
 Michael F. Sheets2
Michael F. Sheets2
- 1 Cardiac Electrophysiology Laboratory, Department of Medicine, The University of Chicago, Chicago, IL, USA
- 2 Department of Internal Medicine, Nora Eccles Harrison Cardiovascular Research and Training Institute, University of Utah, Salt Lake City, UT, USA
Na channels are the source of excitatory currents for the nervous system and muscle. They are the target for a class of drugs called local anesthetics (LA), which have been used for local and regional anesthesia and for excitatory problems such as epilepsy and cardiac arrhythmia. These drugs are prototypes for new analgesic drugs. The drug-binding site has been localized to the inner pore of the channel, where drugs interact mainly with a phenylalanine in domain IV S6. Drug affinity is both voltage- and use-dependent. Voltage-dependency is the result of changes in the conformation of the inner pore during channel activation and opening, allowing high energy interaction of drugs with the phenylalanine. LA drugs also reduce the gating current of Na channels, which represents the movement of charged residues in the voltage sensors. Specifically, drug binding to phenylalanine locks the domain III S4 in its outward (activated) position, and slows recovery of the domain IV S4. Although strongly affecting gating, LA drugs almost certainly also block by steric occlusion of the pore. Molecular definition of the binding and blocking interactions may help in new drug development.
Introduction
Local anesthetic (LA) drugs entered clinical use over a 100 years ago for surgical pain control. They continue to be important for local and regional anesthesia as well as for their cardiac antiarrhythmic actions. Their target is the voltage-gated Na channel, and a great deal of research over the recent years has located the binding site within the channel’s inner pore. As the first class of channel-active drugs to be studied extensively, they serve as a model for other drug studies. In addition to cardiac arrhythmias, the voltage-gated Na channel is an important player in many pathophysiological processes, including pain and epilepsy. Many drugs with dissimilar structures resemble LA drugs in their channel action and perhaps also in their binding sites. Consequently, efforts to develop LA-like drugs with specific nerve blocking abilities are widespread. Recently there has been considerable progress in understanding the mechanism of voltage-dependent binding of LA drugs. This article focuses on new insights into LA molecular mechanism of action, which may assist in developing better and more specific drugs.
Clinical Role of Local Anesthetic Drugs Today
The primary use of LA drugs at this time is for local or regional nerve block. Specific examples are dental procedures, eye surgery, epidural anesthesia for lower abdominal or leg surgery, large nerve block in the extremities for orthopedic surgery, and surgical biopsy. High concentrations are achieved by local injection, so that specificity is achieved at the site of injection while systemic effects of the drugs are minimized. Desirable features are rapid onset of nerve block, sufficient block duration, and lack of local reaction. LA drugs are also useful as antiarrhythmic agents for short-term use for suppression of cardiac excitability during rapid arrhythmias, and they can be lifesaving in acute ischemia. However, their action can also establish the conditions for development of arrhythmias, and their long-term use, especially in individuals with ischemic heart disease, must be considered cautiously.
Several nervous system conditions can be treated with LA-type drugs. Some of the commonly used anticonvulsant drugs block Na channels with LA-like characteristics. Because pain is transmitted to the central nervous system via nerve cells that depend on specific isoforms of the Na channel for excitation and conduction, agents that selectively target these Na channel isoforms that transmit pain signals are eagerly sought, in order to avoid the use of analgesic drugs that are addicting or have dangerous side effects.
Structure of the Drug Target
The mammalian Na channel is an intrinsic membrane glycoprotein. It is a molecular complex of a ∼2000 amino acid α-subunit, which contains the main channel functions and drug interaction sites, and a variable number of smaller β-subunits, which modulate membrane expression and channel functional properties (Catterall, 2000; Patino and Isom, 2010). The α-subunit contains 24 transmembrane segments organized into four homologous, but not identical, domains (DI–DIV). The six transmembrane, mostly α-helical segments in each domain (S1–S6) are further divided into a voltage-sensing unit (S1–S4) and a pore-forming unit (S5–S6), connected mainly through the S4–S5 cytoplasmic linker. Much of the protein is not within the membrane, with the glycosylated outside loops involved in toxin interactions, and the larger cytoplasmic loops involved in hormonal and metabolic regulation. There are multiple isoforms of the Na channel α-subunit. This discussion will focus on mammalian brain (NaV1.2) skeletal (NaV1.4), and cardiac (NaV1.5) Na channels, which are very homologous in their intramembrane regions and which have been the major isoforms used for structure–function studies.
No direct detailed three-dimensional structural information for the mammalian Na channel is available to date because of the difficulty in developing crystals of these large amphipathic proteins. The primary and secondary structures of Na channels are similar to the better-studied K channel (e.g., Doyle et al., 1998; Jiang et al., 2002; Long et al., 2005), and it is expected that tertiary structure will be also. The crystal structure of a bacterial voltage-gated Na channel (NaVAb) has been recently reported (Payandeh et al., 2011). In contrast to the mammalian Na channel, it is comprised of four identical subunits, rather than four connected non-identical domains. Nevertheless, its structure will be of great value in the interpretation of experimental studies. The structure of K channels and the bacterial Na channel, combined with studies of mammalian channel chimeras and point mutations, have allowed the development of molecular models of the mammalian Na channel α-subunit (Guy and Seetharamulu, 1986; Lipkind and Fozzard, 2005; Bruhova et al., 2008).
The part of the channel that is best characterized by modeling is the pore region, which has been useful in locating drug and toxin binding sites. The pore is formed mainly by a bundle of the four non-identical S6 α-helical segments, one from each domain. The pore outer vestibule is lined by four infolded hairpin loops of the S5–S6 extracellular regions (P loops) that form the outer vestibule and selectivity filter of the channel, which is located about 40% through the pore from the outside. The outer vestibule and selectivity filter contain several charged amino acid residues, but the inner pore is composed of neutral, mostly hydrophobic amino acid residues.
The voltage-sensing S4 segments are similar to those of voltage-gated K channels, as well as to those of the recently crystalized bacterial Na channel (Payandeh et al., 2011). The S4 segments contain several positively charged residues that are responsive to changes in the transmembrane potential, and their function has been explored by selective mutation (for review, see Bezanilla, 2000). Critical to the function of the Na channel is its ability to open and close its pore in response to changes in the transmembrane potential, a process called voltage-dependent gating. The channel is closed at the resting potential, and opens briefly upon depolarization. Based upon crystal structures of K channels (Doyle et al., 1998; Jiang et al., 2002) and the NaVAb channel (Payandeh et al., 2011), the activation gate of Na channels is located at the cytoplasmic end of the pore, where all four S6 segments converge. The current understanding on opening of the activation gate is that a hinged motion of the S6 segments allows them to separate at their cytoplasmic ends resulting in an open pore (Jiang et al., 2002). Changes in the S6 conformation are linked to movement of the positive charges in the S4 segments, i.e., the voltage sensors, toward the extracellular surface of the membrane during depolarization. Each individual S4 pulls on its S4–S5 cytoplasmic segment, which is adjacent to the S5–S6 pore-wall at its cytoplasmic end, resulting in opening the channel pore (Long et al., 2005). The pore then is occluded (inactivated) by the movement of the intracellular segment formed by the linker between domains III–IV, i.e., the inactivation particle, into the open pore (Patton et al., 1992), and facilitated by the voltage-dependent movement of the domain IV S4 segment (Chahine et al., 1994; Sheets et al., 1999). Upon repolarization these processes appear to be reversed.
Local Anesthetic Drug-Binding Site(s)
The functional characteristics of LA action at concentrations of clinical relevance are complex, but they can be divided into three categories (Sheets et al., 2010). Firstly, when the channel is in the closed conformation, i.e., at very hyperpolarized potentials, block of Na current occurs only at high concentrations (mM range). Secondly, when the channel is opened with batrachotoxin, rapid flicker block can occur by block at the selectivity filter. This flicker block, when evident, is low affinity, i.e., in the millimolar range. The third type is high-affinity block, and it represents interaction of LA’s with the pore conformation that occurs when the voltage sensors are deployed outward and the channel is in its open state and/or open-inactivated state. This occurs at positive potentials and is simply called voltage-dependent block.
The long-standing explanation for high-affinity LA block is called the “modulated receptor hypothesis” (Hille, 2001). It proposes that LA high-affinity depends on structural modification of the binding site (the LA receptor) as a consequence of channel opening and inactivation. Sorting out the types of block using electrophysiology can be less than straight-forward because; (1) channel conformation is voltage dependent, (2) there may be limitations in access and egress to various channel conformations, depending on chemical properties of the drug, and (3) drug on and off rates can be shorter than, in the same order of magnitude as, or longer than channel conformational lifetimes. In voltage clamp assays, block of closed channels is usually tested by holding at very negative potentials, i.e., usually at least −120 mV or more negative with infrequent depolarization for a short duration. This is sometimes called “resting block,” but experimental measures differ greatly, because the holding potential can fail to be sufficiently negative to remove all voltage-dependent block. Resting block (also called closed-state or lipophilic-block) can be confirmed as such when the amount of block is not decreased by further hyperpolarization. Strict open-channel, flicker block can occur at potentials that also support high-affinity block, but it requires much higher drug concentrations; it has been assayed by single channel recordings in channels where inactivation is prevented with batrachotoxin (e.g., Zamponi et al., 1993). Flicker block is not likely to be of clinical significance.
Other electrophysiological assays, which are often of the most physiological relevance, represent a mixture of several types of block in combination with drug access/egress properties. For example, for many LA drugs (and especially those considered to be antiarrhythmic), repetitive test depolarizations show accumulation of block, a property called “use-dependence.” Use-dependent increase in block can occur because drug off rates from the high-affinity site are sufficiently slow that dissociation is not complete between test depolarizing steps either because of high drug-binding affinity (such as lidocaine compared to benzocaine; Hanck et al., 2009) or because of limited egress (such as flecainide; Liu et al., 2003). In fact, protocols with rapid trains of depolarizations are often used as a surrogate for assaying the high-affinity state, since it is non-conducting and therefore amenable to direct measurement only by methods that do depend on measuring ionic current, e.g., binding assays or gating current.
Early information about the location of the high-affinity LA binding site, summarized by Hille (2001), indicated that it was on the Na channel α-subunit and accessible from the cytoplasmic side. Because homology modeling and chemical reactivity studies indicated that the inner pore was most likely lined with residues of the S6 segments, Catterall and associates systematically mutated these residues and determined the effects on LA block at depolarized potentials (Ragsdale et al., 1994; Yarov-Yarovoy et al., 2002). They found only two residues that had a major effect on high-affinity block; Phe-1764 and Tyr-1771 in DIVS6. This is NaV1.2 (brain isoform) numbering; for the skeletal muscle isoform (NaV1.4), it is Phe-1579, and for the cardiac isoform (NaV1.5), it is Phe-1759 or Phe-1760 (depending on which clone is used). So far, no differences in the binding interactions have been identified for these isoforms. Consequently, to simplify further discussion of these residues independent of the isoform studied, they will be identified as IVS6-Phe and IVS6-Tyr. Mutation of Ile-1760 also had effects although it is probably not part of the binding site (see below). Small effects of uncertain mechanism were noted for mutations of Leu-1465, Asn-1466, and Ile-1469 in DIIIS6, and Ile-409 and possible Asn-418 in DIS6. In contrast, the low affinity, closed-state block seen with test depolarizations from very hyperpolarized potentials was not reduced by these mutations, and may have been slightly increased. This demonstrated clearly that the low affinity and the high-affinity sites are clearly different. Indeed, the high-affinity block can be abolished by removal of only IVS6-Phe (Hanck et al., 2009).
Docking of LA drugs into homology models of the open, inner pore of the Na channel (Figure 1) illustrate the structure of a possible high-affinity binding site (Lipkind and Fozzard, 2005). The typical LA drug has a tertiary amine hydrophilic domain and an aromatic ring hydrophobic domain, separated by an intermediate linker containing an amide or an ester group. Consistent with the suggestion by Ragsdale et al. (1994), the Lipkind–Fozzard model had the alkylamine group interact with IVS6-Phe and the aromatic ring interact with IVS6-Tyr. The alkylamine group fitted into a space between IVS6-Phe and the leucine in IIIS6, which was created by opening of the pore, with a van der Waals energy of about −4 to −5 kcal/mol, and the aromatic ring interacted with IVS6-Tyr with an energy of about −2 to −3 kcal/mol. Replacement of IVS6-Phe with alanine in the model reduced the interaction energy about 3 kcal/mol, consistent with experimental evidence that this substitution abolished measurable voltage-dependent LA block. Ahern et al. (2008) systematically reduced the π-electron charge of IVS6-Phe by incorporation of unnatural amino acids at this position, and they demonstrated that the LA interaction with phenylalanine is due to an electrostatic force. Block by benzocaine, which contains the hydrophobic aromatic ring, but not the partially charged alkylamine end, was not influenced by their unnatural amino acid substitutions. The aromatic end of LA drugs interacted in the model with IVS6-Tyr with an energy of about −3 kcal/mol, and the interaction was not an aromatic–aromatic one. Interaction of the drug aromatic moiety with IVS6-Tyr was investigated by Li et al. (1999) using tetracaine. They found that substitution of IVS6-Tyr by hydrophobic residues produced minimal change in use-dependent LA block, consistent with an aliphatic interaction rather than an aromatic-aromatic one. In keeping with experimental data the model did not show interaction with residues in DIS6 or DIIS6.
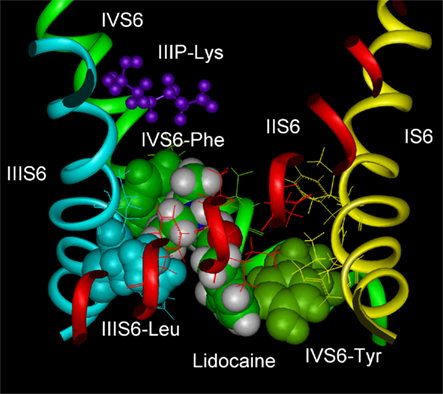
Figure 1. Molecular model of lidocaine in its binding site in the Na channel inner pore. IVS6-Phe and IVS6-Tyr are shown as green space-filled images, and IIIS6-Leu is shown as blue spaced-filled images. IIIP-Lys is part of the DEKA selectivity ring and is shown as purple stick figures. Lidocaine in CPK space-filled images interacts with IVS6-Phe and IVS6-Tyr, almost filling the pore. The figure was prepared by Gregory Lipkind.
Because of their size, charged lidocaine-like molecules did not fit well into the inner pore of the Lipkind–Fozzard model of the closed Na channel. In the closed pore model both IVS6-Phe and IVS6-Tyr face the central pore lumen, but the cleft between IVS6-Phe and the leucine in IIIS6 is too small to allow the alkylamine to fit. IVS6-Tyr is part of the densely packed S6 crossing in the model. In the bacterial NaVAb, the crossover is located lower (Payandeh et al., 2011), but experimental study of accessibility of MTS reagents supports the mammalian model (Sunami and Fozzard, unpublished data). According to the model, charged LA drugs may not be able to enter the inner pore, or if they do (possibly through pore-wall fenestrations, as noted in the NaVAb structure; Payandeh et al., 2011), they do not form strong interactions with the key residues of IVS6-Phe and IVS6-Tyr. Therefore, LA drugs appear to bind with high-affinity to two residues in DIVS6 and possibly with low affinity to one or more in DIIIS6 in the inner pore when the binding site is optimized by the conformational changes that occur during channel voltage-dependent activation and opening. IVS6-Phe is the most important residue for LA binding, and its mutation abolishes voltage-dependent, i.e., use-dependent, block. The site of LA binding for low affinity closed-channel block is not yet clearly resolved, and may be simply a combination of hydrophobic interactions with the closed pore.
Role of the Drug Access Path
The location of the LA binding site has depended upon the study of point mutational data that assume that impairment of drug block of Na current by point mutations involve parts of the drug-binding site. However, there are two other possible explanations for the effects of amino acid substitutions: (1) the mutation could interfere with a molecular sequence of events downstream of the binding step without being part of the binding site itself. All evidence at this point supports the direct role in binding by IVS6-Phe and probably IVS6-Tyr, but roles of the others are less certain. (2) Another possible action of mutations is to alter the drug pathway to and from its binding site deep within the inner pore. Although LA drugs access their high-affinity binding site only from the inside for most Na channel isoforms (Narahashi and Frasier, 1971; Strichartz, 1973), two early experimental studies suggested that access to the inner pore site is possible from the outside. Alpert et al. (1989) used a cardiac cell preparation with independent perfusion of the outside and the inside and found that QX314, a permanently charged quaternary amine that is membrane impermeant, could block cardiac Na current from the outside. At that time, it was not clear whether QX314 could access the inner pore site directly or if there was another binding site. When Ragsdale et al. (1994) observed that replacement of Ile-1760 by alanine in NaV1.2 reduced etidocaine block, they determined that this was not the result of a loss of affinity, but of a more rapid off-rate. This mutation also generated channels that were sensitive to extracellularly applied QX314. Subsequently, Qu et al. (1995) showed that an isoform difference in the upper part of DIVS6 between the nerve channel (NaV1.2) and the cardiac channel (NaV1.5) was responsible for an external LA access path in the cardiac isoform, and Sunami et al. (1997) found that an isoform difference in the selectivity region also allowed extracellular LA to block the cardiac channel. These external access paths can affect lidocaine block, but only in the cardiac isoform (Lee et al., 2001). Bruhova et al. (2008) have explored the possible molecular paths for external access in their model of the Na channel.
Mechanism of LA Block of the Na Channel
Location of the LA binding site in the open-channel inner pore suggests several possible mechanisms for high-affinity LA block. A key to approaching this question is recognition that high-affinity block is voltage dependent over the range that the Na channel activates to open the pore. These voltage-dependent conformational changes appear to create the high-affinity site composed mainly of IVS6-Phe and IVS6-Tyr. Block could occur because the LA molecule binds in the inner pore and physically blocks conductance, because drug-binding interferes with the gating machinery itself, or both. Experimental evidence can be found to support both possibilities.
Physical obstruction of the pore would be voltage dependent because the channel’s conformational changes during activation create the high-affinity site. Molecular modeling is consistent with this idea, with the bound drug located horizontally across the pore (Hanck et al., 2009). LA drugs all produce complete block at the single channel level, i.e., reduction in probability of opening, associated with modest decrease in open times (Grant et al., 1989). The selectivity filter is the narrowest segment of the pore, and direct drug interaction with it can under some circumstances produce fast flicker block at very high concentrations (Zamponi et al., 1993). However, electrostatic interaction of the positively charged lidocaine with the negatively charged residues in the selectivity filter suggests that the high-affinity site is about 10 Å below the selectivity filter (Sunami et al., 1997). For charged LA drugs, the positive charge itself would be expected to interfere with permeation of Na, even if it does not occlude the pore completely (McNulty et al., 2007).
The alternative idea for mechanism of block is that it interferes with channel gating. The long-standing modulated receptor hypothesis for blocking action of LA drugs predicted a stabilization of the inactivated state of the Na channel (Hille, 1977). There is ample evidence of interaction between LA drug binding and gating. Voltage sensor movement, which is the mediator between membrane voltage and conformational changes of gating, can be measured directly by intramembranous charge movement, and LA binding reduces this gating charge as much as 40% (Hanck et al., 1994). The most dramatic effect of lidocaine on gating currents is loss of the entire contribution of the DIIIS4 gating charge, which normally contributes ∼30% to maximal charge (Sheets and Hanck, 2003), along with loss of a part of the DIVS4 charge. It appears that lidocaine stabilizes the DIIIS4 voltage sensor in its activated (depolarized) position, so that it is unable to reset upon repolarization. Muroi and Chanda (2008) studied the effects of lidocaine on fluorescence indicators of individual S6 segment movements in rat skeletal Na channels expressed in Xenopus oocytes. They found a somewhat smaller reduction in total gating charge (S1–S4) of ∼25%, with the greatest change in fluorescence-voltage curve in the DIIIS4. While the mechanism is not understood in detail, it appears that LA drugs interfere allosterically with return of DIIIS6 to its closed configuration and coupling to DIIIS4 via the S4–S5 linker prevents restoration of the DIIIS4.
However, it is the DIVS4 that controls fast inactivation (Chahine et al., 1994; Sheets et al., 1999). Measurement of the movement of the DIVS4 gating charge in drug-bound channels has shown more modest changes; a small (∼10%) reduction in total current (about 1/3 of the normal contribution of DIVS4) associated with a shift to the left of its voltage dependence (Sheets and Hanck, 2003). The shift to the left at more hyperpolarized potentials appears to result from the bound LA drug stabilizing the DIVS4 in a partially depolarized position, so its movement is facilitated upon depolarization (Sheets et al., 2010). The gating charge movements of DIS4 and DIIS4, which are presumably responsible for the fast activation of the Na current, were unchanged.
Does outward movement of DIIIIS4 and DIVS4 simply increase lidocaine affinity for steric block in the pore, or does lidocaine binding cripple the gating process resulting in altered kinetics and a non-conducting channel? Studies on Na channels that have had both their S4’s in DIII and IV pre-stabilized in outward positions using MTSEA-biotin show dramatically increased affinity for lidocaine block, with perhaps the DIVS4 being the more important player, supporting the idea of an increase in steric block by lidocaine (Sheets and Hanck, 2007). Importantly, the pre-stabilization of both the DIIIS4 and DIVS4 did not, by themselves, prevent the channel from opening and closing normally and conducting current in the absence of LA drug. Consequently, the lidocaine-induced effects on gating currents do not directly cause block of channel conductance, but instead, they enhance lidocaine affinity. Arcisio-Miranda et al. (2010) mutated multiple residues in the S4–S5 and terminal S6 regions of DIII and found several mutations that dramatically reduced the stabilizing effect of lidocaine on the DIIIS4 gating charge, but in most cases these mutations failed to prevent high-affinity (use-dependent) lidocaine block. This confirms the important role of the molecular link between DIIIS4–S5 and DIIIS6 in lidocaine effect on DIIIS4, and it also confirms that the DIIIS4 stabilization by lidocaine does not in itself produce the block. Arcisio-Miranda and colleagues have not reported comparable experiments on the DIVS4 movement.
High-affinity block is most likely the result of occlusion of the pore permeation path by the LA drugs (Hanck et al., 2009). This process is voltage dependent because it depends on conformational changes during activation to create the high-affinity site, as predicted by the modulated receptor hypothesis. These conformational changes also favor binding of the DIII–DIV inactivation loop, and its binding may help stabilize the LA binding site in its high-affinity conformation. A detailed understanding of how inactivation is related to the kinetics of LA block remains unclear, and there are conflicting reports on the effects of inactivation on lidocaine block. Vedantham and Cannon (1999) reported that the IFM blocking segment within the intracellular linker between domain III and IV, i.e., the inactivation particle, is not stabilized by lidocaine, but it returns quite normally to its unbound position upon repolarization in drug-bound Na channels. Chemical removal of inactivation has been reported to reduce LA affinity (Bennett et al., 1995). However, a mutation that removes inactivation apparently fails to reduce lidocaine affinity (Wang et al., 2004). It also remains possible that high-affinity LA drug-binding favors some ill-defined closed-state of the inner pore occurring naturally during slow inactivation (Ong et al., 2000) or a unique LA-induced closed-state. Recovery from LA block after repolarization would then depend on unbinding of the LA molecule accompanied by movements of the DIIIS4 and DIVS4 sensors and the inactivation particle, with resulting loss of the high-affinity site. Regardless of the details, which need careful experimental study, it is clear that the link between drug binding in the pore and voltage sensor movements provides a mechanism to control drug affinity dynamically and, thereby, provides for a range of therapeutic options. It would be helpful if future experimentation determined how drug binding to residues in the S6 segment of domain IV is coupled to the movements of the voltage sensor in domain III, and sorted out the relationship of drug affinity to conformational changes induced by movement of IVS4 and its associated inactivation processes. Although useful isoform differences in LA drug action have been difficult to find, many opportunities have not yet been explored.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Gregory Lipkind for discussion of this topic and for the figure. The work was supported by an N.I.H. RO1, HL096476 to Michael F. Sheets and Dorothy A. Hanck.
References
Ahern, C. A., Eastwood, A. L., Dougherty, D. A., and Horn, R. (2008). Electrostatic contributions of aromatic residues in the local anesthetic receptor of voltage-gated sodium channels. Circ. Res. 102, 86–94.
Alpert, L. A., Fozzard, H. A., Hanck, D. A., and Makielski, J. C. (1989). Is there a second external lidocaine binding site on mammalian cardiac cells? Am. J. Physiol. 257, H79–H84.
Arcisio-Miranda, M., Muroi, Y., Chowdhury, S., and Chanda, B. (2010). Molecular mechanism of allosteric modification of voltage-dependent sodium channels by local anesthetics. J. Gen. Physiol. 136, 541–554.
Bennett, P. B., Valenzuela, C., Chen, L.-Q., and Kallen, R. G. (1995). On the molecular nature of the lidocaine receptor of cardiac Na channels: modification of block by alterations in α-subunit III-IV interdomain. Circ. Res. 7, 584–592.
Bezanilla, F. (2000). The voltage sensor in voltage dependent ion channels. Physiol. Rev. 80, 555–592.
Bruhova, I., Tikhonov, D. B., and Zhorov, B. S. (2008). Access and binding of local anesthetics in the closed sodium channel. Mol. Pharmacol. 74, 1033–1045.
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25.
Chahine, M., George, A. L. Jr., Zhou, M., Ji, S., Sun, W., Barchi, R. L., and Horn, R. (1994). Sodium channel mutations in paramyotonia congenital uncouple inactivation from activation. Neuron 12, 281–294.
Doyle, D. A., Cabral, J. M., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., Chait, B. T., and MacKinnon, R. (1998). The structure of the potassium channel: molecular basis of K conduction and selectivity. Science 280, 69–77.
Grant, A. O., Deitz, M. A., Gilliam, F. R. III, and Starmer, C. F. (1989). Blockade of cardiac sodium channels by lidocaine. Circ. Res. 65, 1247–1262.
Guy, H. R., and Seetharamulu, P. (1986). Molecular model of the action potential sodium channel. Proc. Natl. Acad. Sci. U.S.A. 83, 508–512.
Hanck, D. A., Makielski, J. C., and Sheets, M. F. (1994). Kinetic effects of quaternary lidocaineblock of cardiac sodium currents: a gating current study. J. Gen. Physiol. 103, 19–43.
Hanck, D. A., Nikitina, E., McNulty, M. M., Fozzard, H. A., Lipkind, G. M., and Sheets, M. F. (2009). Using lidocaine and benzocaine to link sodium channel molecular configurations to state-dependent antiarrhythmic drug affinity. Circ. Res. 105, 492–499.
Hille, B. (1977). Local anesthetics: hydrophylic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69, 497–515.
Jiang, Y., Lee, A., Chen, J., Cadene, M., Chait, B. T., and MacKinnon, R. (2002). The open pore conformation of potassium channels. Nature 417, 523–526.
Lee, P. J., Sunami, A., and Fozzard, H. A. (2001). Cardiac-specific external paths for lidocaine, defined by isoform-specific residues, accelerate recovery from use-dependent block. Circ. Res. 89, 1014–1021.
Li, H. L., Galue, A., Meadows, L., and Ragsdale, D. S. (1999). A molecular basis for the different local anesthetic affinities of resting versus open and inactivated states of the sodium channel. Mol. Pharmacol. 55, 134–141.
Lipkind, G. M., and Fozzard, H. A. (2005). Molecular modeling of local anesthetic drug binding by voltage-gated sodium channels. Mol. Pharmacol. 68, 1611–1622.
Liu, H., Atkins, J., and Kass, R. S. (2003). Common molecular determinants of flecainide and lidocaine block of heart Na channels: evidence from experiments with neutral and quaternary flecainide analogs. J. Gen. Physiol. 121, 199–214.
Long, S. B., Campbell, E. B., and MacKinnon, R. (2005). Crystal structure of a mammalian voltage-dependent Shaker family K channel. Science 309, 897–903.
McNulty, M. M., Edgerton, G. B., Shah, R. D., Hanck, D. A., Fozzard, H. A., and Lipkind, G. M. (2007). Charge at the lidocaine binding site residue Phe-1759 affects permeation in human cardiac voltage-gated sodium channels. J. Physiol. (Lond.) 581, 741–755.
Muroi, Y., and Chanda, B. (2008). Local anesthetics disrupt energetic coupling between the voltage-sensing segments of a sodium channel. J. Gen. Physiol. 133, 1–15.
Narahashi, T., and Frasier, D. T. (1971). Site of action and active form of local anesthetics. Neurosci. Res. 4, 65–99.
Ong, B. H., Tomaselli, G. F., and Balser, J. R. (2000). A structural rearrangement in the sodium channel pore linked to slow inactivation and use dependence. J. Gen. Physiol. 116, 653–662.
Patino, G. A., and Isom, L. L. (2010). Electrophysiology and beyond: multiple roles of Na channel β subunits in development and disease. Neurosci. Lett. 486, 53–59.
Patton, D. E., West, J. W., Catterall, W. A., and Goldin, A. L. (1992). Amino acid residues required for fast sodium channel inactivation. Proc. Natl. Acad. Sci. USA 89, 10905–10909.
Payandeh, J., Scheuer, T., Zheng, N., and Catterall, W. A. (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358.
Qu, Y., Rogers, J., Tanada, T., Scheuer, T., and Catterall, W. A. (1995). Molecular determinants of drug access to the receptor site for antiarrhythmic drugs in the cardiac Na channel. Proc. Natl. Acad. Sci. U.S.A. 92, 11839–11843.
Ragsdale, D. S., McPhee, J. C., Scheuer, T., and Catterall, W. A. (1994). Molecular determinants of state-dependent block of Na channels by local anesthetics. Science 265, 1724–1728.
Sheets, M. F., Fozzard, H. A., Lipkind, G. M., and Hanck, D. A. (2010). Sodium channel molecular conformations and antiarrhythmic drug affinity. Trends Cardiovasc. Med. 20, 16–21.
Sheets, M. F., and Hanck, D. A. (2003). Molecular action of lidocaine on the voltage sensors of sodium channels. J. Gen. Physiol. 121, 163–175.
Sheets, M. F., and Hanck, D. A. (2007). Outward stabilization of the S4 segments in domains III and IV enhances lidocaine block of sodium channels. J. Physiol. (Lond.) 582, 317–334.
Sheets, M. F., Kyle, J. W., Kallen, R. G., and Hanck, D. A. (1999). The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys. J. 77, 747–757.
Strichartz, G. R. (1973). The inhibition of sodium currents in myelinated nerve by quaternary derivatives of lidocaine. J. Gen. Physiol. 62, 37–57.
Sunami, A., Dudley, S. C. Jr., and Fozzard, H. A. (1997). Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proc. Natl. Acad. Sci. U.S.A. 94, 14126–14131.
Vedantham, V., and Cannon, S. C. (1999). The position of the fast-inactivation gate during lidocaine block of voltage-gated Na channels. J. Gen. Physiol. 113, 7–16.
Wang, S.-Y., Mitchell, J., Moczydlowski, E., and Wang, G. K. (2004). Block of inactivation-deficient Na channels by local anesthetics in stably transfected mammalian cells: evidence for drug binding along the activation pathway. J. Gen. Physiol. 124, 691–701.
Yarov-Yarovoy, V., McPhee, J. C., Idavoog, D., Pate, C., Scheuer, T., and Catterall, W. A. (2002). Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na channel α-subunit in voltage-dependent gating and drug block. J. Biol. Chem. 277, 35393–35401.
Keywords: lidocaine, molecular modeling, Na channel, local anesthetics, gating currents
Citation: Fozzard HA, Sheets MF and Hanck DA (2011) The sodium channel as a target for local anesthetic drugs. Front. Pharmacol. 2:68. doi: 10.3389/fphar.2011.00068
Received: 23 August 2011;
Accepted: 14 October 2011;
Published online: 01 November 2011.
Edited by:
Mohamed Chahine, Laval University, CanadaReviewed by:
Michael E. O’Leary, Jefferson University, USAMotohiro Nishida, Kyushu University, Japan
Copyright: © 2011 Fozzard, Sheets and Hanck. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Harry A. Fozzard, PO Box 574, Dana, NC 28724, USA. e-mail:aGFmb3p6YXJAdWNoaWNhZ28uZWR1