


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 13 January 2012
Sec. Neuropharmacology
volume 2 - 2011 | https://doi.org/10.3389/fphar.2011.00096
This article is part of the Research Topic The link between brain energy homeostasis and neuronal activity View all 8 articles
Ever since it was shown for the first time that lactate can support neuronal function in vitro as a sole oxidative energy substrate, investigators in the field of neuroenergetics have been debating the role, if any, of this glycolytic product in cerebral energy metabolism. Our experiments employed the rat hippocampal slice preparation with electrophysiological and biochemical methodologies. The data generated by these experiments (a) support the hypothesis that lactate, not pyruvate, is the end-product of cerebral aerobic glycolysis; (b) indicate that lactate plays a major and crucial role in affording neural tissue to respond adequately to glutamate excitation and to recover unscathed post-excitation; (c) suggest that neural tissue activation is accompanied by aerobic lactate and NADH production, the latter being produced when the former is converted to pyruvate by mitochondrial lactate dehydrogenase (mLDH); (d) imply that NADH can be utilized as an endogenous scavenger of reactive oxygen species (ROS) to provide neuroprotection against ROS-induced neuronal damage.
Almost a quarter of century ago we demonstrated for the first time the ability of cerebral tissue in vitro to sustain normal neuronal function by utilizing lactate aerobically as its sole energy substrate (Schurr et al., 1988). The debate that ensued following this discovery on whether or not lactate plays a major role in cerebral energy metabolism continues, although much more evidence in favor of such a role has been accumulated over that period (Gladden, 2004; Schurr, 2006; Pellerin et al., 2007; Mangia et al., 2009; Zielke et al., 2009; Barros and Deitmer, 2010; Ivanov et al., 2011; Wyss et al., 2011). It is important to realize that lactate has never been suggested to replace glucose as the main energy substrate, but rather to play an important, maybe even a crucial role, especially in specific situations. However, for many who still hold that lactate is simply the end-product of anaerobic glycolysis, it is difficult to visualize this monocarboxylate as a major player in cerebral energy metabolism. Consequently, the debate over lactate role in energy metabolism continues unabated for almost two decades.
The findings of Pellerin and Magistretti (1994) and the hypothesis they have offered to explain them, i.e., the astrocytic-neuronal lactate shuttle hypothesis (ANLSH), have been supported by many studies over the years (Hu and Wilson, 1997a; Schurr et al., 1997a,b, 1999a,b; Mangia et al., 2003; Kasischke et al., 2004; Schurr, 2006; Atlante et al., 2007; Schurr and Payne, 2007; Larsen et al., 2008; Passarella et al., 2008; Gallagher et al., 2009). Those who disagree with the ANLSH quarrel mainly about the central role attributed to lactate in this hypothesis and the studies supporting it (Chih et al., 2001; Chih and Roberts, 2003; Hertz, 2004; Dienel and Hertz, 2005; Korf, 2006; Hertz et al., 2007; Gandhi et al., 2009). While glucose is, indisputably, the only energy substrate that can enter glycolysis, the fate of glucose in this pathway is in dispute. Many continue to hold that aerobic glycolysis’s end-product is pyruvate, the monocarboxylate that is the substrate for the mitochondrial tricarboxylic acid cycle, while lactate is glycolysis’s end-product only under anaerobic conditions. Accordingly, an increase in oxygen supply should occur to allow for the increase in energy demands upon neural activation. Hence, the detractors of lactate’s role as an energy substrate during neural activation claim that an increase in oxygen supply could be achieved simply by an increase in cerebral blood circulation. Although an increase in microcirculation at the activation region has been correlated with a spike in oxygen consumption (Malonek and Grinvald, 1996; Vanzetta and Grinvald, 1999) and with a similar spike in NADH level measured in vitro (Kasischke et al., 2004), these measurements did not reveal the identity of the oxidized energy substrate. Nevertheless, the prevailing assumption of those who discount the role of lactate is that as long as there are sufficient supplies of oxygen and glucose, glycolysis should produce enough pyruvate to satisfy the heightened energy demands upon activation. This scenario does not take into account the fact that glucose is incapable of increasing glycolytic flux and thus of up-regulating VO2, while lactate is capable of producing a dose-dependent increase in VO2 (Levasseur et al., 2006). The study by Hu and Wilson (1997b) actually alludes to this very phenomenon. Thus, we afforded it a closer reexamination in light of the discount and criticism it received so far (Dienel and Hertz, 2005; Fillenz, 2005; Korf, 2006). Hence, in aiming to clarify some of the contentious issues that continue to divide investigators in the field of cerebral energy metabolism, we consider here several of our own studies and postulates, including one unpublished study, along with the study of Hu and Wilson (1997b). These considerations have led us to contend that most of the disagreements as to lactate’s role in cerebral energy metabolism could be easily settled if and when lactate is “allowed” to be the only end-product of glycolysis, both aerobic and anaerobic.
The experimental portions of the studies already published by us and that are part of the considerations and elaborations herewith can be found in the respective papers. All the experiments in those studies and in the unpublished one we are detailing here employed the rat hippocampal slice preparation, using electrophysiological and biochemical modalities. Therefore, the methods described below apply to the unpublished data on the role of lactate in neuroprotection against glutamate-mediated, reactive oxygen species (ROS)-induced neuronal damage, the results of which are shown in Figures 8–10. Adult (200–350 g) male Sprague-Dawley rats were used with diligent attention to minimizing animal stress. Rats were housed in the animal facility at the University of Louisville School of Medicine, maintained on a 12 h light/12 h dark cycle at 22°C and had unlimited access to food and water. All protocols were approved by the University of Louisville School of Medicine Institutional Animal Care and Use Committee. For each experiment, one rat was decapitated under ether anesthesia, its brain rapidly removed and rinsed with cold (6–8°C) artificial cerebrospinal fluid (aCSF, see composition below) and dissected. Isolated hippocampi were sliced transversely at 400 μm with a McIlwain tissue chopper and the resulting slices (10–12 slices per hippocampus) were placed in a dual linear-flow incubation/recording chamber (Schurr et al., 1985). Each of the two compartments of the chamber was supplied with a humidified gas mixture (95% O2/5% CO2) through separate flow meters (2 l/min) and aCSF via a dual peristaltic pump (1 ml/min). The aCSF composition was (in mM): NaCl 124; KCl 5; NaH2PO4 3; CaCl2 2.5; MgSO4 2; NaHCO3 23; D-glucose 10. The aCSF had a pH of 7.3–7.4 and an osmolality of 300 ± 5 mOSM. The temperature of the incubation chamber was held at 34 ± 0.3°C. After 60 min of incubation, the 10 mM glucose aCSF was replaced either with 2.5 mM glucose-, 5 mM Na-lactate-, or 5 mM Na-pyruvate-aCSF. A 45 min period of equilibration with the replacement medium was allowed before the beginning of electrophysiological recordings of evoked (once/min) CA1 population spike (PS, neuronal function). Each experimental protocol lasted 60 min. Glutamate (Glut; 2.5 mM) and 3-methyl-1-phenyl-2-pyrazolin-5-one (MCI-186, 33 μM), a free radical scavenger (Wu et al., 2006) were supplemented when indicated. In other experiments, 5 mM pyruvate-aCSF was replaced with either (1 mM lactate + 4 mM pyruvate)-aCSF or (0.5 mM glucose + 4 mM pyruvate)-aCSF.
Continuous (45 min) extracellular recordings of electrically evoked PS in the stratum pyramidale of the hippocampal CA1 region were made from one slice in each compartment of the dual chamber using borosilicate micropipettes filled with aCSF (impedance 2–5 MΩ). A two-channel preamplifier (×100) and two field-effect transistor head stages (custom-made) were used. Bipolar stimulating electrodes were placed in the Schaffer collaterals (orthodromic stimulation) and stimulus pulses (0.1 ms in duration) of an amplitude twice threshold (~10 V) were applied once/min to evoke a response, using two Grass S44 stimulators and two SIU-V constant voltage stimulus isolation units (Grass Technologies, West Warwick, RI, USA). The presence of neuronal function (PS of >3 mV) in each of the slices in both compartments of the dual chamber was verified prior to the beginning of the experimental protocol. The above value of the PS has been determined as our standard, based on hundreds of experiments over 25 years, as the minimum amplitude of a PS that can be accurately measured to indicate the presence of neuronal function that could safely be distinguished from an artifact. Nevertheless, the majority of viable, neuronally functional, slices typically exhibited the maximal PS amplitude in response to the chosen electrical stimulation (8–10 mV). After 45 min of continuous recording in one slice in each compartment, the rest of the slices were tested for the presence and amplitude of their PS by stimulating the Schaffer collaterals and recording the evoked response in the CA1 cell body layer, a test that lasted 10–15 min. Acquisition, analysis and storage of data was done using the CED 1401 mk II data acquisition system with Spike 2 software package (Cambridge Electronic Design corporation, Cambridge, UK). All chemicals were of analytical grade (Sigma Chemical, St. Louis, MO, USA). Each experiment (condition) was repeated three times (30–36 slices). Separate experiments (not shown here) were carried out to determine the optimal concentration of glucose, lactate, pyruvate, Glut, and MCI-186 used in this study.
Lactate and glucose were measured using the enzymatic kits of Sigma Chemical Co. (St. Louise, MO, USA). Slices were taken out of the incubation-recording chamber at the time points indicated, rinsed in ice-cold aCSF containing no glucose and homogenized in 0.2 ml of 8% perchloric acid. The homogenate was then neutralized with 0.1 ml of 2 M KHCO3 and centrifuged for 3 min at 8000 × g. The supernatant (0.1 ml) was used for the analysis of both lactate and glucose. Assays were automated on the Cobas Fara centrifugal analyzer (Roche Diagnostic Systems, Branchburg, NJ, USA). The lactate kit (826-UV) uses lactate dehydrogenase to convert lactate and NAD+ to pyruvate and NADH, respectively. The glucose kit uses hexokinase to convert glucose and ATP to glucose-6-phosphate and ADP. Glucose-6-phosphate is subsequently oxidized by glucose-6-phosphate dehydrogenase and NAD+ to 6-phosphogluconate and NADH. In both assays, NADH was measured fluorometrically (excitation at 340 nm and emission at 450 nm).
Reactive oxygen species generation was assessed using the fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA; Invitrogen, Carlsbad, CA, USA) as described by Liang et al. (2005) with small modifications. The non-fluorescent H2DCFDA, which crosses the cell membrane, is deacetylated intracellularly by non-specific endogenous esterases to its membrane impermeable form, dichlorodihydrofluorescein (H2DCF). H2DCF, when oxidized by ROS, is converted to the fluorescent product dichlorofluorescein (DCF). Briefly, for each experimental sample, five slices were perfused (34°C) for 45 min with aCSF containing 20 μM H2DCFDA and the selected energy substrate (pyruvate or lactate) with or without Glut. After 45 min, slices were perfused for an additional 10 min with their respective aCSF without H2DCFDA, then rinsed in cold aCSF and homogenized in 1 ml cold 0 mM glucose aCSF containing 40 μl of the detergent Non-idet-P40 (Roche Applied Science, Indianapolis, IN, USA). Homogenates were then spun for 20 min at 12,000 rpm (13.4 g) in 4°C. All steps and procedures of the experiments involving H2DCFDA were performed in the dark. Excitation at 485 nm and emission at 535 nm was read using a SpectraFluor Plus spectrophotometer (Tecan, San Jose, CA, USA). Fluorescent readings from slices incubated with pyruvate (5 mM) and Glut (2.5 mM) were considered to yield the maximum DCF fluorescence level (100%; control). H2DCFDA was dissolved in methanol as stock solution. The final concentration of methanol in the H2DCFDA-aCSF was less than 0.2%. All samples were read for fluorescence in duplicates and each experiment was repeated at least three times.
Measurements are expressed as means ± SEM. For the electrophysiological data, statistically significant differences between groups were determined using the Student’s paired t-test by comparing control slices to glutamate-treated slices within each of the three groups supplemented either with glucose, lactate, or pyruvate. For the fluorescence measurements of ROS, data were assessed for statistically significant differences using paired-sample Wilcoxon signed rank test. For both tests, a value of p ≤ 0.05 was considered to be statistically significant.
Over a decade ago, we performed two sets of experimental paradigms in which hippocampal slices were exposed to Glut for 15 min. Slices were placed in aCSF containing 4 mM glucose for the first paradigm (A) and 10 mM for the other paradigm (B). Accordingly, slices in paradigm A were exposed to a lower Glut concentration (5 mM) than slices in paradigm B (20 mM), since the higher the glucose concentration in the aCSF, the higher Glut concentration slices could tolerate and the higher the level of lactate slices could produce during exposure to Glut (see Schurr et al., 1999a,b). It is important to emphasize that either concentration of Glut by far exceeds the intrinsic physiological concentrations of this neurotransmitter, where synaptic and extrasynaptic spaces are concerned and is thus, excitotoxic to the hippocampal slice preparation. Consequently, the PS amplitude measured during the exposure to Glut quickly falls to 0 mV, only to fully recover after 30 min of Glut washout under control conditions (see samples of PS traces in Figure 1). In contrast, high frequency electrical stimulation had a much less dramatic effect on the PS amplitude, as we have shown previously (Schurr et al., 1986). In each paradigm, the effect of blocking lactate transport with the monocarboxylate transporter (MCT) inhibitor α-cyano-4-hydroxycinnamate (4-CIN, Halestrap and Denton, 1975) was assessed. The blocker concentration was adjusted according to the concentration of glucose (0.25 mM 4-CIN when glucose was supplied at 4 mM, 0.5 mM 4-CIN when glucose was supplied at 10 mM). As can be seen from Figure 1, blockade of MCT by 4-CIN prevented the recovery of neuronal function after exposure to Glut in both paradigms. Although not shown in Figure 1, it is important to mention that under the experimental conditions of paradigm A some diminishment in the CA1 PS amplitude occurred during the exposure to Glut, while under the conditions of paradigm B the CA1 PS amplitude quickly disappeared during the 15 min exposure to Glut regardless of the presence or absence of 4-CIN (see sample traces in Figure 1, paradigm B), indicating that Glut is excitotoxic at both concentrations. However, the relatively short time of exposure to Glut (15 min) did not lead to an irreversible damage, since over 90% of the slices under control conditions in both paradigms exhibited almost full recovery of neuronal function (green columns on the right side of Figure 1). Nevertheless, in the presence of 4-CIN, none of the slices exhibited recovery of neuronal function when supplied with 4 mM glucose and exposed to 5 mM Glut for 15 min (Paradigm A). Less than 10% of the slices showed recovery of neuronal function when supplied with 10 mM glucose and exposed to 20 mM Glut for 15 min (paradigm B; yellow columns on the right side of Figure 1). While the lactate content of control slices (green-filled circles in Figure 1) did not appear to be elevated much during Glut exposure in either paradigm, it may reflect the high rate by which neurons utilized the astrocritically produced lactate (Pellerin and Magistretti, 1994) rather than a reduction in lactate production. The significant elevation in lactate tissue content of slices treated with 4-CIN during Glut exposure demonstrates that astrocytic or more likely neuronal (Galeffi et al., 2007) lactate transport is blocked and thus was unavailable to neurons in these slices, blockade that prevented recovery of neuronal function at the end of the washout period under both paradigms.
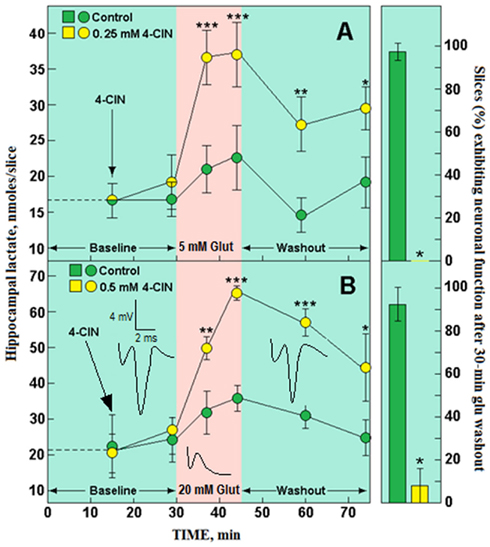
Figure 1. The effect of 15 min exposure to glutamate (Glut) on the ability of hippocampal slices to recover their neuronal function following a 30 min Glut washout and on the content of tissue lactate at several time points during two experimental paradigms. (A) Perfusion of slices with 4 mM glucose aCSF for 30 min, followed by a 15 min exposure to 5 mM Glut, followed by a 30 min washout with 4 mM glucose aCSF, either in the presence of 0.25 mM 4-CIN (yellow symbols) or in the absence of 4-CIN (green symbols); (B) perfusion of slices with 10 mM glucose aCSF, followed by a 15 min exposure to 20 mM Glut, followed by a 30 min washout with 10 mM glucose aCSF in the presence of 0.5 mM 4-CIN (yellow symbols) or in the absence of 4-CIN (green symbols). Also shown are sample traces of PS recorded from one hippocampal slice before and during exposure to 20 mM Glut and after Glut washout. Each data point was repeated three times (30–36 slices over all). Bars are means ± SD; significantly different from control (*p < 0.003; **p < 0.01; ***p < 0.004). For additional methodological details see Schurr et al., 1999a,b).
Since lactate could not be utilized in the presence of 4-CIN, its tissue content remained elevated throughout the washout period (yellow-filled circles in Figure 1). As expected, the content of lactate in slices supplied with 10 mM glucose during Glut exposure in the presence of 4-CIN was almost twice as high as the content of slices supplied with 4 mM glucose, indicating that most of the glucose utilized during Glut exposure is converted to lactate. In essence, the outcome depicted in Figure 1 strongly supports the premise of the ANLSH. These experiments with Glut (see also Schurr et al., 1999a,b) confirm that activation of neural tissue with an excitatory neurotransmitter increases glucose utilization via aerobic glycolysis, which leads to a large elevation in lactate production. Moreover, as indicated by the effects of 4-CIN in the presence of Glut, lactate is the glycolytic product that becomes the oxidative energy substrate, securing the recovery of neuronal function post-Glut activation.
In the debate that has ensued following Pellerin and Magistretti (1994) proposed ANLSH (Tsacopoulos and Magistretti, 1996; Magistretti and Pellerin, 1999; Magistretti et al., 1999; Magistretti, 2000; Chih et al., 2001; Chih and Roberts, 2003; Pellerin and Magistretti, 2003, 2004a,b; Hertz, 2004; Schurr, 2006), the skeptics tend to reject it mainly on the premise that lactate is postulated to be a major oxidative energy substrate, not necessarily on the proposed shuttle per se. The debate is not limited only to this hypothesis; it is still raging about a similar hypothesis concerning lactate shuttles in other tissues that preceded the one offered by Pellerin and Magistretti (see Gladden, 2004, for review). The objection to the idea of a major role for lactate in energy metabolism beyond just being a waste product or, at best, a minor player, is understandable. The dogma of glucose’s obligatory role in energy metabolism in all tissues, organs, and most aerobic organisms is an inseparable part of our understanding and acceptance of this process as formulated during the first half of the twentieth century. However, a trove of experimental data have emerged over the past quarter of a century, all pointing to a major role for lactate in oxidative energy metabolism in brain, skeletal muscle, heart, and probably many other mammalian tissues. These data (Brooks, 1985, 1998, 2000, 2002a,b; Schurr et al., 1988, 1997a,b, 1999a,b; Larrabee, 1995, 1996; Hu and Wilson, 1997b; Brooks et al., 1999; Mangia et al., 2003; Kasischke et al., 2004; Ivanov et al., 2011), in addition to those found in several forgotten studies from the first half of the twentieth century (Ashford and Holmes, 1929, 1931; Holmes, 1930; Holmes and Ashford, 1930; Flock et al., 1938), prompted us to hypothesize that lactate is the end-product of both aerobic and anaerobic glycolysis and, consequently, is the ultimate cerebral oxidative energy substrate in the brain (Schurr, 2006) and possibly in other organs and tissues. The experiments described in Figure 1 contributed greatly to the formulation of this hypothesis.
Among the citations cited above, the one by Hu and Wilson (1997b) carries greater importance than has been realized by both its supporters and detractors. These authors have developed both glucose and lactate, enzyme-based micro-sensors with rapid response time that allowed them direct and continuous in vivo measurements of lactate and glucose concentrations with high temporal resolution in rat brain extracellular fluid. Several authors (Pellerin and Magistretti, 2003; Kasischke et al., 2004; Aubert et al., 2005; Medina and Tabernero, 2005; Serres et al., 2005; Schurr, 2006) have concluded that Hu and Wilson’s findings lend support to the notion that lactate is being utilized aerobically upon neuronal activation. Others (Dienel and Hertz, 2005; Fillenz, 2005; Korf, 2006) have concluded that Hu and Wilson’s observations do not represent oxidative metabolism of lactate. In his commentary, Korf (2006) states that the initial drop in the hippocampal dentate gyrus lactate level following a single stimulation of the perforant pathway (Hu and Wilson, 1997b) is very small in comparison with its subsequent increase, and thus, this minor decrease cannot serve as an argument to support aerobic metabolism of lactate. As can be seen from Figure 2, after the first stimulation, the dip in lactate concentration was 7%, while glucose level dipped by 14%. Based on Hu and Wilson’s (1997a) estimates, the lactate baseline level before stimulation was 1.19 mM and that of glucose was 2.60 mM. Hence, following the first stimulation, the levels of lactate and glucose fell by 0.08 and 0.36 mM, respectively.
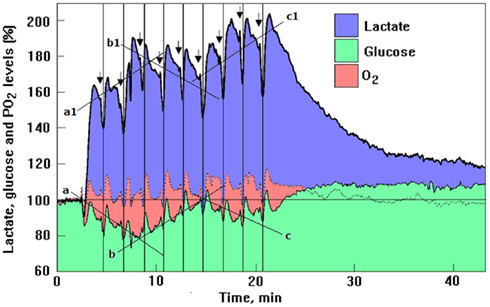
Figure 2. Profiles of time course and dynamic relationships of local extracellular lactate, glucose, and PO2 levels in the rat hippocampal dentate gyrus during a series of 5 s electrical stimulations (arrows) of the perforant pathway at 2 min rest intervals (reproduced with permission from Hu and Wilson, 1997b; copyright 1997, Blackwell, Oxford). The different diagonal lines show the trend in the mean analyte concentration during the series of stimulations. During the second to the fourth stimulations as glucose mean level gradually decreased (line a), lactate mean level gradually increased (line a1). During the next three stimulations the trend was inverted (lines b and b1) and during the 7th to the 10th stimulations the trend inverted itself again (lines c and c1). The changes in the mean concentration of glucose were always in opposite direction to the changes in mean lactate concentration. The vertical lines were drawn to indicate the simultaneous dip in all three analytes in response to each of the 10 electrical stimulations. For additional details see Hu and Wilson (1997b).
One could surmise then that upon the first stimulus, a significant portion of the energy required for this activation is supplied via glycolysis (the conversion of glucose to lactate) as 0.36 mM glucose were consumed, while a smaller portion of that energy is supplied via aerobic consumption of lactate, since only 0.08 mM of the monocarboxylate were consumed. These quantities, however, could be misleading, since 0.36 mM glucose produces a net amount of 0.72 mM ATP (1 mol of glucose produces 2 mol of ATP glycolytically), while 0.08 mM lactate produces 2.72 mM ATP [1 mol of lactate produced ~34 mol of ATP via the mitochondrial tricarboxylic acid (TCA) cycle]. Consequently, the “small” amount of lactate consumed upon the first stimulation actually produced 3.8 times more ATP than the amount produced by the consumption of glucose upon the first stimulation. A significant fact that has been ignored by the detractors of Hu and Wilson’s studies is the sharp increase in tissue lactate level immediately after the first stimulation; from 1.19 (baseline level) to 1.95 mM, a rise of 0.76 mM. Concomitantly, glucose level after the first stimulation decreased from 2.60 (baseline level) to 2.22 mM, a drop of 0.38 mM. Hence, the rise in lactate can be fully attributed to the fall in glucose, as every mole of the sugar is converted glycolytically to 2 mol of lactate. In essence, all the glucose consumed in response to the first stimulation was converted to lactate. This very stoichiometry was seen by us (Figure 1). Aerobic conversion of glucose to lactate rather than to pyruvate is the central theme of our hypothesis (Schurr, 2006). After the second stimulation (Figure 2), the amplitude of the dip in lactate level measured 0.22 mM (from 1.87 mM or 157% of the baseline level, to 1.65 mM or 139% of the baseline level), was almost threefold larger than the one that followed the first stimulation and with an estimated ATP production of 7.48 mM. Glucose dipped after the second stimulation from 2.34 (90% of the baseline level) to 2.13 mM (82% of the baseline level), a dip of 0.21 mM, an approximately 0.15 mM smaller dip (42% smaller) than the dip observed after the first stimulation and with an estimated ATP production of 0.42 mM. Thus, on the second stimulation, ATP production via lactate utilization was 18-fold greater than ATP production via glycolytic glucose utilization. Accordingly, we calculated the size of the dip for each of the ten stimulations applied by Hu and Wilson (1997b) and also estimated the resulted ATP production from glucose and lactate after each of them, calculations that are summarized in Figure 3. Despite the significant increases in lactate “utilization” and decreases in glucose “utilization” beginning with the second stimulation on, Korf (2006) states that there is no less glucose metabolized after repeated stimulation, since the concomitant early dips in glucose remain the same, independent of the changes in lactate. This statement is inaccurate and could be misleading, since it does not take into account the simple fact that every mole of lactate oxidized produces approximately 17 times more ATP than a mole of glucose. The breakdown of 1 mol of glucose glycolytically requires two phosphorylation steps (2 mol of ATP) to produces two trioses, each of which later produces 2 mol of ATP for a net production of 2 mol of ATP for each mole of glucose that passes through glycolysis (Stryer, 1995; see also Figure 4). In contrast, upon entering the mitochondrial TCA cycle and the oxidative phosphorylation chain, the 2 mol of lactate (pyruvate) created from 1 mol of glucose via glycolysis, produce approximately 32–34 mol of ATP (Stryer, 1995) or 16–17 times more ATP than is produced from 1 mol of glucose via glycolysis. Furthermore, over the first four stimulations glucose tissue level continued to drop as lactate tissue level increased (Figure 2, lines a and a1), while during the next four stimulations as lactate tissue level decreased, glucose tissue level increased (Figure 2, lines b and b1). Hu and Wilson (1997b) found this trend noteworthy in their original paper. Although we have not analyzed quantitatively the oxygen profile produced by Hu and Wilson (1997b) in their Figure 2, the authors indicated that unlike the levels of glucose, O2 levels showed essentially no decrease. Thus, one could surmise that each oxygen dip in response to an electrical stimulation signals a concomitant O2 utilization in the mitochondrial electron transport chain with lactate utilization (via pyruvate) in the TCA cycle, the very lactate that is originating glycolytically from glucose.
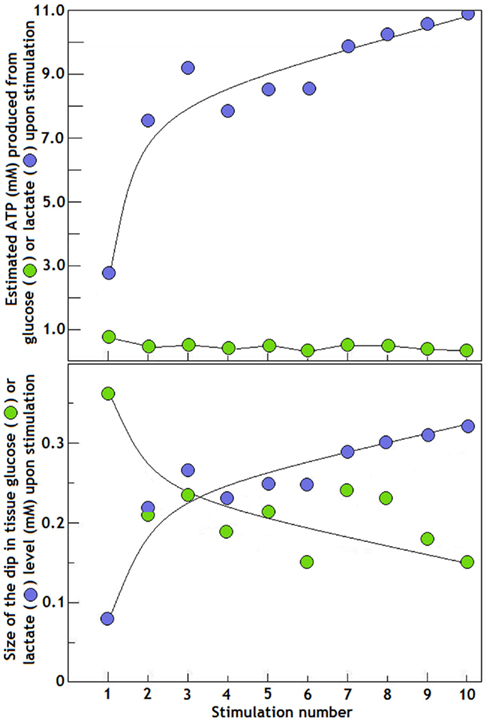
Figure 3. The time course of changes in the amplitude of the dip in tissue glucose and lactate levels in the rat hippocampal dentate gyrus after each of the 10 electrical stimulations applied to the perforant pathway at intervals of 2 min (bottom panel). The amplitude of each dip (in mM) was calculated from the data of Hu and Wilson (1997b) as reproduced in Figure 2. The upper panel illustrates the estimated ATP amount produced based on the size of the dip (in mM) in tissue glucose and lactate levels as shown in the bottom panel. The estimated ATP levels were calculated as follows: the glucose measured dip (in mM) was multiplied by 2, which is the net formation of 2 mol of ATP for each mole of glycolytically metabolized glucose; the lactate measured dip (in mM) was multiplied by 34, which is the net formation of 34 mol of ATP for every 2 mol of lactate (formed glycolytically from 1 mol of glucose) metabolized via the mitochondrial TCA and the oxidative phosphorylation chain.
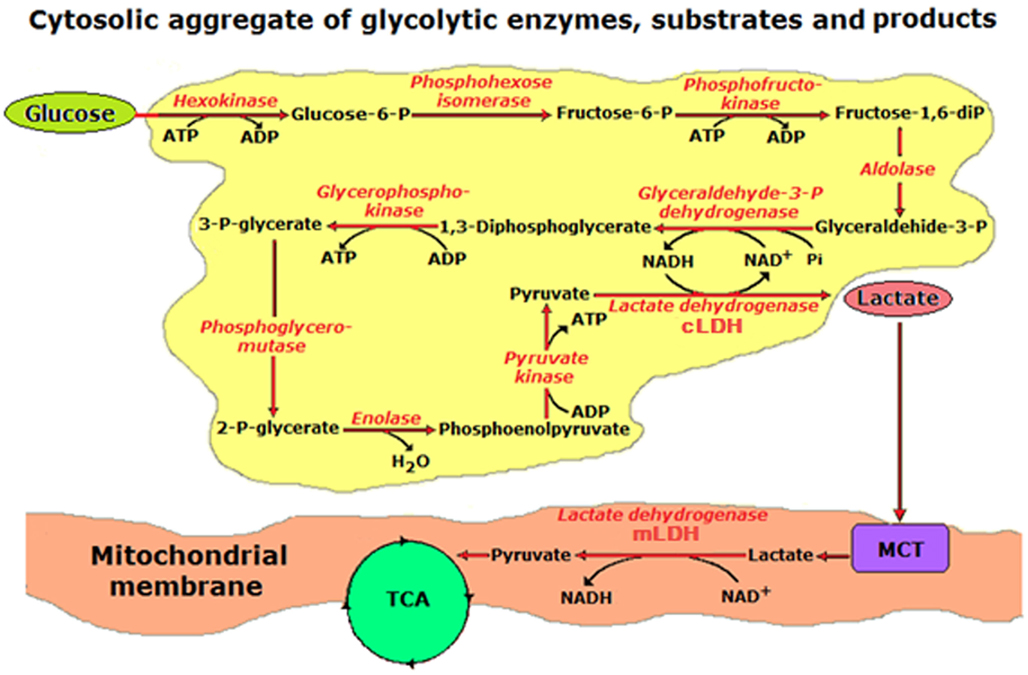
Figure 4. A schematic illustration of a hypothesis first presented elsewhere (Schurr, 2006), postulating lactate, not pyruvate, to be the end-product of aerobic glycolysis. This hypothesis is founded on thermodynamic and other considerations, including data from many studies spanning almost eight decades of research by scientists in laboratories all around the world. The illustration shows the glycolytic apparatus as an aggregate of the glycolytic enzymes, all in close proximity to each other, as required by such a pathway for a maximized efficient output, with the entry of glucose into this pathway on the upper left side and an exit of lactate from the pathway on the right side through the action of cytosolic lactate dehydrogenase (cLDH). Lactate is shuttled via an intracellular shuttle to the mitochondrial membrane, where it is converted back into pyruvate via mitochondrial lactate dehydrogenase (mLDH) and into the tricarboxylic acid (TCA) cycle.
A study by Mangia et al. (2003), using a time-resolved proton magnetic resonance (1H-MRS) in humans, found an approximately 40% dip in lactate tissue concentration 5 s after a 1 s visual stimulation with a recovery back to baseline levels by 12 s. Due to the experimental conditions adopted by Mangia et al. (2003), any increase in lactate concentration, had it occurred, could not be measured. Nevertheless, it is interesting that both Mangia et al. (2003) and Fillenz (2005) interpreted the results of the MRS study as refutation of the ANLSH (Pellerin and Magistretti, 1994), which postulates that an increased astrocytic lactate production supports the bulk of the energy needs of Glut-activated neurons. Hence, according to Mangia et al. (2003) and Fillenz (2005) the short-lived dip in lactate concentration, which, in essence, signals lactate consumption, is sufficient to reject the ANLSH. Moreover, Fillenz (2005) takes Hu and Wilson’s (1997a,b) data to demonstrate “that glucose is the main substrate used by activated neurons.” In the general scheme of things, it is true that glucose is used during neural activation, but for the purpose of producing lactate, most likely by astrocytes. Considering the lopsided difference in the amount of ATP formed via lactate aerobic utilization compared to that produced via glucose glycolytic utilization (see Figure 3, top panel), it is apparent that more than 95% of the ATP produced and consumed by the activated neural tissue is supplied by lactate oxidation. Since the debate over the ANLSH has arguably become the basis on which the idea of lactate as an oxidative energy substrate stands or falls, some participants in this debate set aside data that could neither directly support nor refute this hypothesis. Thus, in their mathematical model, Aubert et al. (2005) selected only the lactate measurements from the data of Hu and Wilson (1997b), leaving out the measurements of glucose and oxygen that were performed concomitantly with those of lactate. Therefore, on the basis of ours and others analyses we believe that the studies of Hu and Wilson(1997a,b) should be reevaluated by those who have discounted them for one reason or another. The principle thrust of their studies is not whether they support or discredit the ANLSH, but rather that they clearly indicate that lactate, the glycolytic product of glucose, is the only substrate capable of answering all the energy needs of activated neural tissue.
Considering our own work over the past two decades and the accumulating data from a multitude of studies, the concept of lactate as an integral intermediate of normal glucose metabolism and ATP production, i.e., aerobic glycolysis and oxidative phosphorylation, has become more main stream and less controversial. Consequently, we hypothesized, as mentioned earlier, that in the brain lactate is the principal product of both aerobic and anaerobic glycolysis (Figure 4; Schurr, 2006).
We have tested this proposed hypothesis in a study that has provided additional evidence in its support (Schurr and Payne, 2007). A newly discovered lactate dehydrogenase inhibitor, malonate, a known competitive inhibitor of succinate dehydrogenase, has been shown to inhibit the conversion of lactate to pyruvate by Saad et al. (2006). This succinate analog is transported into neurons, astrocytes, and mitochondria via the succinate transporter (Aliverdieva et al., 2006; Yodoya et al., 2006). We used malonate in a set of experiments to test the hypothesis that lactate is the end-product of aerobic glycolysis in cerebral tissue (Schurr and Payne, 2007). As can be seen from Figure 5, malonate (M) drastically attenuated the amplitude of lactate-supported neuronal function (evoked hippocampal CA1 PS), as evident from the two representative traces before (L) and after 75 min exposure to the inhibitor (L + M). Lactate-supplemented slices were susceptible to LDH inhibition by malonate, exhibiting a time-dependent diminishment in the PS amplitude. However, malonate was innocuous when slices were supplemented with pyruvate (P, P + M). Thus, this LDH inhibitor appeared to be a more efficient inhibitor than any other known one, especially of mLDH, the mitochondrial form, which converts lactate to pyruvate. The inhibition of the mLDH by malonate significantly slowed down the conversion of lactate to pyruvate, resulting in the observed suppression of neuronal function. Since pyruvate can directly enter the mitochondrial TCA cycle, as it is transported by the same transporter (MCT) that transports lactate, pyruvate-supported neuronal function was unaffected by malonate. With glucose-supplemented slices the effect of malonate was significantly different. Although initially malonate suppressed the PS amplitude of both lactate-supplemented slices by 48% at 45 min incubation (45′ L + M) and of glucose-supplemented slices by 36% (45′ G + M), at 75 min however, the suppression of the PS amplitude of the lactate-supplemented slices increased to 75% (75′ L + M), while that of the glucose-supplemented slices was subsided to only 14% (75′, G + M). While all LDH inhibitors, without exception, exhibit differential inhibition toward either cLDH, the cytosolic form of the enzyme, or mLDH, the mitochondrial form, they do inhibit both forms if allowed long enough incubation time. Hence, the biphasic effect of malonate on the neuronal function of glucose-supplemented slices is predictable, since this inhibitor has stronger inhibitory potency toward mLDH than toward cLDH. Therefore, in the short run, malonate is expected to block mainly the conversion of lactate to pyruvate, which indicates that lactate is the end-product of aerobic glycolysis. If this were not the case, inhibition of cLDH by malonate should have no effect on glucose-supplemented slices, since according to the classic understanding of aerobic glycolysis, inhibition of cLDH should have no effect on the main aerobic glycolytic product, pyruvate, and its entry into the mitochondrial TCA cycle. Hence, the PS amplitude of glucose-supplemented slices should not at all be affected by malonate. The fact that malonate initially inhibited neuronal function of glucose-supplemented slices clearly indicates that glucose must first be glycolytically converted to lactate, a product that must then be converted by mLDH to pyruvate, a LDH form more sensitive to malonate than the cLDH form. Over a longer period of time the degree of inhibition of the cLDH form became more pronounced, such that it blocked glycolysis from continuing beyond the formation of pyruvate, practically preventing lactate production. At this point in time (75′, G + M), the accumulated glycolytic pyruvate became the main product available for mitochondrial use, which resulted in the observed recovery from malonate-induced suppression of glucose-supplemented neuronal function. The somewhat stronger inhibition of neuronal function by malonate (48%) in lactate-supported slices at 45 min (45′ L + M) than that observed in glucose-supplemented slices (36%) also indicates that already at this point in time, there was some inhibition of cLDH and thus accumulation of pyruvate, allowing glucose-supplemented slices to be affected less by malonate than the lactate-supplemented slices.
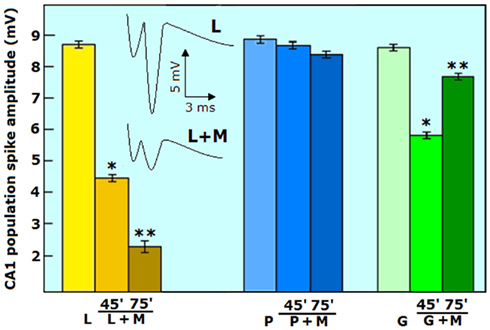
Figure 5. The effect of the LDH inhibitor, malonate (M, 10 mM) on evoked CA1 population spike amplitude (neuronal function) of rat hippocampal slices maintained is aCSF containing either lactate (L, 5 mM), pyruvate (P, 5 mM), or glucose (G, 2.5 mM). Malonate progressively inhibited lactate-supported neuronal function over time and was innocuous against pyruvate-supported neuronal function. Malonate initially inhibited glucose-supported neuronal function, inhibition that was later mostly relieved. Each data point was repeated three times (30–36 slices over all). Bars are means ± SEM; *significantly different from energy substrate alone; **significantly different from energy substrate or energy substrate + malonate at 45 min (p < 0.0001). For additional methodological details see Schurr and Payne (2007).
These results support the hypothesis, which postulates that lactate is the end-product of cerebral aerobic glycolysis and also confirm that its aerobic utilization first requires the conversion of lactate to pyruvate via mLDH. We performed an additional set of experiments (Schurr and Payne, 2007) that provided further support for lactate’s major role in cerebral energy metabolism (Figure 6). In that set we combined Glut and malonate to ascertain lactate’s role as a mitochondrial energy substrate for the maintenance of neuronal function during neural activation. That malonate is detrimental to neuronal viability of glutamate-activated, glucose-supported hippocampal slices was apparent from the partial recovery (50%) of the energy-dependent PS amplitude (Figure 6). This outcome indicates that lactate, whether neuronal or astrocytic in origin, is crucial for neuronal viability upon activation. These results confirm both our previous findings (Figure 1; Schurr et al., 1999a,b) and the outcome of Hu and Wilson’s (1997b) experiments. Alternatively, the outcome of our experiments could be explained by postulating an increase in astrocytic glucose utilization and lactate production in response to Glut uptake, whereupon lactate becomes a major neuronal energy substrate (Pellerin and Magistretti, 1994; Schurr et al., 1999a). Any interference with neuronal utilization of astrocytic lactate under this scenario, i.e., LDH inhibition by malonate, would suppress normal neuronal function. However, such suppression would be abolished if astrocytes, although incapable of lactate production in the presence of malonate, could produce enough pyruvate. That product, upon Glut washout, should be able to fuel the recovery of neuronal function, similar to the observed recovery in slices supplied with exogenous pyruvate (Figure 6). Nevertheless, such recovery did not occur, which indicates that astrocytic pyruvate, if produced during exposure to Glut and malonate, was not readily available to neurons, in contrast to the availability of astrocytic lactate and neuronal pyruvate and to the findings of Yoshioka et al. (2000).
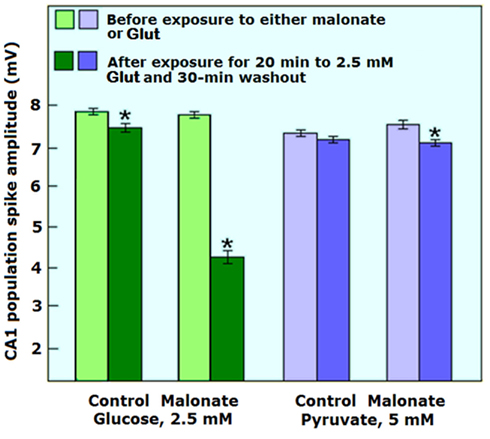
Figure 6. The effect of 20 min exposure to glutamate (Glut, 2.5 mM) on hippocampal CA1 evoked population spike (PS, neuronal function) in the absence or presence of the LDH inhibitor, malonate (10 mM) when either glucose (2.5 mM) or pyruvate (5 mM) was the sole energy substrate. Slices maintained in glucose aCSF could not recover their PS amplitude following Glut washout in the presence of malonate as compared to those maintained in the absence of malonate or those maintained with pyruvate whether malonate was absent or present. Slices were exposed to malonate throughout the experimental protocol for a total of 80 min (30 min before the exposure to Glut, 20 min during the exposure to Glut and 30 min during Glut washout). Each data point was repeated three times (30–36 slices). Bars are means ± SEM; *significantly different from the mean values before exposure to either malonate or Glut (p < 0.01). For additional methodological details see Schurr and Payne (2007).
Why was 2.5 mM glucose, in contrast to 5 mM pyruvate, unable to sustain neuronal viability in slices treated with both malonate and Glut? It is possible that not all the available glucose is converted glycolytically to pyruvate under these conditions. However, glucose-supplemented slices incubated for 20 min with both Glut and malonate should produce enough glycolytic lactate without a significant inhibition of glycolysis by malonate (see Figure 5). Thus, malonate, under these conditions, appears to mainly inhibit lactate conversion to pyruvate by mLDH, which would prevent the full recovery of neuronal function (Figure 6). One should also consider the fact that in order to meet the activated neural tissue increased energy needs, glucose cannot attenuate the rate of mitochondrial respiration through a concomitant increase in the glycolytic flux. In contrast, lactate, and most likely pyruvate, can easily do so, since they both enter the mitochondria directly. Levasseur et al. (2006) have demonstrated that glucose sustained mitochondrial respiration at low, “fixed” rate, since, despite increasing the glucose concentration nearly 100-fold, oxygen consumption was not up-regulated. However, an increase in lactate concentration did elevate mitochondrial oxygen consumption, plausibly allowing mitochondria to meet heightened energy demands. Consequently, in our experiments, glucose-supplemented and oxygenated hippocampal slices were incapable of increasing their mitochondrial respiration rate in response to activation by Glut, since they were unable to up-regulate their glycolytic flux, while at the same time could not utilize lactate directly because of the presence of malonate. Interestingly, the findings of Hu and Wilson (1997b) also agree with the interpretation of Levasseur et al. (2006). More recently, Ivanov et al. (2011) have demonstrated that lactate can cover the energy needs of activated neonatal hippocampal slices and that lactate utilization augmented oxidative phosphorylation. Normally, both neuronal and astrocytic lactate produced from glucose would overcome this limitation (see Figure 5, control glucose, 2.5 mM), but in the presence of malonate this avenue is unavailable (Figure 5, malonate, glucose, 2.5 mM). Obviously, exogenous lactate would be useless in the presence of malonate, which is the reason no results with lactate-supported slices are shown. Nonetheless, results obtained when slices supplemented with 2.5 mM glucose were also supplemented with malonate and exposed to Glut (for 20 min; Figure 6) suggest an augmented shortfall in lactate (or pyruvate) supply under these conditions. This outcome emphasizes the importance of lactate both at rest and during neural activation, an outcome that cannot be explained by the classic depiction of aerobic glycolysis, where pyruvate is positioned as the glycolytic end-product. Using electrical stimulation of several durations instead of Glut application to hippocampal slices, Brennan et al. (2006) have reached a similar conclusion to ours as to the role of mitochondrial energy metabolism, not glycolysis, in supporting the need for the increase in energy supplies. Kann et al. (2011) have recently demonstrated that the induction of gamma oscillations by acetylcholine in hippocampal slices are especially energy demanding and require both high complex I expression and strong functional performance of mitochondria. These authors found that the gamma oscillation power, oxygen consumption, and Complex I expression are higher in the hippocampal subfield CA3 than in subfields CA1 and dentate gyrus and that they utilize mitochondrial oxidative capacity near its maximum limit. These studies along with other cited above and the results of our experiments with the LDH inhibitor, malonate, as summarized in Figure 7, strongly support our hypothesis (Schurr, 2006).
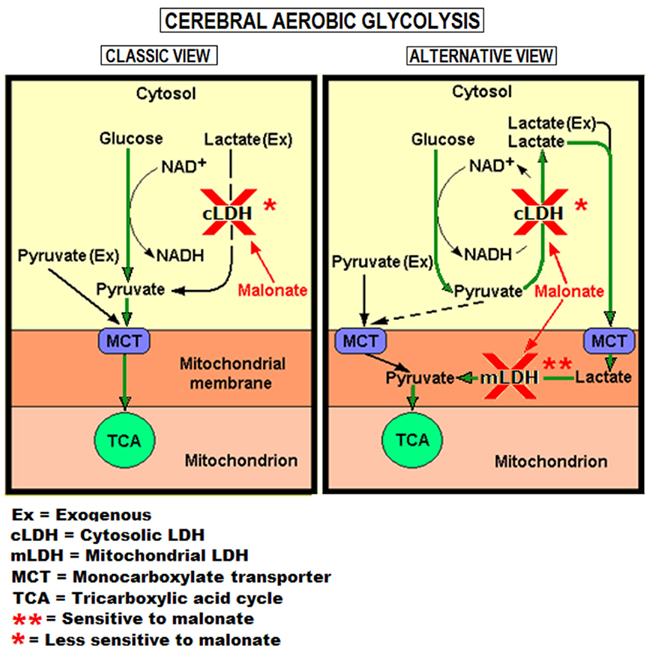
Figure 7. Two views of aerobic glycolysis. The classic view depicts pyruvate as the glycolytic pathway’s end-product (green arrows, left panel) and thus as the pathway that should not be affected by an LDH inhibitor such as malonate in supplying pyruvate to mitochondria. The results shown in Figures 5 and 6 cannot be explained by this view. The alternative view of the aerobic glycolytic pathway postulates lactate to be its end-product (green arrows, right panel, Schurr, 2006). Since glycolytically produced lactate must be converted to pyruvate to allow the latter to enter the TCA cycle, this alternative view explains the ability of malonate to interfere with glucose-supported neuronal function as shown in Figure 5 (right side histograms). However, over time, malonate’s weaker inhibiting activity of the reaction converting pyruvate to lactate shifts the glycolytic conversion of glucose from lactate to pyruvate until the latter becomes the main glycolytic end-product and the mitochondrial substrate (broken arrow, right panel), relieving the suppression of neuronal function observed earlier when glucose is the energy substrate (Figure 5).
The ANLSH, as proposed by Pellerin and Magistretti (1994), postulates that Glut plays a central role in the coupling between neuronal activity and energy metabolism. Accordingly, Glut, released during synaptic activity, is taken up by astrocytes via specific Glut transporters. This transport, which is sodium-dependent, triggers astrocytic aerobic glycolysis and lactate production. The ANLSH further postulates that the astrocytic lactate thus produced is transported via specific monocarboxylate transporters from astrocytes to neurons, where it is utilized as an oxidative mitochondrial energy substrate (Pellerin et al., 1998; Magistretti and Pellerin, 1999). Nevertheless, Yoshioka et al. (2000), using 31P-NMR and electrophysiology of granule cells in rat hippocampal slices, have concluded that, upon high K+ stimulation, the astrocytic main product is pyruvate, not lactate, and the former, not the latter is the neuronal substrate shuttled from astrocytes to neurons. Our results, as described in the previous section refute Yoshioka et al. (2000) conclusion.
Several cellular mechanisms have been suggested over the years to explain the excitotoxicity of Glut. It is now believed that calcium uptake and overload via various glutamate receptor subtypes, a common thread of several neurodegenerative diseases where mitochondrial dysfunction and cellular apoptosis occur, leads to an oxidative stress due to elevated mitochondrial production of ROS (Krieger and Duchen, 2002; Rego and Oliveira, 2003; Emerit et al., 2004; Rosenstock et al., 2004; Lin and Beal, 2005). Acute and severe hypoxia in ischemic disease induce the release of Glut via several mechanisms and down regulate Glut transporters expression, resulting in elevated extracellular Glut (Rossi et al., 2000; Aliprandi et al., 2005; Dallas et al., 2007; Kiewert et al., 2010). The role of excitotoxicity in the pathogenesis of ischemic brain disease has been widely reported (Dirnagl et al., 1999; Kiewert et al., 2010) and elevated Glut levels have been found in plasma and in CSF of stroke patients (Aliprandi et al., 2005). The details of glial Glut sequestration have been extensively studied: GLT1 and GLAST are sodium-dependent Glut transporters that couple inward transport of one Glut molecule with inward entry of 3 Na+, and one H+, and outward movement of one K+. The resulting intracellular Na+ elevation stimulates the Na+/K+ ATPase pump to extrude Na+ and the resulting energy requirement leads to enhanced glucose uptake (Pellerin and Magistretti, 1994). During ischemia, the mitochondrial respiration chain is compromised, leading to energy failure that could be aggravated by the initial increase in Glut uptake. Ischemia-induced energy failure leading to Na+/K+ imbalance and membrane depolarization may also reverse the transporter, carrying Glut to the extracellular space and further contributing to excitotoxicity (Nicholls and Atwell, 1990; Grewer et al., 2008). The hippocampal slice preparation has been used in our laboratory for many years to investigate and elucidate several cellular mechanisms of cerebral ischemia/hypoxia and excitotoxicity as well as in developing neuroprotective modalities against these disorders. To study the ability of each of the three energy substrates, glucose, lactate, and pyruvate, in equicaloric concentrations, to sustain neuronal function in activated neural tissue, we exposed rat hippocampal slices to a relatively low concentration of Glut (2.5 mM). In addition, we aimed to determine whether lactate or pyruvate is the astrocytic product shuttled to neurons and whether or not ROS are involved in the neuronal excitotoxicity of Glut. Hippocampal slices supplemented with either glucose (2.5 mM), pyruvate (5 mM), or lactate (5 mM) sustained their neuronal function uninterrupted throughout the duration of the experiment (60 min). Thus, a relevantly physiological concentration of glucose (2.5 mM) or an equicaloric concentration (5 mM) of either lactate or pyruvate was sufficient to support hippocampal neuronal function in vitro without diminution. However, when exposed to Glut (2.5 mM), pyruvate-supplemented slices (Figures 8 and 9) could not sustain their neuronal function, as was evidenced by the diminution in the PS amplitude. In contrast, neuronal function was diminished only slightly when glucose- or lactate-supplemented slices were exposed to Glut. Replacing a fraction of pyruvate (1/5) with lactate (1 mM) or glucose (0.5 mM) or adding the membrane-permeable ROS scavenger (MCI-186, 33 μM; Wu et al., 2006) to pyruvate-supplemented slices during exposure to Glut, allowed the sustenance of neuronal function without any significant Glut-induced diminution in PS amplitude.
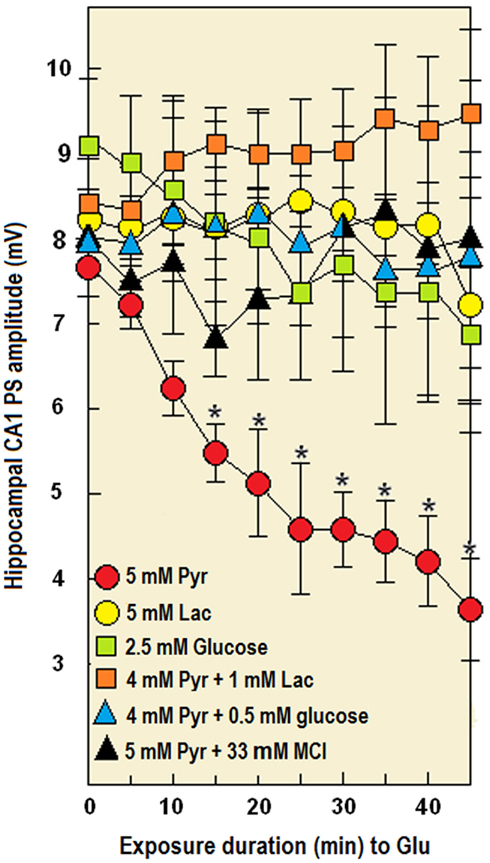
Figure 8. The time-dependent effect of Glut (2.5 mM) on the amplitude of electrically evoked CA1 population spike (PS, neuronal function) in rat hippocampal slices perfused with either 5 mM pyruvate (Pyr), 5 mM lactate (Lac), a mixture of pyruvate (4 mM) with lactate (1 mM), or MCI-186 (33 μM, MCI). Pyruvate alone could not support neuronal function in the presence of glutamate, but mixing this monocarboxylate with a low concentration of either lactate, glucose, or MCI-186 overcame this inability. Each point is the mean amplitude value recorded from three separate hippocampal slices prepared from three different rat brains. Bars are SE of the mean. *Significantly different from the PS amplitude in slices perfused with pyruvate in the absence of glutamate (p < 0.02).
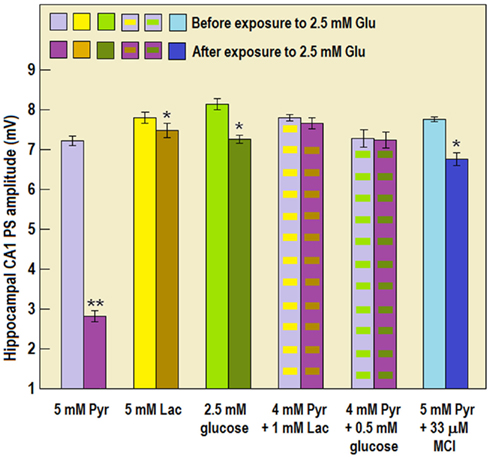
Figure 9. The mean, electrically evoked, population spike (PS, neuronal function) amplitude in rat hippocampal slices perfused for at least 45 min with aCSF containing either 5 mM pyruvate (Pyr), 5 mM lactate (Lac), 2.5 mM glucose, 4 mM Pyr + 1 mM Lac, 4 mM Pyr + 0.5 mM glucose, or 5 mM Pyr + 33 μM MCI-186 (MCI) in the absence or presence of 2.5 mM Glut. Pyruvate alone could not sustain neuronal function in the presence of Glut. Pyruvate inability to sustain normal neuronal function during exposure to Glut appears to be in contrast to the results shown in Figure 6. However, the difference stems from the fact that in this set of experiments slices were exposed to Glut for 45 min, while in the experiments described in Figure 6 they were exposed to Glut for only 20 min. All other treatments overcame the excitotoxicity of Glut. Each column is the mean PS amplitude recorded from at least 23 slices prepared from at least three different rat brains. Bars are SE of the mean. *Significantly different from the PS amplitude in control slices not exposed to Glut (p < 0.004); **significantly different from the PS amplitude in control slices not exposed to Glut (p < 0.00006).
To verify that the formation of ROS contributes to the effect of Glut, ROS levels in hippocampal slices were measured using the fluorescent probe DCFH2-DA, which is deacetylated in cells by non-specific endogenous esterases and oxidized by intracellular oxidant species to form the fluorescent DCF. The results shown in Figure 10 are expressed as percent of maximum ROS production measured in slices supplemented with pyruvate (5 mM) and Glut (2.5 mM). The ROS production in these “control” slices was arbitrarily assigned a value of 100%. In the absence of Glut the level of ROS in pyruvate-supplemented slices was approximately 20% of the level measured when Glut was present (p < 0.05). In contrast, ROS levels in lactate-supplemented slices treated with Glut were only 25% of the levels measured in pyruvate-supplemented, Glut-treated slices (p < 0.05). No difference in the level of ROS was found between lactate-supplemented slices in the presence or absence of Glut. Adding the ROS scavenger MCI-186 (33 μM) to pyruvate-supplemented, Glut-treated slices reduced the level of ROS to less than 10% of control (p < 0.05). Replacement of only 1/5 of the pyruvate concentration in pyruvate-supplemented, Glut-treated slices with lactate (4 mM pyruvate + 1 mM lactate) was sufficient to reduce the level of ROS to 44% of control (p < 0.05). Thus, pyruvate when used as the sole energy substrate was unable to rescue neuronal function in rat hippocampal slices from Glut-induced, ROS-mediated damage, as was evidenced by the large diminution in PS amplitude. In contrast, lactate, even at a concentration one fifth of that of pyruvate was able to prevent Glut excitotoxicity and reduce tissue ROS levels similar to the reduction seen with the ROS scavenger, MCI-186. ROS production in neurons during a toxic Glut challenge is believed to play a central role in neuronal damage from hypoxia/ischemia and reperfusion. Glut receptor-activated neuronal cell death is attributed to a massive influx of Ca2+ and subsequent (within minutes) mitochondrial formation of ROS (Reynolds and Hastings, 1995; Vergun et al., 2001). In the hippocampal slice preparation, Glut-induced excitotoxic damage is expressed as a fall in PS amplitude, i.e., diminished neuronal function (Schurr et al., 1999a). Pyruvate was unable to sustain neuronal function in slices exposed to a relatively low concentration of Glut (2.5 mM), as indicated by a greater than 50% fall in PS amplitude (Figure 9). These results differ from the result shown in Figure 6 only because the exposure to Glut here (45 min) was more than double the length slices in Figure 6 were exposed to the excitotoxin (20 min). This inability of pyruvate to sustain normal neuronal function suggests that this monocarboxylate is not the substrate shuttled from astrocytes to neurons upon excitation by Glut, a finding contrasting the claim that pyruvate is the monocarboxylate shuttled from astrocytes to neurons (Yoshioka et al., 2000).
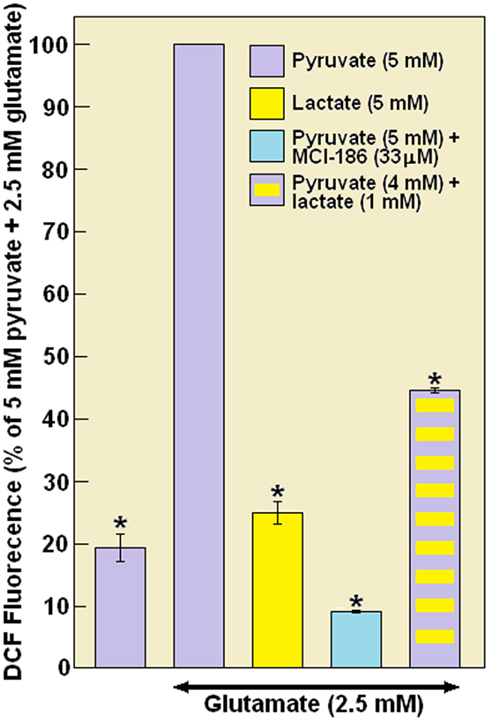
Figure 10. The level of reactive oxygen species (ROS) as measured using the fluorescence of dichlorofluorescein (DCF) extracted from slices perfused with aCSF containing glutamate (2.5 mM) and either pyruvate (5 mM), lactate (5 mM), the combination of [pyruvate (4 mM) + lactate (1 mM)], or the combination of [pyruvate (5 mM) + MCI-186 (33 μM)] (see details in Materials and Methods). Also shown is a set of experiments in which Glut was omitted from the aCSF of pyruvate-supplemented slices. Glut-treated, lactate-, or MCI-186-supplemented slices exhibited significantly lower levels of ROS than pyruvate-supplemented, Glut-treated slices. Each sample for measurement of fluorescence used five slices and was prepared in duplicates. Measurements were repeated at least three times. Bars are SE of the mean. *Significantly different from pyruvate + Glut (p < 0.05).
The ability of both lactate and glucose to sustain neuronal function in the presence of Glut indicates that the metabolism of these two substrates produces a by-product, which affords neuronal tissue to withstand excitotoxicity. Clearly, that by-product is not produced during pyruvate metabolism. Our results concur with the ANLSH and its postulated lactate shuttle. In light of pyruvate’s inability to sustain neuronal function in the presence of Glut, we considered the only difference between the mitochondrial metabolism of pyruvate and lactate, i.e., the conversion of the latter to pyruvate via the action of the mitochondrial lactate dehydrogenase (mLDH). Reduced nicotinamide adenine dinucleotide (NADH), produced as a by-product of lactate conversion to pyruvate by both neurons (Schurr and Payne, 2007) and astrocytes, could provide the necessary reducing power to neutralize Glut-induced ROS and prevent their detrimental effect. Actually, Kirsch and De Groot (2001) were first to suggest such a role for NADH. We could not test this postulate by simply adding exogenous NADH to slices, since NADH is membrane impermeable. Nonetheless, we employed two alternative approaches to address this question. In one we replaced a small fraction of the supplied pyruvate with lactate (1 mM) or glucose (0.5 mM), fractions that by themselves could not sustain neuronal function (Schurr et al., 1988) and then exposed pyruvate-supplemented slices to Glut. In the second we treated pyruvate-supplemented slices with the membrane-permeable ROS scavenger, MCI-186 (33 μM, as determined by dose response experiments, not shown), during the exposure to Glut (Figures 8 and 9). When lactate (1 mM) or glucose (0.5 mM) was mixed with pyruvate (4 mM) in the aCSF, the excitotoxicity of Glu was completely abolished, since neuronal function under these conditions remained unaffected. Adding MCI-186 (33 μM) to pyruvate-supplemented, Glut-treated slices also prevented the Glut-induced excitotoxic damage. We deem both approaches supportive of the ANLSH original postulate, according to which lactate is the astrocytic product shuttled to and utilized by neurons during activation by Glut, as lactate, not pyruvate, metabolism yields NADH that can neutralize ROS and abolish their toxicity.
To ascertain that increased ROS formation is correlated with the degree of neuronal damage measured by electrophysiological means, we employed the fluorescent probe H2DCFDA to quantitatively measure ROS levels in hippocampal slices under several of the conditions used in the electrophysiological portion of the study. Such correlation, if exists, would support the central premise of this investigation, i.e., that lactate aerobic conversion to pyruvate and the accompanied production of the reducing agent NADH could be responsible for the neutralization of ROS. Our findings (Figure 10) demonstrate that pyruvate-supplemented slices treated with Glut contained ROS levels significantly higher than any other treatment combination tested. In the absence of Glut, ROS levels in pyruvate-supplemented slices were only 20% of the levels measured when Glut was present. Lactate-supported, Glut-treated slices produced significantly less ROS (25%) than their pyruvate-supported, Glut-treated counterparts, indicating that a by-product of lactate metabolism is capable of neutralizing ROS, even when the lactate concentration was as low as 1 mM, although not as efficient as the ROS scavenger, MCI-186. When interpreting these findings, it is important to consider that ROS were measured in whole slices, measurements that may have included regions where (astrocytic) lactate was not utilized during Glut excitation, but where ROS were still being produced. In contrast, the electrophysiological assessment of neuronal function was limited to the CA1 region of the hippocampal slice. Several studies have shown a correlation between ROS production in in vitro models of epilepsy and the ability of ROS scavengers to reduce or prevent ROS-induced neurodegeneration (Frantseva et al., 2000) and the possible role NADH [NAD(P)H] plays in neuronal survival during neurodegenerative disease and status epilepticus (Kirsch and De Groot, 2001; Heinemann et al., 2002; Kovács et al., 2002). Interestingly, Herrero-Mendez et al. (2009) have shown that neurons, unlike astrocytes, have low levels of the enzyme Pfkfb3 (6-phosphfructo-2-kinase/fructose-2,6-bisphosphatase-3), responsible for generating fructose-2,6-bisphosphate, the glycolytic product that activates the enzyme 6-phophofructo-1-kinase, the regulator of the glycolytic flux. These investigators have demonstrated that in neurons Pfkfb3 is continuously being degraded by E3 ubiquitin ligase, disabling any significant up-regulation of neuronal glycolysis. In astrocytes, the low activity of the degrading enzyme results in higher levels of Pfkfb3 that enable up-regulation of the glycolytic flux. Herrero-Mendez et al. (2009) also suggested that the low Pfkfb3 levels in neurons allow them to utilize most of their glucose via the pentose phosphate pathway, a pathway known to regenerate reduced glutathione through the oxidation of NADPH(H+), and thus, supply the necessary neuroprotective ROS scavenger. This neuropotective mechanism could play a role in our own experimental paradigm and that of Hu and Wilson (1997b). This mechanism could also work in parallel with the neuroprotective action of NADH produced during the conversion of lactate to pyruvate by mLDH. It is possible that the mitochondrial NADH thus formed is also being used to generate reduced glutathione as the eventual ROS scavenger.
Taking together, the results of the electrophysiological measurements and those of the direct ROS formation measurements in hippocampal slices under the various treatment combinations, suggest that ROS, at least in part, are responsible for Glut excitotoxicity and neuronal damage when energy substrate availability is restricted and that pyruvate supplementation cannot overcome such restriction. Additionally, the results corroborate the premise of the ANLSH, according to which lactate, the astrocytic by-product of Glut uptake, is shuttled to neurons where it is utilized by mitochondria as an energy substrate. We have already provided evidence in support of aerobic neuronal production and utilization of lactate that necessitate an accompanied NADH production (Figures 4 and 7; Schurr, 2006; Schurr and Payne, 2007). These results provide additional support to our previous findings and indicate that the reducing agent, NADH, is a potential endogenous ROS scavenger that could protect neurons from Glut excitotoxicity (Figure 11).
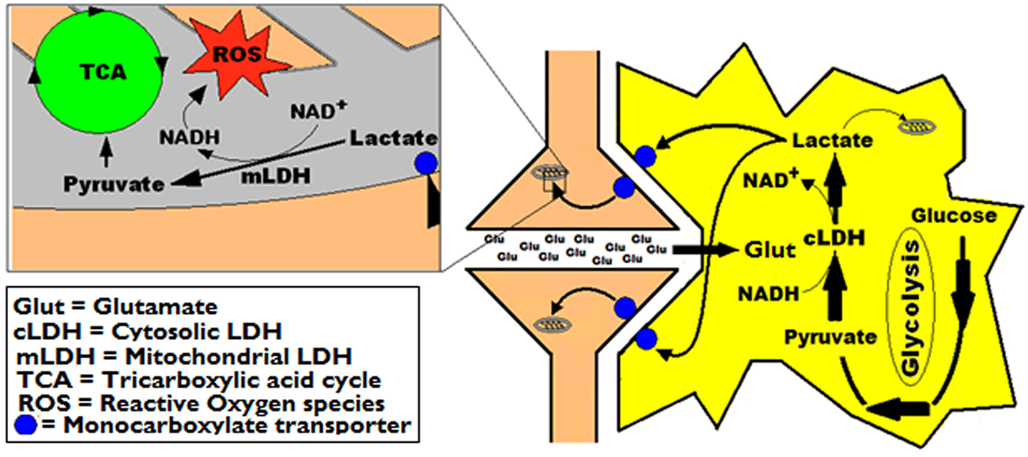
Figure 11. A simplified schematic representation of the astrocytic-neuronal lactate shuttle hypothesis (ANLSH, Pellerin and Magistretti, 1994) combined with our own hypothesis that postulates lactate as the end-product of aerobic glycolysis (Figures 4 and 7). In addition, the left panel illustrate the ability of NADH, formed during lactate conversion to pyruvate in the neuronal mitochondrial membrane, to neutralize Glut-induced ROS-produced damage.
Here we accounted for several of our studies of the last decade or so and revisited the studies of Hu and Wilson(1997a,b), proposing that their outcomes are best explained via the ANLSH (Pellerin and Magistretti, 1994). Furthermore, these experimental investigations, along with many of the cited studies by other investigators, strongly support our own hypothesis (Schurr, 2006) according to which lactate is the real glycolytic end-product both aerobically and anaerobically. Aside from the theoretical and thermodynamic considerations in support of this hypothesis (Schurr, 2006), the experimental results presented here provide scientific evidence in its support. Last, but not least, the postulate that lactate is the end-product of aerobic glycolysis means that this monocarboxylate must be converted to pyruvate by the mitochondrial LDH (mLDH) to be useful as an energy substrate. This conversion is coupled to a formation of the reducing cofactor, NADH, which we have shown here to act as a neutralizing agent of ROS, thus providing neuroprotection against Glut excitotoxicity.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aliprandi, A., Longoni, M., Stanzani, L., Remolizzo, L., Vaccaro, M., Begni, B., Galimberti, G., and Garofolo, R. (2005). Increased plasma glutamate in stroke patients might be linked to altered platelet release and uptake. J. Cereb. Blood Flow Metab. 25, 513–519.
Aliverdieva, D. A., Manaev, D. V., Bondarenko, D. I., and Sholtz, K. F. (2006). Properties of yeast Saccharomyces cerevisiae plasma membrane dicarboxylate transporter. Biochemistry (Mosc.) 71, 1161–1169.
Ashford, C. A., and Holmes, E. G. (1929). Contributions to the study of brain metabolism. V. Role of phosphates in lactic acid production. Biochem. J. 23, 748–759.
Ashford, C. A., and Holmes, E. G. (1931). Further observations on the oxidation of lactic acid by brain tissue. Biochem. J. 25, 2028–2049.
Atlante, A., de Bari, L., Bobba, A., Marra, E., and Passarella, S. (2007). Transport and metabolism of L-lactate occur in mitochondria from cerebellar granule cells and are modified in cells undergoing low potassium dependent apoptosis. Biochim. Biophys. Acta 1767, 1285–1299.
Aubert, A., Costalat, R., Magistretti, P. J., and Pellerin, L. (2005). Brain lactate kinetics: modeling evidence for neuronal lactate uptake upon activation. Proc. Nat. Acad. Sci. U.S.A. 102, 16448–16453.
Barros, L. F., and Deitmer, J. W. (2010). Glucose and lactate supply to the synapse. Brain Res. Rev. 63, 149–159.
Brennan, A. M., Connor, J. A., and Shuttleworth, C. W. (2006). NAD(P)H fluorescence transients after synaptic activity in brain slices: predominant role of mitochondrial function. J. Cereb. Blood Flow Metab. 26, 1389–1406.
Brooks, G. A. (1985). Lactate: glycolytic product and oxidative substrate during sustained exercise in mammals – “the lactate shuttle.” in Comparative Physiology and Biochemistry – Current Topics and Trends, Vol. A, Respiration-Metabolism-Circulation, ed. R. Gilles (Berlin: Springer-Verlag), 208–218.
Brooks, G. A. (1998). Mammalian fuel utilization during sustained exercise. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 120, 89–107.
Brooks, G. A. (2000). Intra- and extra-cellular lactate shuttles. Med. Sci. Sports Exerc. 32, 790–799.
Brooks, G. A. (2002a). Lactate shuttle – between but not within cells? J. Physiol. (Lond.) 541, 333.
Brooks, G. A., Dubouchaud, H., Brown, M., Sicurello, J. P., and Butz, C. E. (1999). Role of mitochondrial lactate dehydrogenase and lactate oxidation in the intracellular lactate shuttle. Proc. Natl. Acad. Sci. U.S.A. 96, 1129–1134.
Chih, C.-P., Lipton, P., and Roberts, E. L. Jr. (2001). Do active cerebral neurons really use lactate than glucose? Trends Neurosci. 24, 573–578.
Chih, C.-P., and Roberts, E. L. Jr. (2003). Energy substrates for neurons during neural activity: a critical review of the astrocyte-neuron lactate shuttle hypothesis. J. Cereb. Blood Flow Metab. 23, 1263–1281.
Dallas, M., Boycott, H. E., Atkinson, L., Miller, A., Boyle, J. P., Pearson, H. A., and Peers, C. (2007). Hypoxia suppresses glutamate transport in astrocytes. J. Neurosci. 27, 3946–3955.
Dienel, G. A., and Hertz, L. (2005). Astrocytic contributions to bioenergetics of cerebral ischemia. Glia 50, 362–388.
Dirnagl, U., Iadecola, C., and Moskowitz, M. A. (1999). Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 22, 391–397.
Emerit, J., Edeas, M., and Bricaire, F. (2004). Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 58, 39–46.
Flock, E., Bollman, J. L., and Mann, D. C. (1938). The utilization of pyruvic acid by the dog. J. Biol. Chem. 125, 49–56.
Frantseva, M. V., Perez-Velasquez, J. L., Tsorakidis, G., Mendonca, A. J., Adamchik, Y., Mills, L. R., Carlen, P. L., and Burnham, M. W. (2000). Oxidative stress is involved in seizure-induced neurodegeneration in the kindling model of epilepsy. Neuroscience 97, 431–435.
Galeffi, F., Foster, K. A., Sadgrove, M. P., Beaver, C. J., and Turner, D. A. (2007). Lactate uptake contributes to the NAD(P)H biphasic response and tissue oxygen response during synaptic stimulation in area CAI of rat hippocampal slices. J. Neurochem. 103, 2449–2461.
Gallagher, C. N., Carpenter, K. L., Grice, P., Howe, D. J., Mason, A., Teemofeev, I., Menon, D. K., Kirpatrick, P. J., Pickard, J. D., Sutherland, G. R., and Hutchinson, P. J. (2009). The human brain utilizes lactate via the tricarboxylic acid cycle: a 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain 132, 2839–2849.
Gandhi, G. K., Cruz, N. F., Ball, K. K., and Dienel, G. A. (2009). Astrocytes are poised for lactate trafficking and release from activated brain and for supply of glucose to neurons. J. Neurochem. 111, 522–536.
Gladden, L. B. (2004). Lactate metabolism: a new paradigm for the third millennium. J. Physiol. (Lond.) 558, 5–30.
Grewer, C., Gameiro, A., Zhang, Z., Tao, Z., Braams, S., and Rauen, T. (2008). Glutamate forward and reverse transport: from molecular mechanism to transporter-mediated release after ischemia. IUBMB Life 60, 609–619.
Halestrap, A. P., and Denton, R. M. (1975). The specificity and metabolic implications of the inhibition of pyruvate transport in isolated mitochondria and intact tissue preparations by α-cyano-4-hydroxycinnamate and related compounds. Biochem. J. 148, 97–106.
Heinemann, U., Buchheim, K., Gabriel, S., Kann, O., Kovács, R., and Schuchmann, S. (2002). Cell death and the metabolic activity during epileptiform discharges and status epilepticus in the hippocampus. Prog. Brain Res. 135, 197–210.
Herrero-Mendez, A., Almeida, A., Fernández, E., Maestre, C., Moncada, S., and Bolanos, J. P. (2009). The bioenergetic and antioxidant statusof neurons is controlled by continuous degredation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 11, 747–752.
Hertz, L. (2004). The astrocyte-neuron lactate shuttle: a challenge of a challenge. J. Cereb. Blood Flow Metab. 24, 1241–1248.
Hertz, L., Peng, L., and Dienel, G. A. (2007). Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Neurosci. Res. 27, 219–249.
Holmes, E. G., and Ashford, C. A. (1930). Lactic acid oxidation in brain with reference to the “Meyerhof cycle.” Biochem. J. 24, 1119–1127.
Hu, Y., and Wilson, G. S. (1997a). Rapid changes in local extracellular rat brain glucose observed with an in vivo glucose sensor. J. Neurochem. 68, 1745–1752.
Hu, Y., and Wilson, G. S. (1997b). A temporary local energy pool coupled to neuronal activity: fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J. Neurochem. 69, 1484–1490.
Ivanov, A., Mukhtarov, M., Bregestovski, P., and Zilberter, Y. (2011). Lactate effectively covers energy demands during neuronal network activity in neonatal hippocampal slices. Front. Neuroenerg. 3:2. doi:10.3389/fnene.2011.00002
Kann, O., Huchzermeyer, C., Kovács, R., Wirtz, S., and Schuelke, M. (2011). Gamma oscillations in the hippocampus require high complex I gene expression and strong functional performance of mitochondria. Brain 134(Pt 2), 345–358.
Kasischke, K. A., Vishwasrao, H. D., Fisher, P. J., Zipfel, W. R., and Webb, W. W. (2004). Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 305, 99–103.
Kiewert, C., Mdzinarishvile, A., Hartmann, J., Bickel, U., and Klein, J. (2010). Metabolic and transmitter changes in core and penumbra after middle cerebral artery occlusion in mice. Brain Res. 1312, 101–107.
Kirsch, M., and De Groot, H. (2001). NAD(P)H, a directly operating antioxidant? FASEB J. 15, 1569–1574.
Korf, J. (2006). Is brain lactate metabolized immediately after neuronal activity through the oxidative pathway? J. Cereb. Blood Flow Metab. 26, 1584–1586.
Kovács, R., Schuchmann, S., Gabriel, S., Kann, O., Kardos, J., and Heinemann, U. (2002). Free radical-mediated cell damage after experimental status epilepticus in hippocampal slice cultures. J. Neurophysiol. 88, 2909–2918.
Krieger, C., and Duchen, M. R. (2002). Mitochondria, Ca2+ and neurodegenerative disease. Eur. J. Pharmacol. 447, 177–188.
Larrabee, M. G. (1995). Lactate metabolism and its effects on glucose metabolism in the excised neural tissue. J. Neurochem. 64, 1734–1741.
Larrabee, M. G. (1996). Partitioning of CO2 production between glucose and lactate in excised sympathetic ganglia, with implications for brain. J. Neurochem. 67, 1726–1734.
Larsen, T. S., Rasmussen, P., Overgaard, M., Secher, N. H., and Nielsen, H. B. (2008). Non-selective β-adrenergic blockade prevents reduction of the cerebral metabolic ratio during exhaustive exercise in humans. J. Physiol. (Lond.) 586, 2807–2815.
Levasseur, J. E., Alessandri, B., Reinert, M., Clausen, T., Zhou, Z., Altememi, N., and Bullock, M. R. (2006). Lactate, not glucose, up-regulates mitochondrial oxygen consumption both in sham and lateral fluid percussed rat brains. Neurosurgery 59, 1122–1131.
Liang, H.-W., Xia, Q., and Bruce, I. C. (2005). Reactive oxygen species mediate the neuroprotection conferred by mitochondrial ATP-sensitive potassium channel opener during ischemia in the rat hippocampal slice. Brain Res. 1042, 169–175.
Lin, M. T., and Beal, M. F. (2005). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 19, 787–795.
Magistretti, P. J. (2000). Cellular bases of functional brain imaging: insights from neuron-glia metabolic coupling. Brain Res. 886, 108–112.
Magistretti, P. J., and Pellerin, L. (1999). Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 1155–1163.
Magistretti, P. J., Pellerin, L., Rothman, D. L., and Shulamn, R. G. (1999). Energy on demand. Science 283, 496–497.
Malonek, D., and Grinvald, A. (1996). Interactions between electrical activity and cortical microcirculation revealed by imaging spectroscopy: implications for functional brain mapping. Science 272, 551–554.
Mangia, S., Garreffa, G., Bianciardi, M., Giove, F., Di Salle, F., and Maraviglia, B. (2003). The aerobic brain: lactate decrease at the onset of neural activity. Neuroscience 118, 7–10.
Mangia, S., Giove, F., Tkac, I., Logotetethetis, N. K., Henry, P. G., Olman, C. A., Maraviglia, B., Di Salle, F., and Ugurbil, K. (2009). Metabolic and hemodynamic events after changes in neuronal activity: current hypotheses, theoretical predictions and in vivo NMR experimental findings. J. Cereb. Blood Flow Metab. 29, 441–463.
Medina, J. M., and Tabernero, A. (2005). Lactate utilization by brain cells and its role in CNS development. J. Neurosci. Res. 79, 2–10.
Nicholls, D., and Atwell, D. (1990). The release and uptake of excitatory amino acids. Trends Pharmacol. Sci. 11, 462–468.
Passarella, S., de Bari, L., Valenti, D., Pizzuto, R., Paventi, G., and Atlante, A. (2008). Mitochondria and L-lactate metabolism. FEBS Lett. 582, 3569–3576.
Pellerin, L., Bouzier-Sore, A. K., Aubert, A., Serres, S., Merle, M., Costalat, R., and Magistretti, P. J. (2007). Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55, 1251–1262.
Pellerin, L., and Magistretti, P. J. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. U.S.A. 91, 10625–10629.
Pellerin, L., and Magistretti, P. J. (2003). Food for thought: challenging the dogmas. J. Cereb. Blood Flow Metab. 23, 1282–1286.
Pellerin, L., and Magistretti, P. J. (2004b). Empiricism and rationalism: two paths toward the same goal. J. Cereb. Blood Flow Metab. 24, 1240–1241.
Pellerin, L., Pellegri, G., Bittar, P. G., Charnay, Y., Bouras, C., Martin, J. L., Stella, N., and Magistretti, P. J. (1998). Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci. 20, 291–299.
Rego, A. C., and Oliveira, C. R. (2003). Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: implications for the pathogenesis of neurodegenerative diseases. Neurochem. Res. 28, 1563–1574.
Reynolds, I. J., and Hastings, T. G. (1995). Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. 15, 3318–3327.
Rosenstock, T. R., Carvalho, A. C., Jurkiewicz, A., Frussa-Filho, R., and Smaili, S. S. (2004). Mitochondrial calcium, oxidative stress and apoptosis in a neurodegenerative disease model induced by 3-nitropropionic acid. J. Neurochem. 88, 1220–1228.
Rossi, D. J., Oshima, T., and Attwell, D. (2000). Glutamate release in severe brain ischemia is mainly by reversed uptake. Nature 403, 316–321.
Saad, L. O., Mirandola, S. R., Maciel, E. N., and Castiho, R. F. (2006). Lactate dehydrogenase activity is inhibited by methyl malonate in vitro. Neurochem. Res. 31, 541–548.
Schurr, A. (2006). Lactate: the ultimate cerebral oxidative energy substrate? J. Cereb. Blood Flow Metab. 26, 142–152.
Schurr, A., Miller, J. J., Payne, R. S., and Rigor, B. M. (1999a). An increase in lactate output by brain tissue serves to meet the energy needs of glutamate-activated neurons. J. Neurosci. 19, 34–39.
Schurr, A., Payne, R. S., Miller, J. J., and Rigor, B. M. (1999b). Study of cerebral energy metabolism using the rat hippocampal slice preparation. Methods 18, 117–126.
Schurr, A., and Payne, R. S. (2007). Lactate, not pyruvate, is neuronal aerobic glycolysis end product: and in vitro electrophysiological study. Neuroscience 147, 613–619.
Schurr, A., Payne, R. S., Miller, J. J., and Rigor, B. M. (1997a). Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: an in vitro study. Brain Res. 744, 105–111.
Schurr, A., Payne, R. S., Millet, J. J., and Rigor, B. M. (1997b). Brain lactate is an obligatory aerobic energy substrate for functional recovery after hypoxia: further in vitro validation. J. Neurochem. 69, 423–426.
Schurr, A., Reid, K. H., Tseng, M. T., Edmonds, H. L. Jr., and Rigor, B. M. (1985). A dual chamber for comparative studies using the brain slice preparation. Comp. Biochem. Physiol. 82A, 701–704.
Schurr, A., Reid, K. H., Tseng, M. T., Edmonds, H. L. Jr., West, C., and Rigor, B. M. (1986). Effect of electrical stimulation on the viability of the hippocampal slice preparation. Brain Res. Bull. 16, 299–301.
Schurr, A., West, C. A., and Rigor, B. M. (1988). Lactate-supported synaptic function in the rat hippocampal slice preparation. Science 240, 1326–1328.
Serres, S., Bezancon, E., Franconi, J. M., and Merle, M. (2005). Ex vivo NMR study of lactate metabolism in rat brain under various depressed states. J. Neurosci. Res. 79, 19–25.
Stryer, L. (1995). “Metabolic energy: generation and storage,” in Biochemistry, 4th Edn (New York, NY: W. H. Freeman), 441–558.
Tsacopoulos, M., and Magistretti, P. J. (1996). Metabolic coupling between glia and neurons. J. Neurosci. 16, 877–885.
Vanzetta, I., and Grinvald, A. (1999). Increased cortical oxidative metabolism due to sensory stimulation: implications for functional brain imaging. Science 286, 1555–1558.
Vergun, O., Sobolevsky, A. I., Yelshansky, M. V., Keelan, J., Khodorov, B. I., and Duchen, M. R. (2001). Exploration of the role of reactive oxygen species in glutamate neurotoxicity in rat hippocampal neurones in culture. J. Physiol. (Lond.) 531, 147–163.
Wu, T., Ding, X.-S., Wang, W., and Wu, J. (2006). MCI-186 (3-methyl-1-phenyl-2-pyrazolin-5-one) attenuated ischemia/reperfusion injury in cultured rat hippocampal cells. Biol. Pharm. Bull. 29, 1613–1617.
Wyss, M. T., Jolivet, R., Buck, A., Magistretti, P. J., and Weber, B. (2011). In vivo evidence for lactate as a neuronal energy source. J. Neurosci. 31, 7477–7485.
Yodoya, E., Wada, M., Shimada, A., Katsukawa, H., Okada, N., Yamamoto, A., Ganaphaty, V., and Fujita, T. (2006). Functional and molecular identification of sodium-coupled decarboxylate transporters in rat primary cultured cerebrocortical astrocytes and neurons. J. Neurochem. 97, 162–173.
Yoshioka, K., Nishimaru, N., Yanai, S., Shimoda, H., and Yamada, K. (2000). Characteristics of monocarboxylates as energy substrates other than glucose in rat brain slices and the effect of selective glial poisoning – a 31P NMR study. Neurosci. Res. 36, 215–226.
Keywords: lactate, glycolysis, LDH, hippocampal slice, neuronal function, reactive oxygen species, NADH, neuroprotection
Citation: Schurr A and Gozal E (2011) Aerobic production and utilization of lactate satisfy increased energy demands upon neuronal activation in hippocampal slices and provide neuroprotection against oxidative stress. Front. Pharmacol. 2:96. doi: 10.3389/fphar.2011.00096
Received: 07 October 2011;
Accepted: 23 December 2011;
Published online: 13 January 2012.
Edited by:
Yuri Zilberter, Faculté de Médecine Timone, FranceReviewed by:
Oliver Kann, University of Heidelberg, GermanyCopyright: © 2012 Schurr and Gozal. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Avital Schurr, Department of Anesthesiology and Perioperative Medicine, School of Medicine, University of Louisville, 530 South Jackson Street, CCB-ULH, Room C2A03, Louisville, KY, USA. e-mail:YTBzY2h1MDFAbG91aXN2aWxsZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.