

- Department of Pharmacology and Therapeutics, Faculty of Medicine, University of British Columbia, Vancouver, BC, Canada
Diabetes mellitus is a multi-faceted metabolic disorder where there is increased oxidative stress that contributes to the pathogenesis of this debilitating disease. This has prompted several investigations into the use of antioxidants as a complementary therapeutic approach. Alpha lipoic acid, a naturally occurring dithiol compound which plays an essential role in mitochondrial bioenergetic reactions, has gained considerable attention as an antioxidant for use in managing diabetic complications. Lipoic acid quenches reactive oxygen species, chelates metal ions, and reduces the oxidized forms of other antioxidants such as vitamin C, vitamin E, and glutathione. It also boosts antioxidant defense system through Nrf-2-mediated antioxidant gene expression and by modulation of peroxisome proliferator activated receptors-regulated genes. ALA inhibits nuclear factor kappa B and activates AMPK in skeletal muscles, which in turn have a plethora of metabolic consequences. These diverse actions suggest that lipoic acid acts by multiple mechanisms, many of which have only been uncovered recently. In this review we briefly summarize the known biochemical properties of lipoic acid and then discussed the oxidative mechanisms implicated in diabetic complications and the mechanisms by which lipoic acid may ameliorate these reactions. The findings of some of the clinical trials in which lipoic acid administration has been tested in diabetic patients during the last 10 years are summarized. It appears that the clearest benefit of lipoic acid supplementation is in patients with diabetic neuropathy.
Introduction
Lipoic acid (LA) or α-lipoic acid (ALA) is a naturally occurring compound that is also known as 1,2-dithiolane-3-pentanoic acid or thioctic acid (Busby et al., 1999). It is synthesized enzymatically in plant and animal mitochondria from octanoic acid and cysteine (as a sulfur source). ALA acts as a cofactor for pyruvate dehydrogenase and α-keto-glutarate dehydrogenase activity (Schmidt et al., 1994), and is also required for the oxidative decarboxylation of pyruvate to acetyl-CoA, a critical step bridging glycolysis and the citric acid cycle (Reed, 1998). The presence of an asymmetric carbon produces two optical isomers R-LA and S-LA. Only the naturally occurring R isomer is bound to protein and acts as an essential cofactor in biological systems (Reed, 1998). However, synthetic LA is a racemic mixture of R and S isoforms, where S-LA can prevent the polymerization of R-LA to enhance its bioavailability (Shay et al., 2009). In cells containing mitochondria, ALA is reduced in an NADH-dependent reaction with lipoamide dehydrogenase to form dihydrolipoic acid (DHLA), whereas in cells that lack mitochondria, ALA can instead be reduced to DHLA via NADPH with glutathione (GSH) and thioredoxin reductases (Jones et al., 2002). Unlike GSH, for which only the reduced form is an antioxidant, both the oxidized and reduced forms of LA are powerful antioxidants whose functions include: (1) quenching of reactive oxygen species (ROS), (2) regeneration of exogenous and endogenous antioxidants such as vitamins C and E, and GSH, (3) chelation of metal ions, and (4) reparation of oxidized proteins (5) regulation of gene transcription (6) inhibition of the activation of nuclear factor kapp B (NF-κB; Biewenga et al., 1997; Packer, 1998; Zhang and Frei, 2001; Figure 1). This impressive array of cellular and molecular functions has raised considerable interest for the use of this substance as a nutritive supplement and also as a therapeutic agent. In view of this growing interest, this review provides an update on the therapeutic role(s) of this substance in ameliorating diabetes and diabetes-induced complications.
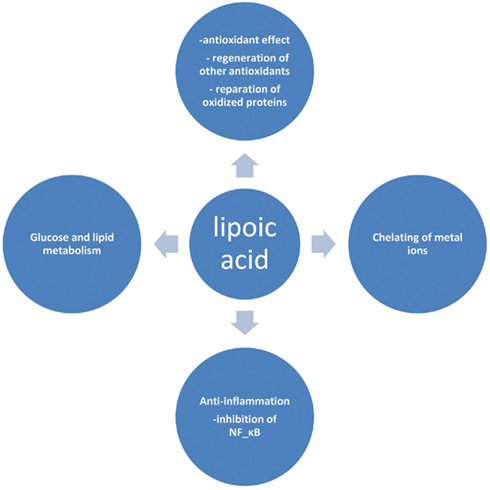
Figure 1. Selected biological actions of lipoic acid.
Biological Function of Lipoic Acid
Quenching Reactive Oxygen Species
Based on the definition of an antioxidant provided by Halliwell and Gutteridge as being “any substance that when present at low concentrations compared to those of an oxidizable substrate significantly delays or prevents oxidation of that substrate,” LA has several unique characteristics among other natural antioxidants to fulfill this criterion (Petersen Shay et al., 2008). Common antioxidants are either water-soluble or lipid soluble agents. In contrast, LA has both hydrophilic and hydrophobic properties. Being both water and fat-soluble means that ALA is widely distributed in plants and animals in both cellular membranes and in the cytosol (Wada et al., 1997). Therefore, it can elicit its antioxidant action in both the cytosol and plasma membrane in contrast to vitamin C (which is lipophobic) and vitamin E (which is lipophilic). Both LA and DHLA scavenge hydroxyl radicals and hypochlorous acid and prevent protein carbonyl formation. In addition DHLA may also be able to regenerate other endogenous antioxidants such as vitamins C and E, and has the beneficial property of neutralizing free radicals without itself becoming one in the process (Biewenga et al., 1997; Bast and Haenen, 2003).
Regeneration of Other Antioxidants
When an antioxidant molecule reacts with an unstable free radical molecule, the antioxidant molecule itself becomes oxidized and loses its benefits until it is reduced again. DHLA has the ability of reducing the oxidized forms of other antioxidants such as vitamin C and E, and GSH.
Glutathione is one of the most important low molecular weight cellular antioxidants, buffering the thiol redox state. The level of GSH is strictly controlled by either substrate availability or transcriptional regulation of specific gene (Petersen Shay et al., 2008). A critical role has recently been described for a transcription factor “nuclear factor erythroid 2-related factor 2 (Nrf2)” against oxidative stress. Normally, Nrf2 is located in the cytoplasm and kept dormant by a cytoplasmic repressor Kelch-like ECH-associated protein 1 (Keap1). A variety of activators release and translocate Nrf2 into the nucleus, where it can regulate the expression of antioxidant enzymes (Lee et al., 2011). LA is one such inducer of Nrf-2-mediated antioxidant gene expression and by so doing is able to significantly increase the cellular capacity of GSH synthesis (Petersen Shay et al., 2008). Furthermore LA also elevates GSH levels through its ability to increase cysteine uptake (Busse et al., 1992). In a variety of cultured cells such as human Jurkat T cells human erythrocytes, C6 glial cells, NB41A3 neuroblastoma cells, and peripheral blood lymphocyte, DHLA also increases GSH synthesis by reducing the ratio of cystine to cysteine (as cysteine is the rate-limiting substrate for this reaction; Han et al., 1997).
Dihydrolipoic acid also regenerates vitamin E either as a result of the direct reaction with tocopheroxyl radical or indirectly by reducing dehydroascorbate, which in turn reduces alpha tocopherol. DHLA is also able to reduce ubiquinone (CoQ10) to ubiquinol, which is an important component of the mitochondrial electron transport chain (Bast and Haenen, 2003). DHLA also reduces the oxidized form of CoQ10, which can additionally reduce the alpha-tocopheroxyl radical (Smith et al., 2004).
Chelation of Metal Ions
Because of the presence of two thiol groups, LA and DHLA both have metal chelating properties. In fact, ALA is a potent chelator of divalent metal ions in vitro and forms stable complexes with Mn2+, Cu2+, Fe2+, Pb2+, and Zn2+ (Ou et al., 1995). Decreased iron uptake and its diminished cytosolic reactive pool have been shown in cultured lens epithelial cells following LA administration. These changes were associated with increased cell resistance to a H2O2 challenge, thus allowing LA to reduce the risk of iron induced oxidative stress (Goralska et al., 2003). In another animal study, R-LA prevented age related cortical iron accumulation and improved antioxidant status (Suh et al., 2005). DHLA also regenerates ascorbate, which in turn reduces iron (Bonomi et al., 1989).
Reparation of Oxidized Proteins
Accumulation of oxidized proteins is thought to be the hallmark of the cell’s aging process (Berlett and Stadtman, 1997; Beckman and Ames, 1998). This can be due to either an increase in the rate of protein oxidation or a decrease in the rate of oxidized protein repair, or a combination of both. Cysteine and methionine, both sulfur-containing amino acids, are among the most sensitive to oxidation; however they are the only amino acids in which the oxidation products can be repaired by specific enzymatic systems (Mary et al., 2004).
Glutathione (GSH) as a versatile antioxidant that has a critical role in maintaining protein thiols in a reduced form and so in regulating the cell cycle (Dickinson and Forman, 2002a,b). Age related decreases in tissue GSH and/or reduced GSH/GSSG (oxidized glutathione) ratios occur in different tissues such as brain, heart (Suh et al., 2004), and liver (Hagen et al., 2000). LA reversed these age related changes and significantly boosted antioxidant system. The beneficial use of LA in reducing the age-associated alterations in GSH can be explained in different ways. First, delivery of exogenous GSH to tissues such as the heart and brain is impossible since it is not transported into these tissues. Second, conventional cysteine delivery agents (e.g., N-acetylcysteine) have low bioavailability to the brain. However, LA is easily taken up into neural tissues and can thus modulate the age related alteration in GSH levels (McLellan et al., 1995; Suh et al., 2005).
Regulation of Gene Transcription
Further to the effects of LA on Nrf-2-mediated antioxidant gene expression, LA has also been shown to modulate peroxisome proliferator activated receptors (PPARs)-regulated genes. The PPARs are a group of nuclear receptor proteins that function as transcription factors regulating the expression of genes (Michalik et al., 2006), and have essential roles in the regulation of cellular differentiation, development and metabolism (carbohydrate, lipid, protein), and tumorigenesis (Belfiore et al., 2009) of higher organisms (Berger and Moller, 2002). The discovery of the effects of thiazolidinediones and fibrates on PPAR-γ and PPAR-α respectively, confirms the importance of ligand-dependent modulation of gene transcription as a pharmacological target in human metabolic diseases. LA activates both PPAR-α (Butler et al., 2009) and -γ (McCarty et al., 2009). PPAR-α regulates the expression of carnitine palmitoyltransferase 1A and acetyl-CoA synthase and PPAR-γ increases the expression of fatty acid translocase/CD36, adipocyte fatty acid binding protein, and lipoprotein lipase. These enzymes are key regulators of glucose and lipid metabolism (Pershadsingh et al., 2005). Some of the metabolic effects of LA are secondary to its regulatory effect on these pathways.
Inhibition of the Activation of NF-κB
Nuclear factor kapp B is a protein complex that controls the transcription of DNA. NF-κB is found in almost all animal cell types and is involved in cellular responses to stimuli such as stress, cytokines, free radicals, ultraviolet irradiation, oxidized low density lipoprotein (LDL), and bacterial or viral antigens (Brasier, 2006; Gilmore, 2006). NF-κB has a key role in regulating the immune response to infection. Incorrect regulation of NF-κB has been linked to cancer, inflammatory and autoimmune diseases, septic shock, viral infection, and improper immune development. NF-κB has also been implicated in processes of synaptic plasticity and memory (Meffert et al., 2003; Tian and Brasier, 2003). The NF-κB protein complex is normally located in the cytoplasm in an inactive form by virtue of its binding to a family of inhibitor of NF-κB (IκB) proteins. Upon cell stimulation by a wide variety of stimuli, signal responsive IKK α and β (TNF-α-inducible IκB kinase complex also known as IKK1 and IKK2) are activated, resulting in the phosphorylation of IκB and its proteasomal degradation. IκB degradation liberates NF-κB, allowing it to translocate to the nucleus and induce gene expression (Baldwin, 1996; Barnes and Karin, 1997).
It is been shown that LA inhibits IκB degradation and NF-κB-dependent gene expression by inhibition of IKK2, suggesting that LA inhibits NF-κB activation independent of its antioxidant function (Ying et al., 2011). ALA inhibits NF-κB activation at the level of or upstream of, IKK-α and IKK-β, by modulating the MEKK1–MKK4–IKK pathway (Lee et al., 2008). This system is a signaling pathway of intracellular protein kinases that can transduce signals from the cell surface to changes in gene expression. Mitogen activated protein kinases (MAPKs) are the most important group of protein kinases in this regard (Zeigler et al., 1999). MAPKs are phosphorylated by upstream MAPK kinases (MEKs), which in turn are activated by MEK kinases (MEKKs; Widmann et al., 1999; Kyriakis and Avruch, 2001). The MEKK system is extraordinary diverse and complex and consists of several members, including MEKK1, 2, 3, 4, ASK1, TAK1, and MLK3. Among these MEKK1 is the best-characterized member and is present in spleen, heart, brain, lung, and kidney (Gardner et al., 1994). The hierarchy of these pathways varies depending on the extracellular stimuli, the specific enzymes expressed in the cell, and the cell types (Packer et al., 1995).
Clinical Uses of ALA
Alpha lipoic acid is effective in many pathological conditions where ROS have been implicated, including diabetes mellitus. Before considering these studies, however, it may be helpful to briefly discuss the pharmacokinetics of this compound.
In humans, LA is synthesized in liver and other tissues and is also obtained from both animal and plant sources in the diet. In animal tissue, higher concentrations of LA are found in the heart, liver, and kidney while spinach, broccoli, tomato, garden peas, brussel sprouts, and rice bran are among rich plant sources. Naturally occurring R-LA in foods covalently bound to lysine in proteins (lipoyllysine), while LA in supplements is free (Singh and Jialal, 2008). An oral dose of LA is rapidly absorbed from the gastrointestinal tract and appreciably increases plasma LA levels. Typically, 20–40% of orally administered racemic ALA is absorbed with peak plasma concentrations higher in the R isomer compared to the S-isomer (Gleiter et al., 1996; Hermann et al., 1996) suggesting better absorption in the former isomer. The amount of a given dose and the presence or absence of food may explain absorption variability. Rapid gastrointestinal uptake is followed by an equally rapid clearance, reflecting both transport into tissues as well as glomerular filtration and renal exertion. Experiments using radiolabeled ALA indicate that when 14C-labeled LA was administered to rats (intraperitoneal, oral, or by a nasogastric tube) 80% of the administered dose was either excreted or found in the tissues (Harrison and McCormick, 1974). LA is subject to extensive catabolism. The most common metabolites are bisnorlipoate, tetranorlipoate, and β-hydroxy-bisnorlipoate (Shay et al., 2009). In addition to its catabolism, some is rapidly reduced to DHLA, which is equally rapidly excreted from cells (Jones et al., 2002). In all animal models studied, LA and its metabolites are readily excreted, primarily in the urine (Biewenga et al., 1997). However, ALA is safe in patients with end stage renal failure as well as in liver disease (Bustamante et al., 1998; Teichert et al., 2005), and in fact, has been used in the treatment of several hepatic diseases with encouraging results (Berkson, 1999; Poh and Goh, 2009).
Diabetes and Oxidative Stress
Diabetes is a chronic metabolic disorder that continues to be a major worldwide health issue. It is characterized by absolute or relative deficiencies in insulin secretion and/or insulin action and is associated with chronic hyperglycemia and disturbances of carbohydrate, lipid, and protein metabolism. As a consequence of the metabolic derangements in diabetes, various complications develop including both macro- and micro-vascular dysfunctions (Duckworth, 2001). Many studies have shown that diabetes mellitus (type 1 and 2) is associated with increased formation of free radicals and decreased antioxidant potential, leading to oxidative damage of cell components (Bashan et al., 2009). Direct evidence of oxidative stress in diabetes is provided by the measurement of oxidative stress markers such as plasma and urinary F2-isoprostane as well as plasma and tissue levels of nitrotyrosine and superoxide radicals ( ; Ceriello et al., 2001; Oberg et al., 2004; Vega-Lopez et al., 2004). There are multiple sources of oxidative stress in diabetes, including non-enzymatic, enzymatic, and mitochondrial pathways.
; Ceriello et al., 2001; Oberg et al., 2004; Vega-Lopez et al., 2004). There are multiple sources of oxidative stress in diabetes, including non-enzymatic, enzymatic, and mitochondrial pathways.
Non-enzymatic sources of oxidative stress originate from the oxidative biochemistry of glucose. Hyperglycemia directly increases ROS generation since glucose undergoes autoxidation to generate • OH radicals (Turko et al., 2001). In addition, glucose reacts with proteins in a non-enzymatic manner leading to the development of Amadori products followed by formation of advanced glycation end products (AGEs). ROS is generated at multiple steps during this process. In hyperglycemia, there is enhanced metabolism of glucose through the polyol (sorbitol) pathway, which also results in enhanced production of 
Enzymatic sources of augmented generation of ROS in diabetes include nitric oxide synthase (NOS), NAD(P)H oxidase, and xanthine oxidase (Guzik et al., 2000, 2002; Aliciguzel et al., 2003). All isoforms of NOS require five cofactors/prosthetic groups such as flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), heme, tetrahydrobiopterin (BH4), and Ca2+-calmodulin. If NOS lacks its substrate L-arginine or one of its cofactors (“uncoupled” NOS), NOS produces  instead of • NO (Aliciguzel et al., 2003; Maritim et al., 2003). A major source of
instead of • NO (Aliciguzel et al., 2003; Maritim et al., 2003). A major source of 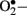 production is NAD(P)H oxidase which is a membrane associated enzyme that consists of five subunits (Guzik et al., 2000, 2002; Etoh et al., 2003; Kitada et al., 2003). Guzik et al. (2002) investigated
production is NAD(P)H oxidase which is a membrane associated enzyme that consists of five subunits (Guzik et al., 2000, 2002; Etoh et al., 2003; Kitada et al., 2003). Guzik et al. (2002) investigated 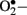 levels in vascular specimens from diabetic patients and probed sources of
levels in vascular specimens from diabetic patients and probed sources of 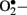 using inhibitors of NOS, NAD(P)H oxidase, xanthine oxidase, and the mitochondrial electron transport chain and reported that the enhanced production of
using inhibitors of NOS, NAD(P)H oxidase, xanthine oxidase, and the mitochondrial electron transport chain and reported that the enhanced production of 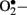 in diabetic patients is predominantly mediated by NAD(P)H oxidase.
in diabetic patients is predominantly mediated by NAD(P)H oxidase.
The mitochondrial respiratory chain is a non-enzymatic source of reactive species. During oxidative phosphorylation, electrons are transferred from the electron carriers NADH and FADH2, through four complexes in the inner mitochondrial membrane, to oxygen, and generate ATP in the process (Green et al., 2004). Under normal conditions, 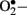 is immediately eliminated by natural antioxidant defense mechanisms. Cells have evolved highly complex antioxidant systems (enzymic and non-enzymatic), which work synergistically to protect cells and organ systems against free radical-induced damage. The most efficient enzymatic antioxidants involve glutathione peroxidase, catalase, and superoxide dismutase. Non-enzymatic antioxidants include vitamins E and C and thiol antioxidants (glutathione, thioredoxin; Golbidi and Laher, 2010). Each of these antioxidants can combine with reactive oxidants to produce other less reactive species that generally has reduced or no toxicity. Superoxide dismutase promotes the dismutation of the superoxide radical to form hydrogen peroxide (H2O2) and oxygen. Enzymes such as glutathione peroxidase (GPx) use reduced glutathione (GSH) as a reducing equivalent to reduce H2O2 to form oxidized glutathione and water. Furthermore, GSH can remove selected oxygen radicals directly and assist in the recycling of vitamin C and E. Catalase converts H2O2 to water and oxygen. The newly identified peroxiredoxin family, which has six isoforms in mammalian cells, represents a group of peroxidases that also catalyze the reduction of H2O2 (Cox et al., 2009). Among the six mammalian isoforms known so farm, peroxiredoxin III is synthesized with a mitochondrial targeting sequence, as is the case for MnSOD.
is immediately eliminated by natural antioxidant defense mechanisms. Cells have evolved highly complex antioxidant systems (enzymic and non-enzymatic), which work synergistically to protect cells and organ systems against free radical-induced damage. The most efficient enzymatic antioxidants involve glutathione peroxidase, catalase, and superoxide dismutase. Non-enzymatic antioxidants include vitamins E and C and thiol antioxidants (glutathione, thioredoxin; Golbidi and Laher, 2010). Each of these antioxidants can combine with reactive oxidants to produce other less reactive species that generally has reduced or no toxicity. Superoxide dismutase promotes the dismutation of the superoxide radical to form hydrogen peroxide (H2O2) and oxygen. Enzymes such as glutathione peroxidase (GPx) use reduced glutathione (GSH) as a reducing equivalent to reduce H2O2 to form oxidized glutathione and water. Furthermore, GSH can remove selected oxygen radicals directly and assist in the recycling of vitamin C and E. Catalase converts H2O2 to water and oxygen. The newly identified peroxiredoxin family, which has six isoforms in mammalian cells, represents a group of peroxidases that also catalyze the reduction of H2O2 (Cox et al., 2009). Among the six mammalian isoforms known so farm, peroxiredoxin III is synthesized with a mitochondrial targeting sequence, as is the case for MnSOD.
Hyperglycemia-induced generation of 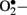 at the mitochondrial level is thought to be the major driver of the vicious cycle of oxidative stress in diabetes (Nishikawa et al., 2000; Brownlee, 2001). There is increased generation of ROS (especially
at the mitochondrial level is thought to be the major driver of the vicious cycle of oxidative stress in diabetes (Nishikawa et al., 2000; Brownlee, 2001). There is increased generation of ROS (especially 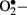 ) when endothelial cells are exposed to clinically relevant hyperglycemic conditions. The augmented generation of pyruvate via accelerated glycolysis under hyperglycemic conditions is thought to flood the mitochondria and thus generates
) when endothelial cells are exposed to clinically relevant hyperglycemic conditions. The augmented generation of pyruvate via accelerated glycolysis under hyperglycemic conditions is thought to flood the mitochondria and thus generates 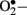 formation at the level of Complex II in the respiratory chain (Nishikawa et al., 2000).
formation at the level of Complex II in the respiratory chain (Nishikawa et al., 2000).
Reactive oxygen species stimulates oxidation of LDL; ox-LDL is not recognized by the LDL receptor and is subsequently taken up by scavenger receptors in macrophages to form foam cells and so lead to atherosclerotic plaques (Boullier et al., 2001). Superoxide anions can activate several damaging pathways in diabetes including accelerated formation of AGE’s, polyol pathway, hexosamine pathway, and protein kinase C (PKC), all of which are involved in micro and macrovascular complications. Both 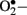 and H2O2 stimulate stress-related signaling mechanisms such as NF-κB, p38-mitogen activated protein kinases (p38-MAPK) and signal transducers and activators of transcription-Janus kinases (STAT-JAK), resulting in vascular smooth muscle cell migration and proliferation. In endothelial cells, H2O2 mediates apoptosis and pathological angiogenesis (Taniyama and Griendling, 2003). Furthermore,
and H2O2 stimulate stress-related signaling mechanisms such as NF-κB, p38-mitogen activated protein kinases (p38-MAPK) and signal transducers and activators of transcription-Janus kinases (STAT-JAK), resulting in vascular smooth muscle cell migration and proliferation. In endothelial cells, H2O2 mediates apoptosis and pathological angiogenesis (Taniyama and Griendling, 2003). Furthermore, 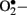 immediately reacts with
immediately reacts with 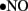 to generate cytotoxic peroxynitrite (ONOO−) and this reaction itself has several consequences. First, ONOO− alters the function of biomolecules by protein nitration as well as by causing lipid peroxidation (Turko et al., 2001). For example, potassium channels, which regulate vasorelaxation, are inhibited by nitration (Liu and Gutterman, 2002; Liu et al., 2002). As reviewed by Turko et al. (2001) increased levels of nitrotyrosine are associated with apoptosis of myocytes, endothelial cells, and fibroblasts in diabetes. Importantly, ONOO− causes single-strand DNA breakage, which in turn activates nuclear enzyme poly(ADP-ribose) polymerase (PARP; a nuclear DNA-repair enzyme that is able to cause a depletion of NAD+; Soriano et al., 2001). Additionally, ONOO− decreases
to generate cytotoxic peroxynitrite (ONOO−) and this reaction itself has several consequences. First, ONOO− alters the function of biomolecules by protein nitration as well as by causing lipid peroxidation (Turko et al., 2001). For example, potassium channels, which regulate vasorelaxation, are inhibited by nitration (Liu and Gutterman, 2002; Liu et al., 2002). As reviewed by Turko et al. (2001) increased levels of nitrotyrosine are associated with apoptosis of myocytes, endothelial cells, and fibroblasts in diabetes. Importantly, ONOO− causes single-strand DNA breakage, which in turn activates nuclear enzyme poly(ADP-ribose) polymerase (PARP; a nuclear DNA-repair enzyme that is able to cause a depletion of NAD+; Soriano et al., 2001). Additionally, ONOO− decreases 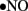 bioavailability causing impaired relaxation and inhibition of the anti-proliferative effects of
bioavailability causing impaired relaxation and inhibition of the anti-proliferative effects of  (Maritim et al., 2003). Furthermore, ONOO− oxidizes BH4, an important cofactor for NOS, and causes uncoupling of NOS to produce
(Maritim et al., 2003). Furthermore, ONOO− oxidizes BH4, an important cofactor for NOS, and causes uncoupling of NOS to produce  instead of
instead of  (Maritim et al., 2003). ROS-induced peroxidation of membrane lipids alters the structure and the fluidity of biological membranes, which will have global effects that alter vascular function (Soriano et al., 2001; Liu and Gutterman, 2002; Liu et al., 2002; Griendling and FitzGerald, 2003; Maritim et al., 2003; Taniyama and Griendling, 2003).
(Maritim et al., 2003). ROS-induced peroxidation of membrane lipids alters the structure and the fluidity of biological membranes, which will have global effects that alter vascular function (Soriano et al., 2001; Liu and Gutterman, 2002; Liu et al., 2002; Griendling and FitzGerald, 2003; Maritim et al., 2003; Taniyama and Griendling, 2003).
ALA as a Therapeutic Adjuvant
Lipoic acid has potential applications for many aspects of the pathology of diabetes. One important action of LA is on the expression of AMPK in the hypothalamus and peripheral tissues. The AMPK complex is evolutionally a well-conserved serine/threonine kinase that functions as a fuel sensor in the cell and is activated when cellular energy is depleted and AMP/ADP ratio rises (Kola et al., 2006). The result of AMPK activation is the inhibition of energy-consuming biosynthetic pathways and the activation of ATP producing catabolic pathways. AMPK can also affect transcription of specific genes involved in energy metabolism, thereby exerting long-term metabolic control (Winder, 2001). The heterotrimeric protein AMPK is formed by combination of the catalytic α subunit (with two isoforms α1 and α2) with β (two isoforms) and γ (three isoforms) subunits. The γ subunit includes four particular cystathionine beta synthase domains giving AMPK its ability to sensitively detect shifts in the AMP/ATP ratio (Cheung et al., 2000). The catalytic domain has a site of phosphorylation at a threonine residue (Thr172) that is an important site for AMPK activation by upstream kinases. Binding of AMP to the regulatory γ subunit of AMPK promotes (a) allosteric activation, (b) phosphorylation of Thr172 by upstream kinases and (c) inhibition of dephosphorylation by protein phosphatases. Cellular stresses that cause an increase in the AMP/ATP ratio such as hypoxia, oxidative stress, hypoglycemia, exercise, or nutrient deprivation can affect cellular metabolic conditions partially through this pathway (Kola et al., 2006). After the discovery of the effect of insulin-sensitizing anti-diabetic drugs such as metformin and thiazolidinediones on the activation of AMPK, much interest was generated toward targeting this pathway for the treatment of diabetes (Musi et al., 2002; Saha et al., 2004). In vivo and in vitro studies have shown that activation of AMPK leads to reduce glucose output form the liver (Viollet et al., 2009). Overexpression of hepatic AMPK also leads to mild hypoglycemia in normal mice and prevented hyperglycemia in diabetic mice (Foretz et al., 2005; Viana et al., 2006). This hypoglycemic effect of AMPK activation is accompanied by down regulation of gluconeogenic gene expression (e.g., phosphoenolpyruvate carboxykinase and glucose-6-phosphatase) and reduced glucose production in hepatocytes expressing activated MAPK or treated with AICAR (5-Aminoimidazole-4-carboxamide ribotide, metabolized to ZMP which is an analog of AMP; Lochhead et al., 2000; Foretz et al., 2005; Viana et al., 2006). Activation of AMPK in skeletal muscle, a major regulator of cellular energy metabolism, increases glucose uptake and fatty acid oxidation. AMPK stimulates GLUT4 translocation to the plasma membrane in an insulin independent manner and increases the expression of the GLUT4 gene through enhanced binding of the transcription factor MEF-2 (myocyte enhancer factor-2) to promoters in the GLUT4 gene (Konrad et al., 2001). Some studies have also shown that AMPK regulates glucose transport through GLUT1 (Xi et al., 2001).
Insulin sensitivity is also improved through reduced triglyceride accumulation by skeletal muscles (Evans et al., 2002). This occurs as a result of AMPK phosphorylating, and thus inactivation, of acetyl-CoA carboxylase (ACC), resulting in decreases in malonyl-coenzyme A (Winder and Hardie, 1996; Ruderman and Flier, 2001). ACC is an important rate-limiting enzyme for the synthesis of malonyl-CoA, which in turn is a critical precursor of fatty acids biosynthesis and a potent inhibitor of mitochondrial fatty acid oxidation. Decrease in malonyl-CoA content results in reduction of fatty acid synthesis and increases in fatty acid oxidation. Triglyceride accumulation in skeletal muscle contributes to insulin resistance in obesity associated insulin resistance and type 2 diabetes (Goodpaster and Kelley, 2002). Muscular AMPK is decreased in Obese Long Evans Tokushima Fatty rats, which are prone to diabetes (Lee et al., 2005a). When these rats are administered ALA, there is increased insulin stimulated whole body glucose disposal and also in skeletal muscle. ALA also increased fatty acid oxidation and stimulated AMPK in skeletal muscle (Lee et al., 2005a). Adenovirus mediated administration of dominant negative AMPK into skeletal muscle prevented the ALA induced increases in fatty acid oxidation and insulin stimulated glucose uptake. These results imply that ALA induced improvement of insulin sensitivity is mediated by activation of AMPK and reduced triglyceride accumulation in skeletal muscle (Lee et al., 2005a).
The exact mechanism underlying ALA stimulated AMPK activation remains to be clarified. To date, two upstream AMPK kinases (AMPKK) have been identified: LKB1 (serine–threonine kinase liver kinase B1, also known as STK11) and Ca/calmodulin dependent protein kinase kinase (CaMKK; Shackelford and Shaw, 2009). LKB1 was originally recognized as a tumor suppressor in humans, and may dictate most of the AMPK activation in the majority of tissues (except hypothalamic neurons Anderson et al., 2008, T cells Tamás et al., 2009, and endothelial cells Stahmann et al., 2006). Activation and phosphorylation of AMPK by LKB1 requires an increase in AMP. Binding of MAP to AMPK changes the conformation of AMPK, making it a better substrate for LKB1. Therefore, LKB1 mediates the prolonged and adaptive activation of AMPK following energy stress (Shackelford and Shaw, 2009). CaMKK phosphorylates and activates AMPK in an AMP independent manner; CAMKK is activated by a rise in intracellular Ca2+ concentrations (Hawley et al., 2005; Hurley et al., 2005). CaMKK has two isoforms, CaMKK-α (e.g., in skeletal muscle) and CaMKK-β (in neural tissues; Anderson et al., 1998; Jensen et al., 2007). In an attempt to clarify the mechanism(s) of ALA induced AMPK activation, Shen et al. (2007) reported that LKB1 may not be involved in this process, since ALA treatment of C2C12 myotubes increased the intracellular Ca2+ concentration, suggesting that ALA may activate AMPK by stimulating the CaMKK pathway. Chelation of intracellular free Ca2+ and inhibition of CaMKK abolished ALA induced AMPK activation, with ALA treatment increasing the association of AMPK with CaMKK.
AMPK is also expressed in the hypothalamus where it is implicated in the regulation of appetite (Andersson et al., 2004; Minokoshi et al., 2004). Activation of hypothalamic AMPK increases food intake and body weight, whereas inhibition of it is necessary for leptin to promote satiety and reduce food consumption (Andersson et al., 2004). Increases in energy expenditure, decreases hypothalamic AMPK activity and reduced plasma glucose, insulin, free fatty acid, and leptin have all been demonstrated in ALA treated rats (Kim et al., 2004; Lee et al., 2005b). Furthermore, intracerebroventricular injection of AICAR (an activator of AMPK) reversed the effects of ALA on food consumption. These studies suggest that hypothalamic AMPK is involved in appetite regulation and ALA exerts anorexic, anti-obesity effects by suppressing hypothalamic AMPK activity. ALA suppresses AMPK phosphorylation and α2 MAPK activity in the arcuate nucleus but not in the paraventricular nucleus or the lateral hypothalamic area (Kim et al., 2004). Recently, a report shows that ALA (1800 mg/day) reduced body weight in obese diabetic human subjects (Koh et al., 2011; Table 1).
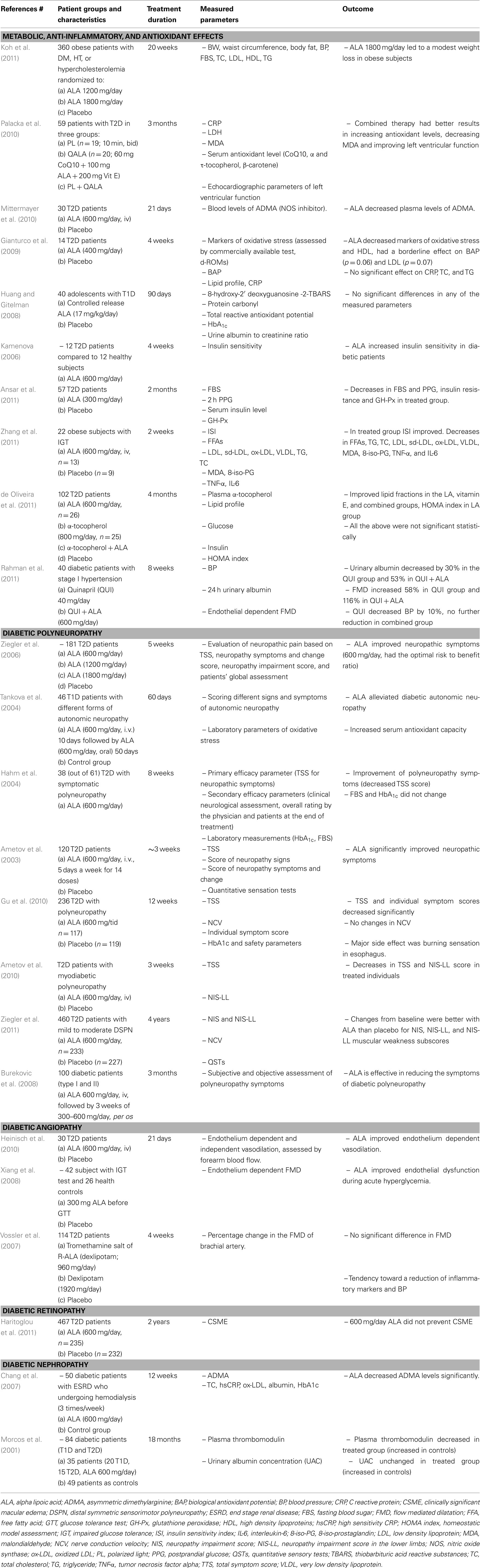
Table 1. Selected clinical trials with ALA in diabetic patients during the last 10 years.
As discussed earlier, some of the metabolic effects of LA could be secondary to its modifying effects on PPAR α/γ. Adenovirus mediated overexpression of PPAR coactivator-1α in the skeletal muscle increases mitochondrial respiration and elevates GLUT4 transcript levels (Michael et al., 2001). ALA increases mRNA expression of PPAR coactivator-1α, which might account for improved glucose homeostasis (Wang et al., 2010). It is also been shown that ALA prevented hyperglycemia-induced reduction of PPAR-γ, hyperinsulinemia, insulin resistance, systolic hypertension, and superoxide production (Midaoui et al., 2003; El Midaoui et al., 2006).
Alpha lipoic acid can have both detrimental and cytoprotective effects on pancreatic beta cells, based on the underlying pathophysiologic state and on the concentration of ALA. In type 2 diabetes, lowering of beta cell numbers is accompanied by a significant increase in beta cell apoptosis where oxidative stress is an important contributor (Butler et al., 2003; Marchetti et al., 2004). The prolonged AMPK activation by ALA leads to enhanced production of mitochondria-derived oxygen radicals and onset of an intrinsic mitochondrial apoptosis pathway (Targonsky et al., 2006; Cai et al., 2007). Using type 1 diabetic mice, Richards et al. (2005) reported that adenovirus mediated activation of AMPK in isolated mouse islets resulted in reduced glucose oxidation and insulin secretion and poorer glycemic control compared with islet cells infected with null or dominant negative AMPK viruses. However, a review of the literature indicates that ALA effects are concentration dependent (Bilska and Wlodek, 2005). For instance, in tumor cells LA induces cell proliferation at low concentrations (1 μmol/l) while it is anti-proliferative at higher concentrations (100 μmol/l; Dovinova et al., 1999). The concentration of ALA required for significant induction of AMPK activation in islet cells is at least 500 μmol/l (Targonsky et al., 2006), which is considerably greater than the plasma concentrations of ALA reached during treatment of diabetic neuropathy (Niebch et al., 1997; Chen et al., 2005). There are also several reports on the pro-oxidant effects of LA – which again, occur at different concentrations than those associated with its beneficial effects (Saris et al., 1998; Mottley and Mason, 2001; Dicter et al., 2002; Moini et al., 2002). However, it is reasonable to conclude that the beneficial/protective effects of ALA are most likely to occur under conditions of increased oxidative stress such as diabetes. For example, Lee et al. (2009) demonstrated dose-dependent, pro-apoptotic effects of LA in rat insulinoma cells, while in a related study, they also reported that pre-treatment with ALA reduced ROS production, mitochondrial membrane depolarization, and c-JNK activation in beta cells. It is believed that while ALA is detrimental to beta cells at high concentrations, it exerts cytoprotective effects on beta cells in diabetes at clinically relevant concentrations.
Another potential application for using LA as an adjuvant in the treatment of diabetes is related to its ability to inhibit glycation reactions. Although several mechanisms have been explained for the pathophysiology of chronic diabetic complications, protein glycation potentially can be an important underlying mechanism (Brownlee et al., 1988; Schalkwijk et al., 2002). Preventive effects of ALA in these reactions have been shown in different studies (Suzuki et al., 1992; Kawabata and Packer, 1994). Both LA and DHLA can protect albumin from glycation, suggesting that the preventive effect is independent of its redox state. It has been suggested that non-covalent hydrophobic binding to serum albumin is involved in this effect (Kawabata and Packer, 1994). Due to the presence of a hydrophobic carbon chain, it is likely that binding between albumin and α-lipoate is by hydrophobic interactions similar to that with fatty acids. This reaction leads to masking the glycation sites of serum albumin (Packer et al., 1995). Protective effects of ALA against LDL glycation have also been investigated in several studies (Kawabata and Packer, 1994; Schepkin et al., 1994). ALA was found not to protect LDL from glycation in the short term in vitro studies.
As stated earlier, ROS-induced vascular dysfunction is one of the main features of diabetic mellitus; this state of ROS accumulation is strongly associated with impaired endothelium dependent NO-mediated vasodilation. Activated PKC has a variety of effects on gene expression, such as decreased expression of eNOS and increases in the expression of endothelin, vascular endothelial growth factor, plasminogen activator inhibitor-1, transforming growth factor-β, NAD(P)H oxidases, and NFκB (which in turn activates many proinflammatory genes in the vasculature; Golbidi et al., 2011). LA improves the redox state of the plasma and endothelium dependent vasodilation (Heitzer et al., 2001; Sena et al., 2008). It is known that insulin receptor tyrosine kinase, phosphatidylinositol 3-kinase (PI 3-kinase), and Akt are essential components of insulin signaling pathways related to production of NO in vascular endothelium. Phosphorylation of endothelial nitric oxide synthase (eNOS) by Akt is also necessary for its activation by insulin (Montagnani et al., 2002). In aged endothelial cells, administration of LA partially restores the reductions in eNOS phosphorylation through Akt (Smith and Hagen, 2003). ALA also induces Akt phosphorylation in human umbilical vascular endothelial cells and the THP-1 human monocyte cell line (Artwohl et al., 2007; Zhang et al., 2007). These studies suggest that improved endothelial function due to ALA is at least partially attributed to recoupling of eNOS and increased NO bioavailability.
Asymmetric dimethylarginine (ADMA) is an endogenous NOS inhibitor that is produced by methylation of protein bound L-arginine in a reaction that is catalyzed by protein arginine methyltransferases (McBride and Silver, 2001). ADMA is excreted via the kidneys following metabolism by the enzyme dimethylarginine dimethylamino hydrolase (DDAH), which is sensitive to oxidative stress (Mittermayer et al., 2010). DDAH is expressed in different tissues such as kidneys, pancreas, brain, liver, lungs, endothelium, and myocardial cells (Kimoto et al., 1993; Nijveldt et al., 2003a,b). ADMA causes endothelial dysfunction in forearm resistance arteries (Calver et al., 1993) and increases the systemic vascular resistance and arterial blood pressure while decreasing cardiac output (Achan et al., 2003). An increased concentration of ADMA predicts cardiovascular events in different populations including T2D patients (Böger et al., 1997, 1998; Surdacki et al., 1999; Abbasi et al., 2001; Zoccali et al., 2001; Mittermayer et al., 2002; Stühlinger et al., 2002; Tarnow et al., 2004; Krzyzanowska et al., 2005, 2006, 2007). Indeed, ADMA may not only be a risk marker but also an etiologic factor in adverse cardiovascular events (Krzyzanowska et al., 2008). Reduced degradation of ADMA due to decreased DDAH activity has been proposed as an important mechanism for endothelial dependent vasodilator dysfunction in hypercholesterolemia (Ito et al., 1999) and diabetes (Lin et al., 2002). It has been reported that ALA decreases ADMA levels in the culture media of endothelial cells by increased expression and activity of DDAH and also of signal transducer and activator of transcription (STAT)3 phosphorylation. ALA also amplified STAT3 transfection-induced increase in DDAH II promoter activity (Lee et al., 2010). In two other studies, ALA was shown to reduce ADMA in diabetic patients with end stage renal disease (Chang et al., 2007) and with normal renal function (Mittermayer et al., 2010). These data provide a novel mechanism by which ALA regulates endothelial function.
Diabetic neuropathy is another potential clinical indication for ALA usage. At present, it is believed that several mechanisms contribute to the pathogenesis of this condition. These mechanisms for diabetic neuropathy are no longer being considered as being separate, but rather as a complex interplay giving rise to multiple interactions. The following mechanisms are thought to be involved in diabetic neuropathy: (a) increased flux through the polyol pathway with resulting sorbitol and fructose accumulation. Sorbitol production produces compensatory depletion of other organic osmolytes such as myo-inositol and taurine. The latter is an endogenous antioxidant, whose reduction leads to attenuation of oxidative defenses (Ziegler, 2006). Depletion of myo-inositol is associated with reduced Na+–K+-ATPase activity (Vallianou et al., 2009). (b) Endoneurial micro-vascular damage and hypoxia due to nitric oxide inactivation (Kishi et al., 1999). (c) Accumulation of AGEs and subsequent activation of NF-κB (Ziegler, 2006). (d) Homocysteinemia (Ambrosch et al., 2001). (e) Increased nerve lipid peroxidation (Low et al., 1997). (f) Modulation of MAPKs (Cavaletti et al., 2007). (g) Abnormal Ca2+ homeostasis and signaling (Hall et al., 2001) and (h) Decreased expression and level of neurotrophic factors, such as nerve growth factor, neurotrophin 3, and insulin like growth factor as well as alterations in axonal transport. LA has direct or indirect corrective effects on nearly all of these mechanisms. In a small study on a group of diabetic patients with neuropathy, ALA increased low pre-treatment level of plasma nitrates and nitrites, which are commonly, used markers of NO production. Increased NO production theoretically increases circulation to the neurons (Strokov et al., 2000). In another in vitro study, addition of ALA resulted in reduction of hyperglycemia-induced lipid peroxidation in both brain and sciatic tissues as measured by TBARS (Nickander et al., 1996). This effect on lipid peroxidation was confirmed in a study of 10 diabetic patients with peripheral neuropathy. A single daily dose of 600 mg LA for 70 days reduced serum lipid peroxidation (Androne et al., 2000). ALA also reduces protein glycosylation (measured by glycated hemoglobin), lower lipid peroxidation, and increase Na+/K+-ATPase activity in human red blood cells exposed to high glucose concentration (Jain and Lim, 2000).
The effects of LA on diabetic neuropathy have also been studied in streptozotocin-induced diabetic rats, where ALA promoted glucose uptake by nerve cells (Kishi et al., 1999), increased nerve myo-inositol (Kishi et al., 1999; Stevens et al., 2000), GSH levels (Böger et al., 1998; Stühlinger et al., 2002), Na/K-ATPase activity (Stevens et al., 2000), and nerve blood flow (Nagamatsu et al., 1995) while normalizing NAD/NADH ratios (Stevens et al., 2000).
Conclusion
The many unique properties of ALA and its interaction with other important antioxidant such as vitamin E, ascorbate, and GSH provide a fertile field for continued research. A number of experimental and clinical studies have shown the beneficial effect of ALA as a therapeutic agent for a diverse spectrum of diseases from heavy metal poisoning, radiation damage, ischemia-reperfusion injury, and neurodegenerative diseases to managing metabolic syndrome, diabetes, and its various complications. ALA/DHLA redox couple affects important biological processes including the regulation of several gene transcriptions and the activity of enzymes and receptors. Specifically in diabetes, it prevents beta cell destruction, enhances glucose uptake, and its antioxidant effects may be particularly useful in slowing the development of diabetic complications such as diabetic neuropathy. Given the growing evidence for increased oxidative stress and inflammatory pathways activated in diabetes, therapeutic paradigms may have to shift to target these processes through the use of compounds such as ALA. So far, the strongest evidence for the clinical use of ALA is from double blind, placebo controlled trials showing that LA administration significantly ameliorates polyneuropathies associated with diabetes. However, further work is necessary to assess the potential therapeutic roles for other diabetes-associated complications such as retinopathy and cataract. The lack of clear-cut benefits in some of the clinical trials to date does not disprove the benefits of ALA. It is believed that better designed experiments with appropriate doses, duration, and the selection of populations based on a specific pathophysiology may provide evidence for some of the hidden therapeutic potential of ALA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbasi, F., Asagmi, T., Cooke, J. P., Lamendola, C., McLaughlin, T., Reaven, G. M., Stuehlinger, M., and Tsao, P. S. (2001). Plasma concentrations of asymmetric dimethylarginine are increased in patients with type 2 diabetes mellitus. Am. J. Cardiol. 88, 1201–1203.
Achan, V., Broadhead, M., Malaki, M., Whitley, G., Leiper, J., MacAllister, R., and Vallance, P. (2003). Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler. Thromb. Vasc. Biol. 23, 1455–1459.
Aliciguzel, Y., Ozen, I., Aslan, M., and Karayalcin, U. (2003). Activities of xanthine oxidoreductase and antioxidant enzymes in different tissues of diabetic rats. J. Lab. Clin. Med. 142, 172–177.
Ambrosch, A., Dierkes, J., Lobmann, R., Kühne, W., König, W., Luley, C., and Lehnert, H. (2001). Relation between homocysteinaemia and diabetic neuropathy in patients with type 2 diabetes mellitus. Diabet. Med. 18, 185–192.
Ametov, A. S., Barinov, A., Dyck, P. J., Hermann, R., Kozlova, N., Litchy, W. J., Low, P. A., Nehrdich, D., Novosadova, M., O’Brien, P. C., Reljanovic, M., Samigullin, R., Schuette, K., Strokov, I., Tritschler, H. J., Wessel, K., Yakhno, N., Ziegler, D., and SYDNEY Trial Study Group. (2003). The sensory symptoms of diabetic polyneuropathy are improved with alpha-lipoic acid: the SYDNEY trial. Diabetes Care 26, 770–776.
Ametov, A. S., Novosadova, M. V., Barinov, A. N., Samigullin, R., and Trischler, H. J. (2010). Long-term effect of 3-week intravenous alpha-lipoic acid administration in symptomatic diabetic polyneutropathy with clinical manifestations. Ter. Arkh. 82, 61–64.
Anderson, K. A., Means, R. L., Huang, Q. H., Kemp, B. E., Goldstein, E. G., Selbert, M. A., Edelman, A. M., Fremeau, R. T., and Means, A. R. (1998). Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2/calmodulin-dependent protein kinase beta. J. Biol. Chem. 273, 31880–31889.
Anderson, K. A., Ribar, T. J., Lin, F., Noeldner, P. K., Green, M. F., Muehlbauer, M. J., Witters, L. A., Kemp, B. E., and Means, A. R. (2008). Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 7, 377–388.
Andersson, U., Filipsson, K., Abbott, C. R., Woods, A., Smith, K., Bloom, S. R., Carling, D., and Small, C. J. (2004). AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 279, 12005–12008.
Androne, L., Gavan, N. A., Veresiu, I. A., and Orasan, R. (2000). In vivo effect of lipoic acid on lipid peroxidation in patients with diabetic neuropathy. In vivo 14, 327–330.
Ansar, H., Mazloom, Z., Kazemi, F., and Hejazi, N. (2011). Effect of alpha-lipoic acid on blood glucose, insulin resistance and glutathione peroxidase of type 2 diabetic patients. Saudi Med. J. 32, 584–588.
Artwohl, M., Muth, K., Kosulin, K., de Martin, R., Hölzenbein, T., Rainer, G., Freudenthaler, A., Huttary, N., Schmetterer, L., Waldhäusl, W. K., and Baumgartner-Parzer, S. M. (2007). R-(+)-alpha-lipoic acid inhibits endothelial cell apoptosis and proliferation: involvement of Akt and retinoblastoma protein/E2F-1. Am. J. Physiol. Endocrinol. Metab. 293, E681–E69.
Baldwin, A. S. Jr. (1996). The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu. Rev. Immunol. 14, 649–683.
Barnes, P. J., and Karin, M. (1997). Nuclear factor-kappa B: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 336, 1066–1071.
Bashan, N., Kovsan, J., Kachko, I., Ovadia, H., and Rudich, A. (2009). Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol. Rev. 89, 27–71.
Bast, A., and Haenen, G. R. (2003). Lipoic acid: a multifunctional antioxidant. Biofactors 17, 207–213.
Beckman, K. B., and Ames, B. N. (1998). The free radical theory of aging matures. Physiol. Rev. 78, 547–581.
Belfiore, A., Genua, M., and Malaguarnera, R. (2009). PPAR-gamma agonists and their effects on IGF-I receptor signaling: implications for cancer. PPAR Res. 2009, 830501.
Berger, J., and Moller, D. E. (2002). The mechanisms of action of PPARs. Annu. Rev. Med. 53, 409–435.
Berkson, B. M. (1999). A conservative triple antioxidant approach to the treatment of hepatitis C. Combination of alpha lipoic acid (thioctic acid), silymarin, and selenium: three case histories. Med. Klin. (Munich); 94(Suppl. 3), 84–89.
Berlett, B. S., and Stadtman, E. R. (1997). Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 272, 20313–20316.
Biewenga, G. P., Haenen, G. R., and Bast, A. (1997). The pharmacology of the antioxidant lipoic acid. Gen. Pharmacol. 29, 315–331.
Böger, R. H., Bode-Böger, S. M., Szuba, A., Tsao, P. S., Chan, J. R., Tangphao, O., Blaschke, T. F., and Cooke, J. P. (1998). Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation 98, 1842–1847.
Böger, R. H., Bode-Böger, S. M., Thiele, W., Junker, W., Alexander, K., and Frölich, J. C. (1997). Biochemical evidence for impaired nitric oxide synthesis in patients with peripheral arterial occlusive disease. Circulation 95, 2068–2074.
Bonomi, F., Cerioli, A., and Pagani, S. (1989). Molecular aspects of the removal of ferritin-bound iron by DL-dihydrolipoate. Biochim. Biophys. Acta 994, 180–186.
Boullier, A., Bird, D. A., Chang, M. K., Dennis, E. A., Friedman, P., Gillotre-Taylor, K., Hörkkö, S., Palinski, W., Quehenberger, O., Shaw, P., Steinberg, D., Terpstra, V., and Witztum, J. L. (2001). Scavenger receptors, oxidized LDL, and atherosclerosis. Ann. N. Y. Acad. Sci. 947, 214–222.
Brownlee, M. (2001). Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820.
Brownlee, M., Cerami, A., and Vlassara, H. (1988). Advanced products of nonenzymatic glycosylation and the pathogenesis of diabetic vascular disease. Diabetes Metab. Rev. 4, 437–451.
Burekovic, A., Terzic, M., Alajbegovic, S., Vukojevic, Z., and Hadzic, N. (2008). The role of alpha-lipoic acid in diabetic polyneuropathy treatment. Bosn. J. Basic Med. Sci. 8, 341–345.
Busby, R. W., Schelvis, J. P. M., Yu, D. S., Babcock, G. T., and Marletta, M. A. (1999). Lipoic acid biosynthesis: LipA is an iron-sulphur protein. J. Am. Chem. Soc. 121, 4706–4707.
Busse, E., Zimmer, G., Schopohl, B., and Kornhuber, B. (1992). Influence of alpha-lipoic acid on intracellular glutathione in vitro and in vivo. Arzneimittelforschung 42, 829–831.
Bustamante, J., Lodge, J. K., Marcocci, L., Tritschler, H. J., Packer, L., and Rihn, B. H. (1998). Alpha-lipoic acid in liver metabolism and disease. Free Radic. Biol. Med. 24, 1023–1039.
Butler, A. E., Janson, J., Bonner-Weir, S., Ritzel, R., Rizza, R. A., and Butler, P. C. (2003). Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102110.
Butler, J. A., Hagen, T. M., and Moreau, R. (2009). Lipoic acid improves hypertriglyceridemia by stimulating triacylglycerol clearance and downregulating liver triacylglycerol secretion. Arch. Biochem. Biophys. 485, 63–71.
Cai, Y., Martens, G. A., Hinke, S. A., Heimberg, H., Pipeleers, D., and Van de Casteele, M. (2007). Increased oxygen radical formation and mitochondrial dysfunction mediate beta cell apoptosis under conditions of AMP-activated protein kinase stimulation. Free Radic. Biol. Med. 42, 64–78.
Calver, A., Collier, J., Leone, A., Moncada, S., and Vallance, P. (1993). Effect of local intra-arterial asymmetric dimethylarginine (ADMA) on the forearm arteriolar bed of healthy volunteers. J. Hum. Hypertens. 7, 193–194.
Cavaletti, G., Miloso, M., Nicolini, G., Scuteri, A., and Tredici, G. (2007). Emerging role of mitogen-activated protein kinases in peripheral neuropathies. J. Peripher. Nerv. Syst. 12, 175–194.
Ceriello, A., Mercuri, F., Quagliaro, L., Assaloni, R., Motz, E., Tonutti, L., and Taboga, C. (2001). Detection of nitrotyrosine in the diabetic plasma: evidence of oxidative stress. Diabetologia 44, 834–838.
Chang, J. W., Lee, E. K., Kim, T. H., Min, W. K., Chun, S., Lee, K. U., Kim, S. B., and Park, J. S. (2007). Effects of alpha-lipoic acid on the plasma levels of asymmetric dimethylarginine in diabetic end-stage renal disease patients on hemodialysis: a pilot study. Am. J. Nephrol. 27, 70–74.
Chen, J., Jiang, W., Cai, J., Tao, W., Gao, X., and Jiang, X. (2005). Quantification of lipoic acid in plasma by high-performance liquid chromatography electrospray ionization mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 824, 249–257.
Cheung, P. C., Salt, I. P., Davies, S. P., Hardie, D. G., and Carling, D. (2000). Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem. J. 346(Pt 3), 659–669.
Cox, A. G., Winterbourn, C. C., and Hampton, M. B. (2009). Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signaling. Biochem. J. 425, 313–325.
de Oliveira, A. M., Rondó, P. H., Luzia, L. A., D’Abronzo, F. H., and Illison, V. K. (2011). The effects of lipoic acid and α-tocopherol supplementation on the lipid profile and insulin sensitivity of patients with type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled trial. Diabetes Res. Clin. Pract. 92, 253–260.
Dickinson, D. A., and Forman, H. J. (2002a). Cellular glutathione and thiols metabolism. Biochem. Pharmacol. 64, 1021–1028.
Dickinson, D. A., and Forman, H. J. (2002b). Glutathione in defense and signaling: lessons from a small thiol. Ann. N. Y. Acad. Sci. 973, 488–504.
Dicter, N., Madar, Z., and Tirosh, O. (2002). Alpha-lipoic acid inhibits glycogen synthesis in rat soleus muscle via its oxidative activity and the uncoupling of mitochondria. J. Nutr. 132, 3001–3006.
Dovinova, I., Novotny, L., Rauko, P., and Kvasnicka, P. (1999). Combined effect of lipoic acid and doxorubicyn in murine leukemia. Neoplasma 46, 237–241.
Duckworth, W. C. (2001). Hyperglycemia and cardiovascular disease. Curr. Atheroscler. Rep. 3, 383–391.
El Midaoui, A., Wu, L., Wang, R., and de Champlain, J. (2006). Modulation of cardiac and aortic peroxisome proliferator-activated receptor-gamma expression by oxidative stress in chronically glucose-fed rats. Am. J. Hypertens. 19, 407–412.
Etoh, T., Inoguchi, T., Kakimoto, M., Sonoda, N., Kobayashi, K., Kuroda, J., Sumimoto, H., and Nawata, H. (2003). Increased expression of NAD(P)H oxidase subunits, NOX4 and p22phox, in the kidney of streptozotocin-induced diabetic rats and its reversibity by interventive insulin treatment. Diabetologia 46, 1428–1437.
Evans, J. L., Goldfine, I. D., Maddux, B. A., and Grodsky, G. M. (2002). Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr. Rev. 23, 599–622.
Foretz, M., Ancellin, N., Andreelli, F., Saintillan, Y., Grondin, P., Kahn, A., Thorens, B., Vaulont, S., and Viollet, B. (2005). Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes 54, 1331–1339.
Gardner, A. M., Lange-Carter, C. A., Vaillancourt, R. R., and Johnson, G. L. (1994). Measuring activation of kinases in mitogen-activated protein kinase regulatory network. Meth. Enzymol. 238, 258–270.
Gianturco, V., Bellomo, A., D’Ottavio, E., Formosa, V., Iori, A., Mancinella, M., Troisi, G., and Marigliano, V. (2009). Impact of therapy with alpha-lipoic acid (ALA) on the oxidative stress in the controlled NIDDM: a possible preventive way against the organ dysfunction? Arch. Gerontol. Geriatr. 49(Suppl.), 129–133.
Gilmore, T. D. (2006). Introduction to NF-kappa B: players, pathways, perspectives. Oncogene 25, 6680–6684.
Gleiter, C. H., Schug, B. S., Hermann, R., Elze, M., Blume, H. H., and Gundert-Remy, U. (1996). Influence of food intake on the bioavailability of thioctic acid enantiomers. Eur. J. Clin. Pharmacol. 50, 513–514.
Golbidi, S., Ebadi, S. A., and Laher, I. (2011). Antioxidants in the treatment of diabetes. Curr. Diabetes Rev. 7, 106–125.
Golbidi, S., and Laher, I. (2010). Antioxidant therapy in human endocrine disorders. Med. Sci. Monit. 16, RA9–RA24.
Goodpaster, B. H., and Kelley, D. E. (2002). Skeletal muscle triglyceride: marker or mediator of obesity-induced insulin resistance in type 2 diabetes mellitus? Curr. Diab. Rep. 2, 216–222.
Goralska, M., Dackor, R., Holley, B., and McGahan, M. C. (2003). Alpha lipoic acid changes iron uptake and storage in lens epithelial cells. Exp. Eye Res. 76, 241–248.
Green, K., Brand, M. D., and Murphy, M. P. (2004). Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 53, S110–S118.
Griendling, K. K., and FitzGerald, G. A. (2003). Oxidative stress and cardiovascular injury: part I: basic mechanisms and in vivo monitoring of ROS. Circulation 108, 1912–1916.
Gu, X. M., Zhang, S. S., Wu, J. C., Tang, Z. Y., Lu, Z. Q., Li, H., Liu, C., Chen, L., and Ning, G. (2010). Efficacy and safety of high-dose α-lipoic acid in the treatment of diabetic polyneuropathy. Zhonghua Yi Xue Za Zhi 90, 2473–2476.
Guzik, T. J., Mussa, S., Gastaldi, D., Sadowski, J., Ratnatunga, C., Pillai, R., and Channon, K. M. (2002). Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 105, 1656–1662.
Guzik, T. J., West, N. E., Black, E., McDonald, D., Ratnatunga, C., Pillai, R., and Channon, K. M. (2000). Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ. Res. 86, E85–E90.
Hagen, T. M., Vinarsky, V., Wehr, C. M., and Ames, B. N. (2000). (R)-alpha-lipoic acid reverses the age-associated increase in susceptibility of hepatocytes to tert-butylhydroperoxide both in vitro and in vivo. Antioxid. Redox Signal. 2, 473–483.
Hahm, J. R., Kim, B. J., and Kim, K. W. (2004). Clinical experience with thioctacid (thioctic acid) in the treatment of distal symmetric polyneuropathy in Korean diabetic patients. J. Diabetes Complicat. 18, 79–85.
Hall, K. E., Liu, J., Sima, A. A., and Wiley, J. W. (2001). Impaired inhibitory G-protein function contributes to increased calcium currents in rats with diabetic neuropathy. J. Neurophysiol. 86, 760–770.
Han, D., Handelman, G., Marcocci, L., Sen, C. K., Roy, S., Kobuchi, H., Tritschler, H. J., Flohé, L., and Packer, L. (1997). Lipoic acid increases de novo synthesis of cellular glutathione by improving cystine utilization. Biofactors 6, 321–338.
Haritoqlou, C., Gerss, J., Hammes, H. P., Kampik, A., Ulbig, M. W., and RETIPON Study Group. (2011). Alpha lipoic acid for the prevention of diabetic macular edema. Ophthalmologica 226, 127–137.
Harrison, E. H., and McCormick, D. B. (1974). The metabolism of dl-(1,6-14C)lipoic acid in the rat. Arch. Biochem. Biophys. 160, 514–522.
Hawley, S. A., Pan, D. A., Mustard, K. J., Ross, L., Bain, J., Edelman, A. M., Frenguelli, B. G., and Hardie, D. G. (2005). Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2, 9–19.
Heinisch, B. B., Francesconi, M., Mittermayer, F., Schaller, G., Gouya, G., Wolzt, M., and Pleiner, J. (2010). Alpha-lipoic acid improves vascular endothelial function in patients with type 2 diabetes: a placebo-controlled randomized trial. Eur. J. Clin. Invest. 40, 148–154.
Heitzer, T., Finckh, B., Albers, S., Krohn, K., Kohlschütter, A., and Meinertz, T. (2001). Beneficial effects of alpha-lipoic acid and ascorbic acid on endothelium-dependent, nitric oxide-mediated vasodilation in diabetic patients: relation to parameters of oxidative stress. Free Radic. Biol. Med. 31, 53–61.
Hermann, R., Hiebch, G., Borbe, H. O., Fieger-Buschgesf, H., Ruus, P., Nowak, H., Riethmuller-Winzen, H., Peukert, M., and Blume, H. (1996). Enantioselective pharmacokinetics and bioavailability of different racemic alpha lipoic acid formulations in healthy volunteers. Eur. J. Pharm. Sci. 4, 167–174.
Huang, E. A., and Gitelman, S. E. (2008). The effect of oral alpha-lipoic acid on oxidative stress in adolescents with type 1 diabetes mellitus. Pediatr. Diabetes 9, 69–73.
Hurley, R. L., Anderson, K. A., Franzone, J. M., Kemp, B. E., Means, A. R., and Witters, L. A. (2005). The Ca2/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 280, 29060–29066.
Ito, A., Tsao, P. S., Adimoolam, S., Kimoto, M., Ogawa, T., and Cooke, J. P. (1999). Novel mechanism for endothelial dysfunction: dysregulation of dimethylarginine dimethylaminohydrolase. Circulation 99, 3092–3095.
Jain, S. K., and Lim, G. (2000). Lipoic acid decreases lipid peroxidation and protein glycosylation and increases (Na(+) + K(+)) and Ca(++)ATPase activities in high glucose-treated human erythrocytes. Free Radic. Biol. Med. 29, 1122–1128.
Jensen, T. E., Rose, A. J., Jorgensen, S. B., Brandt, N., Schjerling, P., Wojtaszewski, J. F., and Richter, E. A. (2007). Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am. J. Physiol. Endocrinol. Metab. 292, E1308–E1317.
Jones, W., Li, X., Qu, Z. C., Perriott, L., Whitesell, R. R., and May, J. M. (2002). Uptake, recycling, and antioxidant actions of alpha-lipoic acid in endothelial cells. Free Radic. Biol. Med. 33, 83–93.
Kamenova, P. (2006). Improvement of insulin sensitivity in patients with type 2 diabetes mellitus after oral administration of alpha-lipoic acid. Hormones (Athens) 5, 251–258.
Kawabata, T., and Packer, L. (1994). Alpha-lipoate can protect against glycation of serum albumin, but not low density lipoprotein. Biochem. Biophys. Res. Commun. 203, 99–104.
Kim, M. S., Park, J. Y., Namkoong, C., Jang, P. G., Ryu, J. W., Song, H. S., Yun, J. Y., Namgoong, I. S., Ha, J., Park, I. S., Lee, I. K., Viollet, B., Youn, J. H., Lee, H. K., and Lee, K. U. (2004). Anti-obesity effects of alpha-lipoic acid mediated by suppression of hypothalamic AMP-activated protein kinase. Nat. Med. 10, 727–733.
Kimoto, M., Tsuji, H., Ogawa, T., and Sasaoka, K. (1993). Detection of NG, NG-dimethylarginine dimethylaminohydrolase in the nitric oxide-generating systems of rats using monoclonal antibody. Arch. Biochem. Biophys. 300, 657–662.
Kishi, Y., Schmelzer, J. D., Yao, J. K., Zollman, P. J., Nickander, K. K., Tritschler, H. J., and Low, P. A. (1999). Alpha-lipoic acid: effect on glucose uptake, sorbitol pathway, and energy metabolism in experimental diabetic neuropathy. Diabetes 48, 2045–2051.
Kitada, M., Koya, D., Sugimoto, T., Isono, M., Araki, S., Kashiwagi, A., and Haneda, M. (2003). Translocation of glomerular p47phox and p67phox by protein kinase C-beta activation is required for oxidative stress in diabetic nephropathy. Diabetes 52, 2603–2614.
Koh, E. H., Lee, W. J., Lee, S. A., Kim, E. H., Cho, E. H., Jeong, E., Kim, D. W., Kim, M. S., Park, J. Y., Park, K. G., Lee, H. J., Lee, I. K., Lim, S., Jang, H. C., Lee, K. H., and Lee, K. U. (2011). Effects of alpha-lipoic acid on body weight in obese subjects. Am. J. Med. 124, 851–858.
Kola, B., Boscaro, M., Rutter, G. A., Grossman, A. B., and Korbonits, M. (2006). Expanding role of AMPK in endocrinology. Trends Endocrinol. Metab. 17, 205–215.
Konrad, D., Somwar, R., Sweeney, G., Yaworsky, K., Hayashi, M., Ramlal, T., and Klip, A. (2001). The antihyperglycemic drug alpha-lipoic acid stimulates glucose uptake via both GLUT4 translocation and GLUT4 activation: potential role of p38 mitogen-activated protein kinase in GLUT4 activation. Diabetes 50, 1464–1471.
Krzyzanowska, K., Mittermayer, F., Krugluger, W., Schnack, C., Hofer, M., Wolzt, M., and Schernthaner, G. (2006). Asymmetric dimethylarginine is associated with macrovascular disease and total homocysteine in patients with type 2 diabetes. Atherosclerosis 189, 236–240.
Krzyzanowska, K., Mittermayer, F., Schnack, C., Hofer, M., Wolzt, M., and Schernthaner, G. (2005). Circulating ADMA concentrations are elevated in hypopituitary adults with and without growth hormone deficiency. Eur. J. Clin. Invest. 35, 208–213.
Krzyzanowska, K., Mittermayer, F., Shnawa, N., Hofer, M., Schnabler, J., Etmüller, Y., Kapiotis, S., Wolzt, M., and Schernthaner, G. (2007). Asymmetrical dimethylarginine is related to renal function, chronic inflammation and macroangiopathy in patients with type 2 diabetes and albuminuria. Diabet. Med. 24, 81–86.
Krzyzanowska, K., Mittermayer, F., Wolzt, M., and Schernthaner, G. (2008). ADMA, cardiovascular disease and diabetes. Diabetes Res. Clin. Pract. 82(Suppl.), :s1226.
Kyriakis, J. M., and Avruch, J. (2001). Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81, 807–869.
Lee, B. W., Kwon, S. J., Chae, H. Y., Kang, J. G., Kim, C. S., Lee, S. J., Yoo, H. J., Kim, J. H., Park, K. S., and Ihm, S. H. (2009). Dose-related cytoprotective effect of alpha-lipoic acid on hydrogen peroxide-induced oxidative stress to pancreatic beta cells. Free Radic. Res. 43, 68–77.
Lee, C. K., Lee, E. Y., Kim, Y. G., Mun, S. H., Moon, H. B., and Yoo, B. (2008). Alpha-lipoic acid inhibits TNF-alpha induced NF-kappa B activation through blocking of MEKK1-MKK4-IKK signaling cascades. Int. Immunopharmacol. 8, 362–370.
Lee, S., Park, Y., Zuidema, M. Y., Hannink, M., and Zhang, C. (2011). Effects of interventions on oxidative stress and inflammation of cardiovascular diseases. World J. Cardiol. 3, 18–24.
Lee, W. J., Kim, S. W., Kim, G. H., Han, S. M., Won, J. C., Jung, C. H., Park, H. S., Choi do, S., Lee, K. U., and Park, J. Y. (2010). Alpha-lipoic acid activates dimethylarginine dimethylaminohydrolase in cultured endothelial cells. Biochem. Biophys. Res. Commun. 398, 653–658.
Lee, W. J., Song, K. H., Koh, E. H., Won, J. C., Kim, H. S., Park, H. S., Kim, M. S., Kim, S. W., Lee, K. U., and Park, J. Y. (2005a). Alpha-lipoic acid increases insulin sensitivity by activating AMPK in skeletal muscle. Biochem. Biophys. Res. Commun. 332, 885–891.
Lee, W. J., Koh, E. H., Won, J. C., Kim, M. S., Park, J. Y., and Lee, K. U. (2005b). Obesity: the role of hypothalamic AMP-activated protein kinase in body weight regulation. Int. J. Biochem. Cell Biol. 37, 2254–2259.
Lin, K. Y., Ito, A., Asagami, T., Tsao, P. S., Adimoolam, S., Kimoto, M., Tsuji, H., Reaven, G. M., and Cooke, J. P. (2002). Impaired nitric oxide synthase pathway in diabetes mellitus: role of asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation 106, 987–992.
Liu, Y., and Gutterman, D. D. (2002). The coronary circulation in diabetes: influence of reactive oxygen species on K channel-mediated vasodilation. Vascul. Pharmacol. 38, 43–49.
Liu, Y., Terata, K., Chai, Q., Li, H., Kleinman, L. H., and Gutterman, D. D. (2002). Peroxynitrite inhibits Ca2+-activated K+ channel activity in smooth muscle of human coronary arterioles. Circ. Res. 91, 1070–1076.
Lochhead, P. A., Salt, I. P., Walker, K. S., Hardie, D. G., and Sutherland, C. (2000). 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes 49, 896–903.
Low, P. A., Nickander, K. K., and Tritschler, H. J. (1997). The roles of oxidative stress and antioxidant treatment in experimental diabetic neuropathy. Diabetes 46, s38–s42.
Marchetti, P., Del Guerra, S., Marselli, L., Lupi, R., Masini, M., Pollera, M., Bugliani, M., Boggi, U., Vistoli, F., Mosca, F., and Del Prato, S. (2004). Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J. Clin. Endocrinol. Metab. 89, 5535–5541.
Maritim, A. C., Sanders, R. A., and Watkins, J. B. III. (2003). Diabetes, oxidative stress, and antioxidants: a review. J. Biochem. Mol. Toxicol. 17, 24–38.
Mary, J., Vougier, S., Picot, C. R., Perichon, M., Petropoulos, I., and Friguet, B. (2004). Enzymatic reactions involved in the repair of oxidized proteins. Exp. Gerontol. 39, 1117–1123.
McBride, A. E., and Silver, P. A. (2001). State of the arg: protein methylation at arginine comes of age. Cell 106, 5–8.
McCarty, M. F., Barroso-Aranda, J., and Contreras, F. (2009). The “rejuvenatory” impact of lipoic acid on mitochondrial function in aging rats may reflect induction and activation of PPAR-gamma coactivator-1alpha. Med. Hypotheses 72, 29–33.
McLellan, L. I., Lewis, A. D., Hall, D. J., Ansell, J. D., and Wolf, C. R. (1995). Uptake and distribution of N-acetylcysteine in mice: tissue-specific effects on glutathione concentrations. Carcinogenesis 16, 2099–2106.
Meffert, M. K., Chang, J. M., Wiltgen, B. J., Fanselow, M. S., and Baltimore, D. (2003). NF-kappa B functions in synaptic signaling and behavior. Nat. Neurosci. 6, 1072–1078.
Michael, L. F., Wu, Z., Cheatham, R. B., Puigserver, P., Adelmant, G., Lehman, J. J., Kelly, D. P., and Spiegelman, B. M. (2001). Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc. Natl. Acad. Sci. U.S.A. 98, 3820–3825.
Michalik, L., Auwerx, J., Berger, J. P., Chatterjee, V. K., Glass, C. K., Gonzalez, F. J., Grimaldi, P. A., Kadowaki, T., Lazar, M. A., O’Rahilly, S., Palmer, C. N., Plutzky, J., Reddy, J. K., Spiegelman, B. M., Staels, B., and Wahli, W. (2006). International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 58, 726–741.
Midaoui, A. E., Elimadi, A., Wu, L., Haddad, P. S., and de Champlain, J. (2003). Lipoic acid prevents hypertension, hyperglycemia, and the increase in heart mitochondrial superoxide production. Am. J. Hypertens. 16, 173–179.
Minokoshi, Y., Alquier, T., Furukawa, N., Kim, Y. B., Lee, A., Xue, B., Mu, J., Foufelle, F., Ferré, P., Birnbaum, M. J., Stuck, B. J., and Kahn, B. B. (2004). AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428, 569–574.
Mittermayer, F., Mayer, B. X., Meyer, A., Winzer, C., Pacini, G., Wagner, O. F., Wolzt, M., and Kautzky-Willer, A. (2002). Circulating concentrations of asymmetrical dimethyl-L-arginine are increased in women with previous gestational diabetes. Diabetologia 45, 1372–1378.
Mittermayer, F., Pleiner, J., Francesconi, M., and Wolzt, M. (2010). Treatment with alpha-lipoic acid reduces asymmetric dimethylarginine in patients with type 2 diabetes mellitus. Transl. Res. 155, 6–9.
Moini, H., Packer, L., and Saris, N. E. (2002). Antioxidant and prooxidant activities of alpha-lipoic acid and dihydrolipoic acid. Toxicol. Appl. Pharmacol. 182, 84–90.
Montagnani, M., Ravichandran, L. V., Chen, H., Esposito, D. L., and Quon, M. J. (2002). Insulin receptor substrate-1 and phosphoinositide-dependent kinase-1 are required for insulin-stimulated production of nitric oxide in endothelial cells. Mol. Endocrinol. 16, 1931–1942.
Morcos, M., Borcea, V., Isermann, B., Gehrke, S., Ehret, T., Henkels, M., Schiekofer, S., Hofmann, M., Amiral, J., Tritschler, H., Ziegler, R., Wahl, P., and Nawroth, P. P. (2001). Effect of alpha-lipoic acid on the progression of endothelial cell damage and albuminuria in patients with diabetes mellitus: an exploratory study. Diabetes Res. Clin. Pract. 52, 175–183.
Mottley, C., and Mason, R. P. (2001). Sulfur-centered radical formation from the antioxidant dihydrolipoic acid. J. Biol. Chem. 276, 42677–42683.
Musi, N., Hirshman, M. F., Nygren, J., Svanfeldt, M., Bavenholm, P., Rooyackers, O., Zhou, G., Williamson, J. M., Ljunqvist, O., Efendic, S., Moller, D. E., Thorell, A., and Goodyear, L. J. (2002). Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 51, 2074–2081.
Nagamatsu, M., Nickander, K. K., Schmelzer, J. D., Raya, A., Wittrock, D. A., Tritschler, H., and Low, P. A. (1995). Lipoic acid improves nerve blood flow, reduces oxidative stress, and improves distal nerve conduction in experimental diabetic neuropathy. Diabetes Care 18, 1160–1167.
Nickander, K. K., McPhee, B. R., Low, P. A., and Tritschler, H. (1996). Alpha-lipoic acid: antioxidant potency against lipid peroxidation of neural tissues in vitro and implications for diabetic neuropathy. Free Radic. Biol. Med. 21, 631–639.
Niebch, G., Bu chele, B., Blome, J., Grieb, S., Brandt, G., Kampa, P., Raffel, H. H., Locher, M., Borbe, H. O., Nubert, I., and Fleischhauer, I. (1997). Enantioselective high-performance liquid chromatography assay of (+)R- and (-)S-alpha-lipoic acid in human plasma. Chirality 9, 32–36.
Nijveldt, R. J., Teerlink, T., Siroen, M. P., van Lambalgen, A. A., Rauwerda, J. A., and van Leeuwen, P. A. (2003a). The liver is an important organ in the metabolism of asymmetrical dimethylarginine (ADMA). Clin. Nutr. 22, 17–22.
Nijveldt, R. J., Teerlink, T., and van Leeuwen, P. A. (2003b). The asymmetrical dimethylarginine (ADMA)-multiple organ failure hypothesis. Clin. Nutr. 22, 99–104.
Nishikawa, T., Edelstein, D., Du, X. L., Yamagishi, S., Matsumura, T., Kaneda, Y., Yorek, M. A., Beebe, D., Oates, P. J., Hammes, H. P., Giardino, I., and Brownlee, M. (2000). Normalizing mitochondrial superoxide production blocks three pathways of hyperglycemic damage. Nature 404, 787–790.
Oberg, B. P., McMenamin, E., Lucas, F. L., McMonagle, E., Morrow, J., Ikizler, T. A., and Himmelfarb, J. (2004). Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 65, 1009–1016.
Ou, P., Tritschler, H. J., and Wolff, S. P. (1995). Thioctic (lipoic) acid: a therapeutic metal-chelating antioxidant? Biochem. Pharmacol. 50, 123–126.
Packer, L. (1998). alpha-Lipoic acid: a metabolic antioxidant which regulates NF-kappa B signal transduction and protects against oxidative injury. Drug Metab. Rev. 30, 245–275.
Packer, L., Witt, E. H., and Tritschler, H. J. (1995). Alpha-lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 19, 227–250.
Palacka, P., Kucharska, J., Murin, J., Dostalova, K., Okkelova, A., Cizova, M., Waczulikova, I., Moricova, S., and Gvozdjakova, A. (2010). Complementary therapy in diabetic patients with chronic complications: a pilot study. Bratisl. Lek. Listy 111, 205–211.
Pershadsingh, H. A., Ho, C. I., Rajamani, J., Lee, C., Chittiboyina, A. G., Deshpande, R., Kurtz, T. W., Chan, J. Y., Avery, M. A., and Benson, S. C. (2005). Alpha lipoic acid is a weak dual PPAR α/γ agonist an ester derivative with increased PPAR α/γ efficacy and antioxidant activity. J. Appl. Res. 5, 510–523.
Petersen Shay, K., Moreau, R. F., Smith, E. J., and Hagen, T. M. (2008). Is alpha-lipoic acid a scavenger of reactive oxygen species in vivo? Evidence for its initiation of stress signaling pathways that promote endogenous antioxidant capacity. IUBMB Life 60, 362–367.
Poh, Z. X., and Goh, K. P. (2009). A current update on the use of alpha lipoic acid in the management of type 2 diabetes mellitus. Endocr. Metab. Immune Disord. Drug Targets 9, 392–398.
Rahman, S. T., Merchant, N., Haque, T., Wahi, J., Bhaheetharan, S., Ferdinand, K. C., and Khan, B. V. (2011). The impact of lipoic acid on endothelial function and proteinuria in quinapril-treated diabetic patients with stage I hypertension: results from the quality study. J. Cardiovasc. Pharmacol. Ther. 12. [Epub ahead of print].
Richards, S. K., Parton, L. E., Leclerc, I., Rutter, G. A., and Smith, R. M. (2005). Over-expression of AMP-activated protein kinase impairs pancreatic {beta}-cell function in vivo. J. Endocrinol. 187, 225–235.
Ruderman, N., and Flier, J. S. (2001). Cell biology. Chewing the fat – ACC and energy balance. Science 291, 2558–2559.
Saha, A. K., Avilucea, P. R., Ye, J. M., Assifi, M. M., Kraegen, E. W., and Ruderman, N. B. (2004). Pioglitazone treatment activates AMP-activated protein kinase in rat liver and adipose tissue in vivo. Biochem. Biophys. Res. Commun. 314, 580–585.
Saris, N. E., Karjalainen, A., Teplova, V. V., and Lindros, K. O. (1998). The stimulation of the mitochondrial permeability transition by dihydrolipoate and alpha-lipoate. Biochem. Mol. Biol. Int. 44, 127–134.
Schalkwijk, C. G., Lieuw-a-Fa, M., van Hinsbergh, V. W., and Stehouwer, C. D. (2002). Pathophysiological role of amadori-glycated proteins in diabetic microangiopathy. Semin. Vasc. Med. 2, 191–197.
Schepkin, V., Kawabata, T., and Packer, L. (1994). NMR study of lipoic acid binding to bovine serum albumin. Biochem. Mol. Biol. Int. 33, 879–886.
Schmidt, A. M., Hori, O., Brett, J., Yan, S. D., Wautier, J. L., and Stern, D. (1994). Cellular receptors for advanced glycation end products. Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arterioscler. Thromb. 14, 1521–1528.
Sena, C. M., Nunes, E., Louro, T., Proença, T., Fernandes, R., Boarder, M. R., and Seiça, R. M. (2008). Effects of alpha-lipoic acid on endothelial function in aged diabetic and high-fat fed rats. Br. J. Pharmacol. 153, 894–906.
Shackelford, D. B., and Shaw, R. J. (2009). The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575.
Shay, K. P., Moreau, R. F., Smith, E. J., Smith, A. R., and Hagen, T. M. (2009). Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta 1790, 1149–1160.
Shen, Q. W., Zhu, M. J., Tong, J., Ren, J., and Du, M. (2007). Ca2+/calmodulin-dependent protein kinase kinase is involved in AMP-activated protein kinase activation by alpha-lipoic acid in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 293, C1395–C1403.
Singh, U., and Jialal, I. (2008). Alpha-lipoic acid supplementation and diabetes. Nutr. Rev. 66, 646–657.
Smith, A. R., and Hagen, T. M. (2003). Vascular endothelial dysfunction in aging: loss of Akt-dependent endothelial nitric oxide synthase phosphorylation and partial restoration by (R)-alpha-lipoic acid. Biochem. Soc. Trans. 31, 1447–1449.
Smith, A. R., Shenvi, S. V., Widlansky, M., Suh, J. H., and Hagen, T. M. (2004). Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr. Med. Chem. 11, 1135–1146.
Soriano, F. G., Virag, L., and Szabo, C. (2001). Diabetic endothelial dysfunction: role of reactive oxygen and nitrogen species production and poly(ADP-ribose) polymerase activation. J. Mol. Med. 79, 437–448.
Stahmann, N., Woods, A., Carling, D., and Heller, R. (2006). Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. Mol. Cell Biol. 26, 5933–5945.
Stevens, M. J., Obrosova, I., Cao, X., Van Huysen, C., and Greene, D. A. (2000). Effects of DL-alpha-lipoic acid on peripheral nerve conduction, blood flow, energy metabolism, and oxidative stress in experimental diabetic neuropathy. Diabetes 49, 1006–1015.
Strokov, I. A., Manukhina, E. B., Bakhtina, L. Y., Malyshev, I. Y., Zoloev, G. K., Kazikhanova, S. I., and Ametov, A. S. (2000). The function of endogenous protective systems in patients with insulin-dependent diabetes mellitus and polyneuropathy: effect of antioxidant therapy. Bull. Exp. Biol. Med. 130, 986–990.
Stühlinger, M. C., Abbasi, F., Chu, J. W., Lamendola, C., McLaughlin, T. L., Cooke, J. P., Reaven, G. M., and Tsao, P. S. (2002). Relationship between insulin resistance and an endogenous nitric oxide synthase inhibitor. JAMA 287, 1420–1426.
Suh, J. H., Moreau, R., Heath, S. H., and Hagen, T. M. (2005). Dietary supplementation with (R)-alpha-lipoic acid reverses the age-related accumulation of iron and depletion of antioxidants in the rat cerebral cortex. Redox Rep. 10, 52–60.
Suh, J. H., Wang, H., Liu, R. M., Liu, J., and Hagen, T. M. (2004). (R)-alpha-lipoic acid reverses the age-related loss in GSH redox status in post-mitotic tissues: evidence for increased cysteine requirement for GSH synthesis. Arch. Biochem. Biophys. 423, 126–135.
Surdacki, A., Nowicki, M., Sandmann, J., Tsikas, D., Boeger, R. H., Bode-Boeger, S. M., Kruszelnicka-Kwiatkowska, O., Kokot, F., Dubiel, J. S., and Froelich, J. C. (1999). Reduced urinary excretion of nitric oxide metabolites and increased plasma levels of asymmetric dimethylarginine in men with essential hypertension. J. Cardiovasc. Pharmacol. 33, 652–658.
Suzuki, Y. J., Tsuchiya, M., and Packer, L. (1992). Lipoate prevents glucose-induced protein modifications. Free Radic. Res. Commun. 17, 211–217.
Tamás, P., Hawley, S. A., Clarke, R. G., Mustard, K. J., Green, K., Hardie, D. G., and Cantrell, D. A. (2009). The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575.
Taniyama, Y., and Griendling, K. K. (2003). Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension 42, 1075–1081.
Tankova, T., Koev, D., and Dakovska, L. (2004). Alpha-lipoic acid in the treatment of autonomic diabetic neuropathy (controlled, randomized, open-label study). Rom. J. Intern. Med. 42, 457–464.
Targonsky, E. D., Dai, F., Koshkin, V., Karaman, G. T., Gyulkhandanyan, A. V., Zhang, Y., Chan, C. B., and Wheeler, M. B. (2006). Alpha-lipoic acid regulates AMP-activated protein kinase and inhibits insulin secretion from beta cells. Diabetologia 49, 1587–1598.
Tarnow, L., Hovind, P., Teerlink, T., Stehouwer, C. D., and Parving, H. H. (2004). Elevated plasma asymmetric dimethylarginine as a marker of cardiovascular morbidity in early diabetic nephropathy in type 1 diabetes. Diabetes Care 27, 765–769.
Teichert, J., Tuemmers, T., Achenbach, H., Preiss, C., Hermann, R., Ruus, P., and Preiss, R. (2005). Pharmacokinetics of alpha-lipoic acid in subjects with severe kidney damage and end-stage renal disease. J. Clin. Pharmacol. 45, 313–328.
Tian, B., and Brasier, A. R. (2003). Identification of a nuclear factor kappa B-dependent gene network. Recent Prog. Horm. Res. 58, 95–130.
Turko, I. V., Marcondes, S., and Murad, F. (2001). Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA: 3-oxoacid CoA-transferase. Am. J. Physiol. Heart Circ. Physiol. 281, H2289–H2294.
Vallianou, N., Evangelopoulos, A., and Koutalas, P. (2009). Alpha-lipoic acid and diabetic neuropathy. Rev. Diabet. Stud. 6, 230–236.
Vega-Lopez, S., Devaraj, S., and Jialal, I. (2004). Oxidative stress and antioxidant supplementation in the management of diabetic cardiovascular disease. J. Investig. Med. 52, 24–32.
Viana, A. Y., Sakoda, H., Anai, M., Fujishiro, M., Ono, H., Kushiyama, A., Fukushima, Y., Sato, Y., Oshida, Y., Uchijima, Y., Kurihara, H., and Asano, T. (2006). Role of hepatic AMPK activation in glucose metabolism and dexamethasone-induced regulation of AMPK expression. Diabetes Res. Clin. Pract. 73, 135–142.
Viollet, B., Lantier, L., Devin-Leclerc, J., Hebrard, S., Amouyal, C., Mounier, R., Foretz, M., and Andreelli, F. (2009). Targeting the AMPK pathway for the treatment of type 2 diabetes. Front. Biosci. 14, 3380–3400.
Vossler, S., Füllert, S., Schneider, F., Haak, E., Haak, T., Samigullin, R., Tritschler, H., Tooke, J. E., and Konrad, T. (2007). Pharmacodynamic effects of orally administered dexlipotam on endothelial function in type 2-diabetic patients. Int. J. Clin. Pharmacol. Ther. 45, 385–393.
Wada, H., Shintani, D., and Ohlrogge, J. (1997). Why do mitochondria synthesize fatty acids? Evidence for involvement in lipoic acid production. Proc. Natl. Acad. Sci. U.S.A. 94, 1591–1596.
Wang, Y., Li, X., Guo, Y., Chan, L., and Guan, X. (2010). Alpha-lipoic acid increases energy expenditure by enhancing adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor-gamma coactivator-1alpha signaling in the skeletal muscle of aged mice. Metabolism 59, 967–976.
Widmann, C., Gibson, S., Jarpe, M. B., and Johnson, G. L. (1999). Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev. 79, 143–180.
Winder, W. W. (2001). Energy-sensing and signaling by AMP-activated protein kinase in skeletal muscle. J. Appl. Physiol. 91, 1017–1028.
Winder, W. W., and Hardie, D. G. (1996). Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am. J. Physiol. 270, E299–E304.
Xi, X., Han, J., and Zhang, J. Z. (2001). Stimulation of glucose transport by AMP-activated protein kinase via activation of p38 mitogen-activated protein kinase. J. Biol. Chem. 276, 41029–41034.
Xiang, G. D., Sun, H. L., Zhao, L. S., Hou, J., Yue, L., and Xu, L. (2008). The antioxidant alpha-lipoic acid improves endothelial dysfunction induced by acute hyperglycemia during OGTT in impaired glucose tolerance. Clin. Endocrinol. (Oxf.) 68, 716–723.
Ying, Z., Kampfrath, T., Sun, Q., Parthasarathy, S., and Rajagopalan, S. (2011). Evidence that α-lipoic acid inhibits NFκ-B activation independent of its antioxidant function. Inflamm. Res. 60, 219–225.
Zeigler, M. E., Chi, Y., Schmidt, T., and Varani, J. (1999). Role of ERK and JNK pathways in regulating cell motility and matrix metalloproteinase 9 production in growth factor-stimulated human epidermal keratinocytes. J. Cell. Physiol. 180, 271–284.
Zhang, W. J., and Frei, B. (2001). Alpha-lipoic acid inhibits TNF-alpha-induced NF-kappa B activation and adhesion molecule expression in human aortic endothelial cells. FASEB J. 15, 2423–2432.
Zhang, W. J., Wei, H., Hagen, T., and Frei, B. (2007). Alpha-lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 104, 4077–4082.
Zhang, Y., Han, P., Wu, N., He, B., Lu, Y., Li, S., Liu, Y., Zhao, S., Liu, L., and Li, Y. (2011). Amelioration of lipid abnormalities by α-lipoic acid through antioxidative and anti-inflammatory effects. Obesity (Silver Spring) 19, 1647–1653.
Ziegler, D. (2006). Treatment of diabetic polyneuropathy: update 2006. Ann. N. Y. Acad. Sci. 1084, 250–266.
Ziegler, D., Ametov, A., Barinov, A., Dyck, P. J., Gurieva, I., Low, P. A., Munzel, U., Yakhno, N., Raz, I., Novosadova, M., Maus, J., and Samigullin, R. (2006). Oral treatment with alpha lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care 29, 2365–2370.
Ziegler, D., Low, P. A., Litchy, W. J., Boulton, A. J., Vinik, A. I., Freeman, R., Samigullin, R., Tritschler, H., Munzel, U., Maus, J., Schütte, K., and Dyck, P. J. (2011). Efficacy and safety of antioxidant treatment with {alpha}-lipoic acid over 4 years in diabetic polyneuropathy: the NATHAN 1 trial. Diabetes Care 34, 2054–2060.
Zoccali, C., Bode-Böger, S., Mallamaci, F., Benedetto, F., Tripepi, G., Malatino, L., Cataliotti, A., Bellanuova, I., Fermo, I., Frölich, J., and Böger, R. (2001). Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet 358, 2113–2117.
Keywords: diabetes, alpha lipoic acid, neuropathy, nephropathy, antioxidant, oxidative stress
Citation: Golbidi S, Badran M and Laher I (2011) Diabetes and alpha lipoic acid. Front. Pharmacol. 2:69. doi: 10.3389/fphar.2011.00069
Received: 27 August 2011; Accepted: 18 October 2011;
Published online: 17 November 2011.
Edited by:
Adolfo Andrade-Cetto, Universidad Nacional Autónoma de México, MexicoReviewed by:
Adolfo Andrade-Cetto, Universidad Nacional Autónoma de México, MexicoL. Pari, Annamalai University, India
Copyright: © 2011 Golbidi, Badran and Laher. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Ismail Laher, Department of Pharmacology and Therapeutics, Faculty of Medicine, University of British Columbia, Vancouver, BC, Canada V6T 1Z3. e-mail:aWxhaGVyQGludGVyY2hhbmdlLnViYy5jYQ==