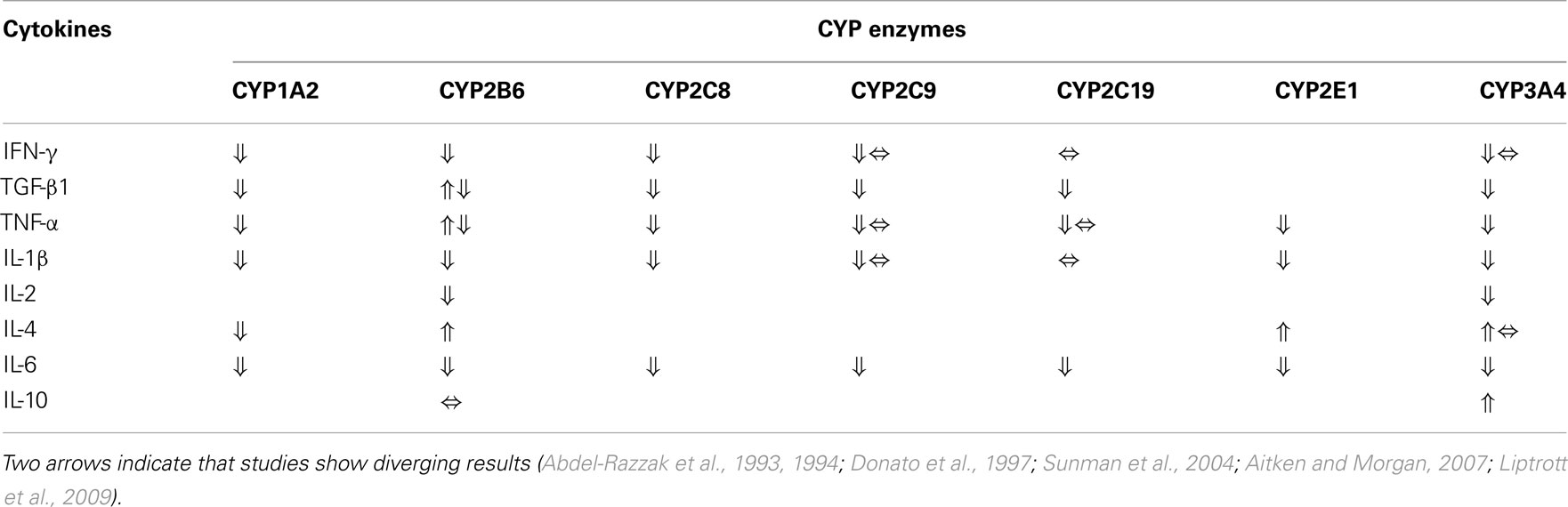
Table 1. Effects of various cytokines on individual drug metabolizing CYP enzyme expression (mRNA or protein) and activity in vitro (no available data for CYP2D6).
Drug Interaction with Therapeutic Proteins
Therapeutic proteins are a group of drugs currently extensively used in the treatment of autoimmune diseases (e.g., rheumatoid arthritis), cancer, and HIV. These drugs include monoclonal antibodies, interferons, and other cytokines among others. Therapeutic proteins are macromolecules, and compared to small-molecule drugs there is still limited knowledge about their pharmacokinetics. For small-molecule drugs problems related to metabolism-based drug–drug interactions have gained extensive attention as a major cause of adverse drug reactions and toxicity problems in general. There are however major differences regarding clearance mechanisms for small-molecule drugs and therapeutic proteins. Proteins are mainly cleared by renal filtration or receptor-mediated clearance, and since they are not metabolized by CYP enzymes, drug–drug interactions involving CYP enzymes have been considered not to be relevant for therapeutic proteins. However, it has recently been clear that these drugs, due to altering the immunological state in patients, can affect the pharmacokinetics of a variety of other drugs by interferences with CYP-mediated metabolism and drug transport (Table 2).
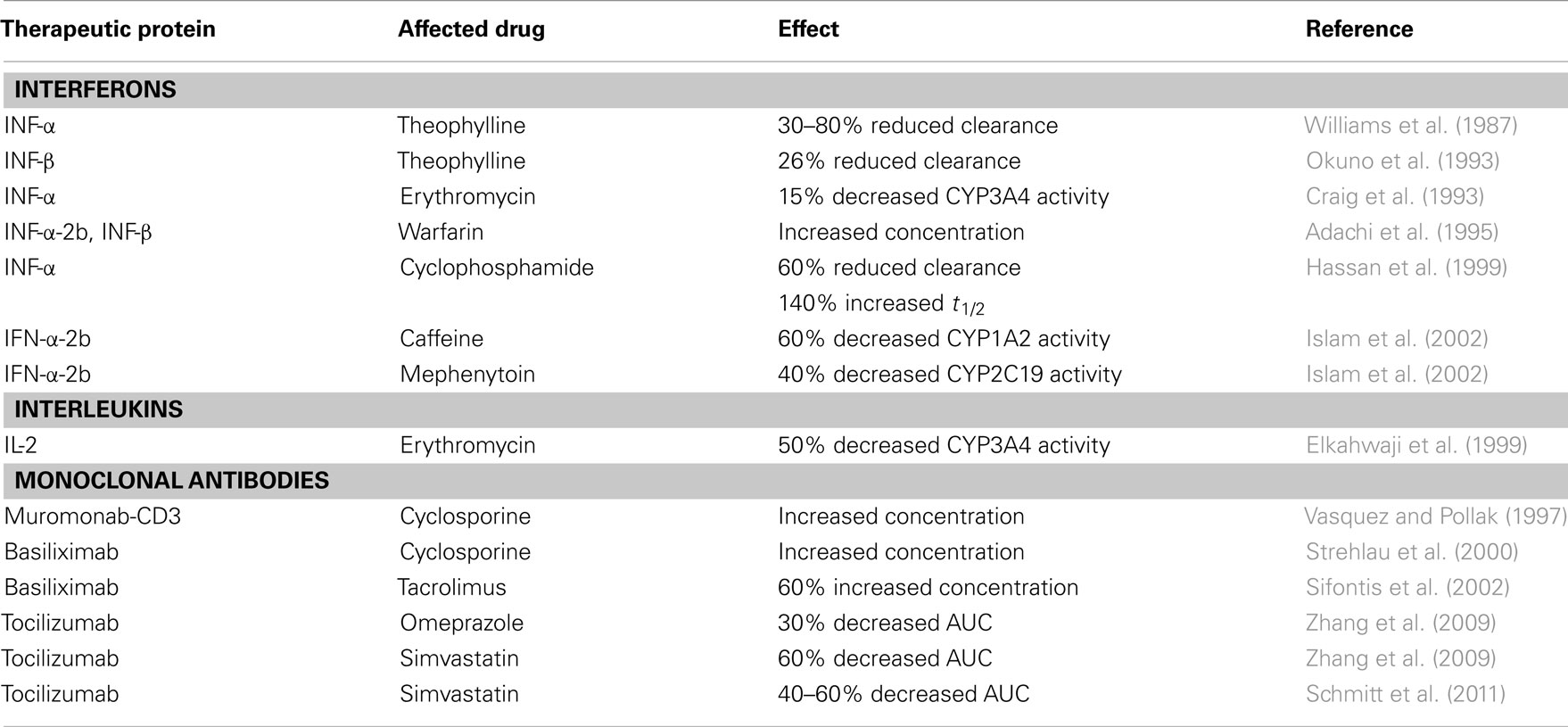
Table 2. Examples of drug interactions caused by therapeutic proteins.
Interferons and Interactions with CYP Metabolism
Interferons (IFNs), produced by the immune system in response to infections and inflammations, have antiviral, antiproliferative, and immunoregulatory effects. INF therapy is extensively used in the treatment of chronic hepatitis C, multiple sclerosis and cancer. In addition to the effect of endogenous cytokines on drug metabolism, therapeutic use of cytokines may therefore additionally contribute to the decreased metabolic ability. Williams et al. (1987) reported already in 1987 that 1 day after a single intramuscular injection of IFN-α in five patients with chronic hepatitis B and four healthy volunteers, clearance of the CYP1A2 substrate theophylline was significantly reduced (30–80%). A 26% reduction in clearance of theophylline was also observed in patients with hepatitis C after IFN-β treatment, with a corresponding increase in terminal half-life of about 40% (Okuno et al., 1993). Additionally IFN-α administration in patients with hepatitis B has been shown to cause a minor decrease of erythromycin metabolism (15%), as determined by ERMBT (Craig et al., 1993), and patients monitored on warfarin needed a dose reduction when IFN-α-2b and IFN-β were given (Adachi et al., 1995).
In cancer patients with multiple myeloma, administration of IFN-α before treatment with cyclophosphamide caused about 60% decreased clearance and 140% increased peak concentration and half-life, accompanied by a decreased concentration of the CYP3A4 metabolite 4-hydroxycyclophosphamide, compared to when IFN-α was administered after cyclophosphamide (Hassan et al., 1999). Also a study in 17 patients with melanoma showed that the activities of CYP1A2 and CYP2C19, measured by the probe drugs caffeine and mephenytoin, were 60 and 40% reduced, respectively, after treatment with high-dose IFN-α-2b (Islam et al., 2002).
On the other hand when administered in lower doses to hepatitis C patients, IFN has been shown to induce a small, statistically non-significant increase in activity of CYP3A4 and CYP2D6 after 1 month of exposure, when administered in combination with ribavirin as antiviral therapy (Becquemont et al., 2002). It is however important to note that these patients had significantly lower pretreatment CYP3A4 and CYP2D6 activities than healthy volunteers. Recently also Gupta et al. (2011) demonstrated that weekly administration of IFN-α-2b to patients with chronic hepatitis C was associated with small increase in CYP2C8/9 and CYP2D6 activities in some individuals, while there was no effect on CYP3A4 activity and a limited inhibitory effect on CYP1A2. IFN-β treatment in patients with multiple sclerosis revealed unaltered CYP2D6 and CYP2C19 activities (Hellman et al., 2003). Several studies have been performed with administration of IFN-α-2b and IFN-α-2a and possible interaction with methadone, which is predominantly metabolized by CYP3A4. A minor increase in methadone exposure in hepatitis C patients after multiple doses of peginterferon-α-2b or peginterferon-α-2a have been reported, but the authors conclude that this may not be of any clinical relevance (Sulkowski et al., 2005; Gupta et al., 2007). However several reports indicate that IFN could cause a clinically relevant interaction when administered with drugs that are CYP substrates, but there might be different effect on the individual CYPs. At least IFN given in high doses for the treatment of cancer seems to decrease the activity of CYP3A4, CYP1A2, and CYP2C19, and it is important to be aware of possible interactions with drugs metabolized through these enzymes. However, a decreased CYP activity due to the disease-state in chronic hepatitis patients may be restored by antiviral therapy involving IFN.
Interleukin and Interactions with CYP Metabolism
Interleukins (ILs) are cytokines mainly synthesized by T lymphocytes, as well as monocytes, macrophages, and endothelial cells. They promote the development and differentiation of T, B, and hematopoietic cells. Therapeutic administration of IL-2 has shown several immunological effects, including activation of cellular immunity and production of cytokines (Winkelhake and Gauny, 1990). Recombinant IL-2 is used to treat advanced cancers (Vlasveld et al., 1992), but there is not much clinical data on the effect of IL-2 on CYP metabolism. However, high-dose administration of IL-2 to patients with liver cancer has been shown to decrease expression of CYP1A2, CYP2C, CYP2E1, and CYP3A4 by approximately 40–60%, and also the CYP1A2 and CYP3A4 activities were 62 and 50% reduced, respectively (Elkahwaji et al., 1999). Thus administration of IL-2 to cancer patients has been proposed to cause clinically important drug interactions (Lee et al., 2010).
Monoclonal Antibodies and Interactions with CYP Metabolism
Human monoclonal antibodies are widely used for treatment of several diseases, e.g., autoimmune diseases (rheumatoid arthritis), cancer, and rejection episodes following transplantation. Since it is now recognized that cytokines induce alterations in CYP metabolism of drugs, it is also evident that cytokine modulators may have an effect on CYP-mediated drug metabolism. However, the prediction of this effect is not straightforward. Use of the monoclonal antibody muromonab-CD3 (OKT3), as antilymphocyte induction therapy, has been shown to cause a significant increase in cyclosporine A (CsA) trough levels in adult renal transplant recipients (Vasquez and Pollak, 1997). The mechanism behind this elevated CsA levels is not known, but it has been hypothesized to be mediated by down-regulation of CYP enzymes by cytokines, because OKT3 administration has been reported to cause cytokine release (especially TNF-α and IFN-γ; Chatenoud et al., 1989). Similarly treatment with the immunosuppressive agent basiliximab, a monoclonal antibody against the IL-2 receptor, in 24 renal transplanted children gave a substantial increase in the whole-blood CsA concentration compared to 15 recipients who received placebo (Strehlau et al., 2000). Also a 63% higher tacrolimus concentration was reported in 12 adult renal transplant recipients 2 days after basiliximab induction therapy compared to eight patients who received antithymocyte globulin therapy (Sifontis et al., 2002). The mechanism for this interaction between basiliximab and CsA or tacrolimus is unclear, but the authors have suggested that the interaction may be mediated via cytokine-induced alterations in CYP3A4 metabolism, when basiliximab binds to the IL-2 receptor on activated T-cells, circulating IL-2 may have an effect on intestinal epithelial cells and hepatocytes and decrease the expression of CYP3A4 (Elkahwaji et al., 1999; Strehlau et al., 2000; Sifontis et al., 2002). Additionally, both tacrolimus and CsA are substrates of P-gp. As P-gp expression is also regulated by cytokines, the observed effect could be caused by a combined effect on CYP3A4 and P-gp.
On the other hand it has recently been shown that treatment with certain monoclonal antibodies can restore cytokine-mediated suppression of hepatic CYP enzymes in inflammatory diseases. The consequence might be therapeutic failure by reduced plasma concentrations of drugs. Tocilizumab, a recombinant human monoclonal antibody directed against the IL-6 receptor, inhibits IL-6 binding to both membrane-expressed and soluble IL-6 receptor (Mihara et al., 2005). Zhang et al. (2009) reported in 2009 that 1 week following administration of a single intravenous dose of tocilizumab the AUCs of omeprazole and simvastatin were about 30 and 60% reduced, indicating an increased activity of CYP2C19 and CYP3A4, respectively. Interestingly the decrease in plasma concentration of simvastatin persisted for 5 weeks. Recently Schmitt et al. (2011) confirmed that tocilizumab administration reversed the IL-6-induced suppression of CYP3A4 activity in rheumatoid arthritis patients. The AUC of simvastatin was approximately 40 and 60% reduced 1 and 5 weeks after tocilizumab treatment, respectively. As a consequence of these recent observations, information about the interaction between tocilizumab and drugs metabolized by CYP enzymes is included in the summary of product characteristics (SPC) of tocilizumab (Box 1). This illustrates a disease–drug interaction, where successful treatment of the disease may result in risk of therapeutic failure, due to increased clearance of drugs metabolized by CYP enzymes.
Box 1. Cited from the SPC of tocilizumab, in the paragraph Interaction with other medicinal products and other forms of interaction (Tocilizumab-SPC)1.
“The expression of hepatic CYP450 enzymes is suppressed by cytokines, such as IL-6, that stimulate chronic inflammation. Thus, CYP450 expression may be reversed when potent cytokine inhibitory therapy, such as tocilizumab, is introduced. In vitro studies with cultured human hepatocytes demonstrated that IL-6 caused a reduction in CYP1A2, CYP2C9, CYP2C19, and CYP3A4 enzyme expression. Tocilizumab normalizes expression of these enzymes. In a study in RA patients, levels of simvastatin (CYP3A4) were decreased by 57% 1 week following a single dose of tocilizumab, to the level similar to, or slightly higher than, those observed in healthy subjects. When starting or stopping therapy with tocilizumab, patients taking medicinal products which are individually adjusted and are metabolized via CYP450 3A4, 1A2, or 2C9 (e.g., atorvastatin, calcium channel blockers, theophylline, warfarin, phenytoin, ciclosporin A, or benzodiazepines) should be monitored as doses may need to be increased to maintain therapeutic effect. Given its long elimination half-life (t1/2), the effect of tocilizumab on CYP450 enzyme activity may persist for several weeks after stopping therapy.”
Therapeutic Proteins and Interactions with Drug Transport
While there has been an increasing awareness on CYP-mediated therapeutic protein-small-molecule drug interactions, the possible effect of therapeutic proteins on drug transporters in humans is far less studied. However, when drug transporter expression is altered by infection and inflammation, one might presume that therapeutic inhibition of infectious or inflammatory stimuli will restore transporter levels and activity and subsequently affect drug disposition. Unfortunately, there is currently not much clinical data in humans to support this hypothesis and only a very limited amount of in vitro studies and in vivo studies in animals.
Ben Reguiga et al. (2005) have studies the effect of IFN-α on the pharmacokinetics of two different P-gp substrates in rats, namely digoxin and docetaxel. IFN-α given daily for 8 days to rats, was found to have a dose-dependent inhibitory effect on intestinal P-gp activity. The pharmacokinetics of oral administered digoxin showed increased bioavailability while i.v. administered digoxin was not affected. Since IFN-α is widely used in cancer therapy and as antiviral therapy, it is combined with anticancer and antiviral drugs, which are P-gp substrates. Increase in bioavailability of P-gp substrates as well as a potential increase in distribution to the brain may therefore lead to increased effect and toxicity problems with these drugs (Goralski et al., 2003). In another study Ben Reguiga et al. (2007) investigated the effect of IFN-α on the pharmacokinetics of another P-gp substrate, namely docetaxel, in rats. They found that the bioavailability of docetaxel was almost threefold increased as compared to control. Additionally, brain levels of docetaxel were increased. Also, in mice, a single injection of IFN-γ resulted in a retardation of plasma elimination of digoxin and increased tissue levels in liver, kidney, and intestine accompanied by reduced excretion into urine and bile. Slight down-regulation of P-gp expression and CYP3A4 expression was also observed (Kawaguchi et al., 2004).
In a recent in vitro study by Chen et al. (2011) OAT2 was the only drug transporter that was down-regulated under the influence of IFN-α2b in human primary hepatocytes, MRP2 was significantly up-regulated, while a panel of other drug efflux and influx transporters were unaffected. These findings are in conflict with several previous reports, but could be a result of species differences.
A clinical study on the interaction between etanercept, an inhibitor of TNF-α, and digoxin concluded on an absence of drug interaction between these drugs (Zhou et al., 2004a). This conclusion could very likely be wrong, as the study was performed in healthy volunteers with no known inflammatory response prior to administration of etanercept. A similar study and conclusion has been performed with the CYP2C9 substrate warfarin (Zhou et al., 2004b). These potentially wrong and harmful conclusions are referred in the SPC of Enbrel where it is stated that “No clinically significant pharmacokinetic drug–drug interactions were observed in studies with digoxin or warfarin” (Etanercept-SPC)2.
Many important questions on drug interactions between therapeutic proteins and small-molecule drugs remain to be investigated. As pointed out by the FDA, future clinical studies need to be designed in order to assure a proper investigation of these issues (Huang et al., 2010).
Clinical Relevance of Immunological Response for Pharmacokinetics
It is thus evident that immunological response in different disease-states as autoimmune diseases, cancer, various infections, and solid organ transplantation contribute to both intra- and inter-individual variability in drug pharmacokinetics, and it might be hypothesized that this underlying mechanism may lead to adverse drug reactions in patients. The effect of endogenous cytokines on drug metabolism may lead to intra-individual variability in pharmacokinetics due to alterations in the disease-state. Additionally, therapeutic use of cytokines may contribute to the decreased metabolic ability. On the other hand, relief of a disease related suppression of drug metabolism by successful treatment may lead to increased metabolism and therapeutic failure of certain drugs.
For most drugs the negative correlation between the activity of inflammatory diseases and CYP activities observed in vivo by use of the cocktail probe approach, indicate a moderate effect of cytokines, i.e., approximately 50% decreased CYP enzyme activity, shifting patients to a slower CYP phenotype in various disease-states. However, there are reports of several-fold increases in drug exposure due to suppression of drug metabolizing enzymes and drug transporters. Also, it must be noted that even a moderate increase in drug exposure, might for some drugs and individuals, be of major clinical relevance. This includes drugs with a narrow therapeutic index and drugs for which clearance depends entirely on one of the metabolic (e.g., theophylline) or transport (e.g., digoxin) routes being affected by cytokines, as well as drugs that are substrates of both CYP enzymes and transporters being regulated by immunological factors (e.g., cyclosporine A). Also, individuals with genetically impaired ability to metabolize by certain CYP enzymes or individuals under treatment with drugs that are strong competitive or mechanism-based inhibitors of certain CYP enzymes, will be especially vulnerable to other sources leading to decreased drug metabolism.
The effect of an immunological response is difficult to predict, since the effect is dependent on the degree or activity of the inflammation and may be changed with successful treatment of the disease. It is also important to mention that the mechanism of regulation of drug metabolizing enzymes and drug transporters by immunological response is complex, as there seems to be cytokine- and organ-specific effects, which obviously complicates the picture. Nevertheless, both treatment with cytokines and anti-cytokines may affect the pharmacokinetics of concomitantly administered drugs (Table 2). While administration of cytokines such as IFNs and ILs may result in depression of CYP activity and consequently increased drug exposure, anti-cytokine treatment may restore the CYP expression and activities, possibly resulting in therapeutic failure of certain drugs.
Alteration in P-gp activity during inflammatory and infectious conditions may have positive or negative clinical implications depending on the disease and drug affected. The recent findings of increased P-gp expression in lymphocytes in autoimmune diseases such as RA and SLE, may be responsible for drug resistance in these patients. It is therefore possible that these patients might benefit from concomitantly treatment with P-gp inhibitors, like tacrolimus or cyclosporine A (Suzuki et al., 2010; Tsujimura and Tanaka, 2011). The effect of inflammatory mediators on P-gp in the brain is still somewhat unclear. However, a possible decreased P-gp activity in the BBB may improve the drug efficacy due to increased concentration within the site of action for treatment of diseases in the CNS.
Conclusion
It is of importance to be aware that disease-states involving cytokines affect the pharmacokinetics of drugs through regulation of expression and activity of drug metabolizing enzymes and drug transporters. The clinical implication is however difficult to predict, since these effects are dependent on the degree of inflammation and may be changed when the diseases are treated. It seems valuable to reconsider the previous impression of drug–drug interactions involving CYP enzymes not being relevant for therapeutic proteins, as both treatment with cytokines and anti-cytokines may affect pharmacokinetics of commonly applied drugs, resulting in altered concentrations in the systemic circulation and also locally at the various sites of action.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Abdel-Razzak, Z., Corcos, L., Fautrel, A., Campion, J. P., and Guillouzo, A. (1994). Transforming growth factor-beta 1 down-regulates basal and polycyclic aromatic hydrocarbon-induced cytochromes P-450 1A1 and 1A2 in adult human hepatocytes in primary culture. Mol. Pharmacol. 46, 1100–1110.
Abdel-Razzak, Z., Loyer, P., Fautrel, A., Gautier, J. C., Corcos, L., Turlin, B., Beaune, P., and Guillouzo, A. (1993). Cytokines down-regulate expression of major cytochrome P-450 enzymes in adult human hepatocytes in primary culture. Mol. Pharmacol. 44, 707–715.
Adachi, Y., Yokoyama, Y., Nanno, T., and Yamamoto, T. (1995). Potentiation of warfarin by interferon. BMJ 311, 292.
Aitken, A. E., and Morgan, E. T. (2007). Gene-specific effects of inflammatory cytokines on cytochrome P450 2C, 2B6 and 3A4 mRNA levels in human hepatocytes. Drug Metab. Dispos. 35, 1687–1693.
Aitken, A. E., Richardson, T. A., and Morgan, E. T. (2006). Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu. Rev. Pharmacol. Toxicol. 46, 123–149.
Anolik, R., Kolski, G. B., Schaible, D. H., and Ratner, J. (1982). Transient alteration of theophylline half-life: possible association with herpes simplex infection. Ann. Allergy 49, 109–111.
Bauer, B., Hartz, A. M., Fricker, G., and Miller, D. S. (2005). Modulation of p-glycoprotein transport function at the blood-brain barrier. Exp. Biol. Med. 230, 118–127.
Bauer, B., Hartz, A. M., and Miller, D. S. (2007). Tumor necrosis factor alpha and endothelin-1 increase P-glycoprotein expression and transport activity at the blood-brain barrier. Mol. Pharmacol. 71, 667–675.
Becquemont, L., Chazouilleres, O., Serfaty, L., Poirier, J. M., Broly, F., Jaillon, P., Poupon, R., and Funck-Brentano, C. (2002). Effect of interferon alpha-ribavirin bitherapy on cytochrome P450 1A2 and 2D6 and N-acetyltransferase-2 activities in patients with chronic active hepatitis C. Clin. Pharmacol. Ther. 71, 488–495.
Belliard, A. M., Tardivel, S., Farinotti, R., Lacour, B., and Leroy, C. (2002). Effect of hr-IL2 treatment on intestinal P-glycoprotein expression and activity in Caco-2 cells. J. Pharm. Pharmacol. 54, 1103–1109.
Ben Reguiga, M., Bonhomme-Faivre, L., and Farinotti, R. (2007). Bioavailability and tissular distribution of docetaxel, a P-glycoprotein substrate, are modified by interferon-alpha in rats. J. Pharm. Pharmacol. 59, 401–408.
Ben Reguiga, M., Bonhomme-Faivre, L., Orbach-Arbouys, S., and Farinotti, R. (2005). Modification of the P-glycoprotein dependent pharmacokinetics of digoxin in rats by human recombinant interferon-alpha. Pharm. Res. 22, 1829–1836.
Benet, L. Z., and Cummins, C. L. (2001). The drug efflux-metabolism alliance: biochemical aspects. Adv. Drug Deliv. Rev. 50(Suppl. 1), S3–S11.
Bentires-Alj, M., Barbu, V., Fillet, M., Chariot, A., Relic, B., Jacobs, N., Gielen, J., Merville, M. P., and Bours, V. (2003). NF-κB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene 22, 90–97.
Blokzijl, H., Vander Borght, S., Bok, L. I., Libbrecht, L., Geuken, M., Van Den Heuvel, F. A., Dijkstra, G., Roskams, T. A., Moshage, H., Jansen, P. L., and Faber, K. N. (2007). Decreased P-glycoprotein (P-gp/MDR1) expression in inflamed human intestinal epithelium is independent of PXR protein levels. Inflamm. Bowel Dis. 13, 710–720.
Chang, K. C., Bell, T. D., Lauer, B. A., and Chai, H. (1978). Altered theophylline pharmacokinetics during acute respiratory viral illness. Lancet 1, 1132–1133.
Chang, T. K., and Waxman, D. J. (2006). Synthetic drugs and natural products as modulators of constitutive androstane receptor (CAR) and pregnane X receptor (PXR). Drug Metab. Rev. 38, 51–73.
Chatenoud, L., Ferran, C., Reuter, A., Legendre, C., Gevaert, Y., Kreis, H., Franchimont, P., and Bach, J. F. (1989). Systemic reaction to the anti-T-cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon-gamma [corrected]. N. Engl. J. Med. 320, 1420–1421.
Chen, C., Han, Y. H., Yang, Z., and Rodrigues, A. D. (2011). Effect of interferon-alpha2b on the expression of various drug-metabolizing enzymes and transporters in co-cultures of freshly prepared human primary hepatocytes. Xenobiotica 41, 476–485.
Chen, Y. L., Le Vraux, V., Leneveu, A., Dreyfus, F., Stheneur, A., Florentin, I., De Sousa, M., Giroud, J. P., Flouvat, B., and Chauvelot-Moachon, L. (1994). Acute-phase response, interleukin-6, and alteration of cyclosporine pharmacokinetics. Clin. Pharmacol. Ther. 55, 649–660.
Cinel, I., and Opal, S. M. (2009). Molecular biology of inflammation and sepsis: a primer. Crit. Care Med. 37, 291–304.
Cordon-Cardo, C., O’Brien, J. P., Casals, D., Rittman-Grauer, L., Biedler, J. L., Melamed, M. R., and Bertino, J. R. (1989). Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. U.S.A. 86, 695–698.
Craig, P. I., Tapner, M., and Farrell, G. C. (1993). Interferon suppresses erythromycin metabolism in rats and human subjects. Hepatology 17, 230–235.
Dickmann, L. J., Patel, S. K., Rock, D. A., Wienkers, L. C., and Slatter, J. G. (2011). Effects of interleukin-6 (IL-6) and an anti-IL-6 monoclonal antibody on drug-metabolizing enzymes in human hepatocyte culture. Drug Metab. Dispos. 39, 1415–1422.
Donato, M. T., Guillen, M. I., Jover, R., Castell, J. V., and Gomez-Lechon, M. J. (1997). Nitric oxide-mediated inhibition of cytochrome P450 by interferon-gamma in human hepatocytes. J. Pharmacol. Exp. Ther. 281, 484–490.
Donnenberg, V. S., Burckart, G. J., Griffith, B. P., Jain, A. B., Zeevi, A., and Berg, A. D. (2001). P-glycoprotein (P-gp) is upregulated in peripheral T-cell subsets from solid organ transplant recipients. J. Clin. Pharmacol. 41, 1271–1279.
Elkahwaji, J., Robin, M. A., Berson, A., Tinel, M., Letteron, P., Labbe, G., Beaune, P., Elias, D., Rougier, P., Escudier, B., Duvillard, P., and Pessayre, D. (1999). Decrease in hepatic cytochrome P450 after interleukin-2 immunotherapy. Biochem. Pharmacol. 57, 951–954.
Forsyth, J. S., Moreland, T. A., and Rylance, G. W. (1982). The effect of fever on antipyrine metabolism in children. Br. J. Clin. Pharmacol. 13, 811–815.
Frye, R. F., Schneider, V. M., Frye, C. S., and Feldman, A. M. (2002). Plasma levels of TNF-alpha and IL-6 are inversely related to cytochrome P450-dependent drug metabolism in patients with congestive heart failure. J. Card. Fail. 8, 315–319.
Ghose, R., White, D., Guo, T., Vallejo, J., and Karpen, S. J. (2008). Regulation of hepatic drug-metabolizing enzyme genes by Toll-like receptor 4 signaling is independent of Toll-interleukin 1 receptor domain-containing adaptor protein. Drug Metab. Dispos. 36, 95–101.
Glassman, A. H., Johnson, L. L., Giardina, E. G., Walsh, B. T., Roose, S. P., Cooper, T. B., and Bigger, J. T. Jr. (1983). The use of imipramine in depressed patients with congestive heart failure. JAMA 250, 1997–2001.
Goralski, K. B., Hartmann, G., Piquette-Miller, M., and Renton, K. W. (2003). Downregulation of mdr1a expression in the brain and liver during CNS inflammation alters the in vivo disposition of digoxin. Br. J. Pharmacol. 139, 35–48.
Gray, J. D., Renton, K. W., and Hung, O. R. (1983). Depression of theophylline elimination following BCG vaccination. Br. J. Clin. Pharmacol. 16, 735–737.
Gu, X., Ke, S., Liu, D., Sheng, T., Thomas, P. E., Rabson, A. B., Gallo, M. A., Xie, W., and Tian, Y. (2006). Role of NF-kappaB in regulation of PXR-mediated gene expression: a mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J. Biol. Chem. 281, 17882–17889.
Gupta, S. K., Kolz, K., and Cutler, D. L. (2011). Effects of multiple-dose pegylated interferon alfa-2b on the activity of drug-metabolizing enzymes in persons with chronic hepatitis C. Eur. J. Clin. Pharmacol. 67, 591–599.
Gupta, S. K., Sellers, E., Somoza, E., Angles, L., Kolz, K., and Cutler, D. L. (2007). The effect of multiple doses of peginterferon alfa-2b on the steady-state pharmacokinetics of methadone in patients with chronic hepatitis C undergoing methadone maintenance therapy. J. Clin. Pharmacol. 47, 604–612.
Gutmann, H., Hruz, P., Zimmermann, C., Straumann, A., Terracciano, L., Hammann, F., Lehmann, F., Beglinger, C., and Drewe, J. (2008). Breast cancer resistance protein and P-glycoprotein expression in patients with newly diagnosed and therapy-refractory ulcerative colitis compared with healthy controls. Digestion 78, 154–162.
Hartmann, G., Kim, H., and Piquette-Miller, M. (2001). Regulation of the hepatic multidrug resistance gene expression by endotoxin and inflammatory cytokines in mice. Int. Immunopharmacol. 1, 189–199.
Hartz, A. M., Bauer, B., Fricker, G., and Miller, D. S. (2006). Rapid modulation of P-glycoprotein-mediated transport at the blood-brain barrier by tumor necrosis factor-alpha and lipopolysaccharide. Mol. Pharmacol. 69, 462–470.
Hassan, M., Nilsson, C., Olsson, H., Lundin, J., and Osterborg, A. (1999). The influence of interferon-alpha on the pharmacokinetics of cyclophosphamide and its 4-hydroxy metabolite in patients with multiple myeloma. Eur. J. Haematol. 63, 163–170.
Hayney, M. S., Hammes, R. J., Fine, J. P., and Bianco, J. A. (2001). Effect of influenza immunization on CYP3A4 activity. Vaccine 20, 858–861.
Hayney, M. S., and Muller, D. (2003). Effect of influenza immunization on CYP3A4 activity in vivo. J. Clin. Pharmacol. 43, 1377–1381.
Hellman, K., Roos, E., Osterlund, A., Wahlberg, A., Gustafsson, L. L., Bertilsson, L., and Fredrikson, S. (2003). Interferon-beta treatment in patients with multiple sclerosis does not alter CYP2C19 or CYP2D6 activity. Br. J. Clin. Pharmacol. 56, 337–340.
Higgins, C. F. (1992). ABC transporters: from microorganisms to man. Annu. Rev. Cell Biol. 8, 67–113.
Hosten, B., Abbara, C., Petit, B., Dauvin, A., Bourasset, F., Farinotti, R., Gonin, P., and Bonhomme-Faivre, L. (2008). Effect of interleukin-2 pretreatment on paclitaxel absorption and tissue disposition after oral and intravenous administration in mice. Drug Metab. Dispos. 36, 1729–1735.
Huang, S. M., Zhao, H., Lee, J. I., Reynolds, K., Zhang, L., Temple, R., and Lesko, L. J. (2010). Therapeutic protein-drug interactions and implications for drug development. Clin. Pharmacol. Ther. 87, 497–503.
Islam, M., Frye, R. F., Richards, T. J., Sbeitan, I., Donnelly, S. S., Glue, P., Agarwala, S. S., and Kirkwood, J. M. (2002). Differential effect of IFNalpha-2b on the cytochrome P450 enzyme system: a potential basis of IFN toxicity and its modulation by other drugs. Clin. Cancer Res. 8, 2480–2487.
Juliano, R. L., and Ling, V. (1976). A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 455, 152–162.
Kantharidis, P., El-Osta, S., Silva, M., Lee, G., Hu, X. F., and Zalcberg, J. (2000). Regulation of MDR1 gene expression: emerging concepts. Drug Resist. Updat. 3, 99–108.
Kawaguchi, H., Matsui, Y., Watanabe, Y., and Takakura, Y. (2004). Effect of interferon-gamma on the pharmacokinetics of digoxin, a P-glycoprotein substrate, intravenously injected into the mouse. J. Pharmacol. Exp. Ther. 308, 91–96.
Kim, R. B., and Wilkinson, G. R. (1996). CYP2E1 activity is not altered by influenza vaccination. Br. J. Clin. Pharmacol. 42, 529–530.
Klimecki, W. T., Futscher, B. W., Grogan, T. M., and Dalton, W. S. (1994). P-glycoprotein expression and function in circulating blood cells from normal volunteers. Blood 83, 2451–2458.
Kojima, K., Nagata, K., Matsubara, T., and Yamazoe, Y. (2007). Broad but distinct role of pregnane X receptor on the expression of individual cytochrome p450s in human hepatocytes. Drug Metab. Pharmacokinet. 22, 276–286.
Kraemer, M. J., Furukawa, C. T., Koup, J. R., Shapiro, G. G., Pierson, W. E., and Bierman, C. W. (1982). Altered theophylline clearance during an influenza B outbreak. Pediatrics 69, 476–480.
Kramer, P., and McClain, C. J. (1981). Depression of aminopyrine metabolism by influenza vaccination. N. Engl. J. Med. 305, 1262–1264.
Langford, D., Grigorian, A., Hurford, R., Adame, A., Ellis, R. J., Hansen, L., and Masliah, E. (2004). Altered P-glycoprotein expression in AIDS patients with HIV encephalitis. J. Neuropathol. Exp. Neurol. 63, 1038–1047.
Le Vee, M., Gripon, P., Stieger, B., and Fardel, O. (2008). Down-regulation of organic anion transporter expression in human hepatocytes exposed to the proinflammatory cytokine interleukin 1beta. Drug Metab. Dispos. 36, 217–222.
Le Vee, M., Jouan, E., Stieger, B., Lecureur, V., and Fardel, O. (2011). Regulation of drug transporter expression by oncostatin M in human hepatocytes. Biochem. Pharmacol. 82, 304–311.
Lee, B. L., Wong, D., Benowitz, N. L., and Sullam, P. M. (1993). Altered patterns of drug metabolism in patients with acquired immunodeficiency syndrome. Clin. Pharmacol. Ther. 53, 529–535.
Lee, J. I., Zhang, L., Men, A. Y., Kenna, L. A., and Huang, S. M. (2010). CYP-mediated therapeutic protein-drug interactions: clinical findings, proposed mechanisms and regulatory implications. Clin. Pharmacokinet. 49, 295–310.
Lee, S. H., and Lee, S. M. (2005). Suppression of hepatic cytochrome p450-mediated drug metabolism during the late stage of sepsis in rats. Shock 23, 144–149.
Lin, J. H., and Lu, A. Y. (2001). Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu. Rev. Pharmacol. Toxicol. 41, 535–567.
Liptrott, N. J., and Owen, A. (2011). The role of cytokines in the regulation of drug disposition: extended functional pleiotropism? Expert Opin. Drug Metab. Toxicol. 7, 341–352.
Liptrott, N. J., Penny, M., Bray, P. G., Sathish, J., Khoo, S. H., Back, D. J., and Owen, A. (2009). The impact of cytokines on the expression of drug transporters, cytochrome P450 enzymes and chemokine receptors in human PBMC. Br. J. Pharmacol. 156, 497–508.
Mayo, P. R., Skeith, K., Russell, A. S., and Jamali, F. (2000). Decreased dromotropic response to verapamil despite pronounced increased drug concentration in rheumatoid arthritis. Br. J. Clin. Pharmacol. 50, 605–613.
Meredith, C. G., Christian, C. D., Johnson, R. F., Troxell, R., Davis, G. L., and Schenker, S. (1985). Effects of influenza virus vaccine on hepatic drug metabolism. Clin. Pharmacol. Ther. 37, 396–401.
Mihara, M., Kasutani, K., Okazaki, M., Nakamura, A., Kawai, S., Sugimoto, M., Matsumoto, Y., and Ohsugi, Y. (2005). Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int. Immunopharmacol. 5, 1731–1740.
Miller, D. S. (2010). Regulation of P-glycoprotein and other ABC drug transporters at the blood-brain barrier. Trends Pharmacol. Sci. 31, 246–254.
Miller, D. S., Bauer, B., and Hartz, A. M. (2008). Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol. Rev. 60, 196–209.
Morgan, E. T. (2009). Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin. Pharmacol. Ther. 85, 434–438.
Morgan, E. T., Goralski, K. B., Piquette-Miller, M., Renton, K. W., Robertson, G. R., Chaluvadi, M. R., Charles, K. A., Clarke, S. J., Kacevska, M., Liddle, C., Richardson, T. A., Sharma, R., and Sinal, C. J. (2008). Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab. Dispos. 36, 205–216.
Ohga, T., Uchiumi, T., Makino, Y., Koike, K., Wada, M., Kuwano, M., and Kohno, K. (1998). Direct involvement of the Y-box binding protein YB-1 in genotoxic stress-induced activation of the human multidrug resistance 1 gene. J. Biol. Chem. 273, 5997–6000.
Okuno, H., Takasu, M., Kano, H., Seki, T., Shiozaki, Y., and Inoue, K. (1993). Depression of drug-metabolizing activity in the human liver by interferon-beta. Hepatology 17, 65–69.
O’Neil, W. M., Gilfix, B. M., Markoglou, N., Di Girolamo, A., Tsoukas, C. M., and Wainer, I. W. (2000). Genotype and phenotype of cytochrome P450 2D6 in human immunodeficiency virus-positive patients and patients with acquired immunodeficiency syndrome. Eur. J. Clin. Pharmacol. 56, 231–240.
Paine, M. F., Hart, H. L., Ludington, S. S., Haining, R. L., Rettie, A. E., and Zeldin, D. C. (2006). The human intestinal cytochrome P450 “pie.” Drug Metab. Dispos. 34, 880–886.
Paine, M. F., Khalighi, M., Fisher, J. M., Shen, D. D., Kunze, K. L., Marsh, C. L., Perkins, J. D., and Thummel, K. E. (1997). Characterization of interintestinal and intraintestinal variations in human CYP3A-dependent metabolism. J. Pharmacol. Exp. Ther. 283, 1552–1562.
Pasanen, M., Rannala, Z., Tooming, A., Sotaniemi, E. A., Pelkonen, O., and Rautio, A. (1997). Hepatitis A impairs the function of human hepatic CYP2A6 in vivo. Toxicology 123, 177–184.
Piquette-Miller, M., Pak, A., Kim, H., Anari, R., and Shahzamani, A. (1998). Decreased expression and activity of P-glycoprotein in rat liver during acute inflammation. Pharm. Res. 15, 706–711.
Renton, K. W. (2004). Cytochrome P450 regulation and drug biotransformation during inflammation and infection. Curr. Drug Metab. 5, 235–243.
Renton, K. W. (2005). Regulation of drug metabolism and disposition during inflammation and infection. Expert Opin. Drug Metab. Toxicol. 1, 629–640.
Renton, K. W., Gray, J. D., and Hall, R. I. (1980). Decreased elimination of theophylline after influenza vaccination. Can. Med. Assoc. J. 123, 288–290.
Renton, K. W., and Mannering, G. J. (1976a). Depression of hepatic cytochrome P-450-dependent monooxygenase systems with administered interferon inducing agents. Biochem. Biophys. Res. Commun. 73, 343–348.
Renton, K. W., and Mannering, G. J. (1976b). Depression of the hepatic cytochrome P-450 mono-oxygenase system by administered tilorone (2,7-bis(2-(diethylamino)ethoxy)fluoren-9-one dihydrochloride). Drug Metab. Dispos. 4, 223–231.
Roberts, D. J., and Goralski, K. B. (2008). A critical overview of the influence of inflammation and infection on P-glycoprotein expression and activity in the brain. Expert Opin. Drug Metab. Toxicol. 4, 1245–1264.
Roberts, D. J., Goralski, K. B., Renton, K. W., Julien, L. C., Webber, A. M., Sleno, L., Volmer, D. A., and Hall, R. I. (2009). Effect of acute inflammatory brain injury on accumulation of morphine and morphine 3- and 6-glucuronide in the human brain. Crit. Care Med. 37, 2767–2774.
Rohlff, C., and Glazer, R. I. (1998). Regulation of the MDR1 promoter by cyclic AMP-dependent protein kinase and transcription factor Sp1. Int. J. Oncol. 12, 383–386.
Schmitt, C., Kuhn, B., Zhang, X., Kivitz, A. J., and Grange, S. (2011). Disease-drug-drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin. Pharmacol. Ther. 89, 735–740.
Seelbach, M. J., Brooks, T. A., Egleton, R. D., and Davis, T. P. (2007). Peripheral inflammatory hyperalgesia modulates morphine delivery to the brain: a role for P-glycoprotein. J. Neurochem. 102, 1677–1690.
Shedlofsky, S. I., Israel, B. C., Mcclain, C. J., Hill, D. B., and Blouin, R. A. (1994). Endotoxin administration to humans inhibits hepatic cytochrome P450-mediated drug metabolism. J. Clin. Invest. 94, 2209–2214.
Shedlofsky, S. I., Israel, B. C., Tosheva, R., and Blouin, R. A. (1997). Endotoxin depresses hepatic cytochrome P450-mediated drug metabolism in women. Br. J. Clin. Pharmacol. 43, 627–632.
Shimada, T., Yamazaki, H., Mimura, M., Inui, Y., and Guengerich, F. P. (1994). Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 270, 414–423.
Shtil, A. A. (2002). Emergence of multidrug resistance in leukemia cells during chemotherapy: mechanisms and prevention. J. Hematother. Stem Cell Res. 11, 231–241.
Sifontis, N. M., Benedetti, E., and Vasquez, E. M. (2002). Clinically significant drug interaction between basiliximab and tacrolimus in renal transplant recipients. Transplant. Proc. 34, 1730–1732.
Strehlau, J., Pape, L., Offner, G., Nashan, B., and Ehrich, J. H. (2000). Interleukin-2 receptor antibody-induced alterations of ciclosporin dose requirements in paediatric transplant recipients. Lancet 356, 1327–1328.
Sugawara, I., Kataoka, I., Morishita, Y., Hamada, H., Tsuruo, T., Itoyama, S., and Mori, S. (1988). Tissue distribution of P-glycoprotein encoded by a multidrug-resistant gene as revealed by a monoclonal antibody, MRK 16. Cancer Res. 48, 1926–1929.
Sulkowski, M., Wright, T., Rossi, S., Arora, S., Lamb, M., Wang, K., Gries, J. M., and Yalamanchili, S. (2005). Peginterferon alfa-2a does not alter the pharmacokinetics of methadone in patients with chronic hepatitis C undergoing methadone maintenance therapy. Clin. Pharmacol. Ther. 77, 214–224.
Sunman, J. A., Hawke, R. L., Lecluyse, E. L., and Kashuba, A. D. (2004). Kupffer cell-mediated IL-2 suppression of CYP3A activity in human hepatocytes. Drug Metab. Dispos. 32, 359–363.
Suzuki, K., Saito, K., Tsujimura, S., Nakayamada, S., Yamaoka, K., Sawamukai, N., Iwata, S., Nawata, M., Nakano, K., and Tanaka, Y. (2010). Tacrolimus, a calcineurin inhibitor, overcomes treatment unresponsiveness mediated by P-glycoprotein on lymphocytes in refractory rheumatoid arthritis. J. Rheumatol. 37, 512–520.
Thiebaut, F., Tsuruo, T., Hamada, H., Gottesman, M. M., Pastan, I., and Willingham, M. C. (1987). Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. U.S.A. 84, 7735–7738.
Thummel, K. E., Shen, D. D., Podoll, T. D., Kunze, K. L., Trager, W. F., Bacchi, C. E., Marsh, C. L., Mcvicar, J. P., Barr, D. M., Perkins, J. D., and Carithers, R. L. Jr. (1994). Use of midazolam as a human cytochrome P450 3A probe: II. Characterization of inter- and intraindividual hepatic CYP3A variability after liver transplantation. J. Pharmacol. Exp. Ther. 271, 557–566.
Tsujimura, S., Saito, K., Nakayamada, S., Nakano, K., Tsukada, J., Kohno, K., and Tanaka, Y. (2004). Transcriptional regulation of multidrug resistance-1 gene by interleukin-2 in lymphocytes. Genes Cells 9, 1265–1273.
Tsujimura, S., and Tanaka, Y. (2011). Treatment strategy based on targeting P-glycoprotein on peripheral lymphocytes in patients with systemic autoimmune disease. Clin. Exp. Nephrol. PMID: 21847519. [Epub ahead of print].
Ufer, M., Hasler, R., Jacobs, G., Haenisch, S., Lachelt, S., Faltraco, F., Sina, C., Rosenstiel, P., Nikolaus, S., Schreiber, S., and Cascorbi, I. (2009). Decreased sigmoidal ABCB1 (P-glycoprotein) expression in ulcerative colitis is associated with disease activity. Pharmacogenomics 10, 1941–1953.
Vasquez, E. M., and Pollak, R. (1997). OKT3 therapy increases cyclosporine blood levels. Clin. Transplant. 11, 38–41.
Vee, M. L., Lecureur, V., Stieger, B., and Fardel, O. (2009). Regulation of drug transporter expression in human hepatocytes exposed to the proinflammatory cytokines tumor necrosis factor-alpha or interleukin-6. Drug Metab. Dispos. 37, 685–693.
Vilaboa, N. E., Galan, A., Troyano, A., De Blas, E., and Aller, P. (2000). Regulation of multidrug resistance 1 (MDR1)/P-glycoprotein gene expression and activity by heat-shock transcription factor 1 (HSF1). J. Biol. Chem. 275, 24970–24976.
Vlasveld, L. T., Rankin, E. M., Hekman, A., Rodenhuis, S., Beijnen, J. H., Hilton, A. M., Dubbelman, A. C., Vyth-Dreese, F. A., and Melief, C. J. (1992). A phase I study of prolonged continuous infusion of low dose recombinant interleukin-2 in melanoma and renal cell cancer. Part I: clinical aspects. Br. J. Cancer 65, 744–750.
Wilkinson, G. R. (2005). Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 352, 2211–2221.
Williams, M. L., Bhargava, P., Cherrouk, I., Marshall, J. L., Flockhart, D. A., and Wainer, I. W. (2000). A discordance of the cytochrome P450 2C19 genotype and phenotype in patients with advanced cancer. Br. J. Clin. Pharmacol. 49, 485–488.
Williams, S. J., Baird-Lambert, J. A., and Farrell, G. C. (1987). Inhibition of theophylline metabolism by interferon. Lancet 2, 939–941.
Winkelhake, J. L., and Gauny, S. S. (1990). Human recombinant interleukin-2 as an experimental therapeutic. Pharmacol. Rev. 42, 1–28.
Yang, J., Hao, C., Yang, D., Shi, D., Song, X., Luan, X., Hu, G., and Yan, B. (2010). Pregnane X receptor is required for interleukin-6-mediated down-regulation of cytochrome P450 3A4 in human hepatocytes. Toxicol. Lett. 197, 219–226.
Zhang, X., Schmitt, C., Grange, S., Terao, K., Miya, K., Kivitz, A., and Marino, M. (2009). Disease-drug interaction studies of tocilizumab with cytochrome P450 substrates in vitro and in vivo. Clin. Pharmacol. Ther. 85, S59–S59.
Zhou, H., Parks, V., Patat, A., Le Coz, F., Simcoe, D., and Korth-Bradley, J. (2004a). Absence of a clinically relevant interaction between etanercept and digoxin. J. Clin. Pharmacol. 44, 1244–1251.
Keywords: cytokines, cytochrome P450 enzymes, P-glycoprotein, therapeutic proteins
Citation: Christensen H and Hermann M (2012) Immunological response as a source to variability in drug metabolism and transport. Front. Pharmacol. 3:8. doi: 10.3389/fphar.2012.00008
Received: 21 December 2011; Paper pending published: 06 January 2012;
Accepted: 16 January 2012; Published online: 10 February 2012.
Edited by:
Jaime Kapitulnik, The Hebrew University of Jerusalem, IsraelReviewed by:
Gert Fricker, University of Heidelberg, GermanyStanislav Yanev, Bulgarian Academy of Sciences, Bulgaria
Copyright: © 2012 Christensen and Hermann. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Hege Christensen, Department of Pharmaceutical Biosciences, School of Pharmacy, University of Oslo, P.O. Box 1068 Blindern, N-0316 Oslo, Norway. e-mail: h.s.christensen@farmasi.uio.no