
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 19 October 2011
Sec. Neuropharmacology
volume 2 - 2011 | https://doi.org/10.3389/fphar.2011.00065
This article is part of the Research Topic Brain effects of Nicotine and derived compounds View all 11 articles
 Mao-Hsien Wang1,2
Mao-Hsien Wang1,2 Hatsumi Yoshiki1
Hatsumi Yoshiki1 Abu Syed Md Anisuzzaman1 Junsuke Uwada1,3 Atsushi Nishimune1,3 Kung-Shing Lee1
Abu Syed Md Anisuzzaman1 Junsuke Uwada1,3 Atsushi Nishimune1,3 Kung-Shing Lee1 Takanobu Taniguchi4
Takanobu Taniguchi4 Ikunobu Muramatsu1,3,5*
Ikunobu Muramatsu1,3,5*Nicotinic acetylcholine receptors (nAChRs) of the cerebral cortex and cerebellum of rats were evaluated by a radioligand binding assay, employing tissue segments, or homogenates as materials. [3H]-epibatidine specifically bound to nAChRs in rat cortex or cerebellum, but the dissociation constants for [3H]-epibatidine differed between segments and homogenates (187 pM for segments and 42 pM for homogenates in the cortex and 160 pM for segments and 84 pM for homogenates in the cerebellum). The abundance of total nAChRs was approximately 310 fmol/mg protein in the segments of cortex and 170 fmol/mg protein in the segments of cerebellum, which were significantly higher than those estimated in the homogenates (115 fmol/mg protein in the homogenates of the cortex and 76 fmol/mg protein in the homogenates of the cerebellum). Most of the [3H]-epibatidine binding sites in the cortex segments (approximately 70% of the population) showed high affinity for nicotine (pKi = 7.9), dihydro-β-erythroidine, and cytisine, but the binding sites in the cerebellum segments had slightly lower affinity for nicotine (pKi = 7.1). An upregulation of nAChRs by chronic administration of nicotine was observed in the cortex segments but not in the cerebellum segments with [3H]-epibatidine as a ligand. The upregulation in the cortex was caused by a specific increase in the high-affinity sites for nicotine (probably α4β2). The present study shows that the native environment of nAChRs is important for a precise quantitative as well as qualitative estimation of nAChRs in rat brain.
Nicotinic acetylcholine receptors (nAChRs) in the nervous system are members of a family of ligand-gated ion channels, pentametric in structure, with at least 11 different subunits (α2–α10 and β2–β4) characterized (Lindstrom et al., 1996; Alexander et al., 2009). Because different subunit compositions produce distinct nAChR subtypes, the potential nAChR subtype diversity is vast (Lindstrom et al., 1996). However, in general, α4β2 and α7 are the predominant subtypes expressed in the central nervous system (CNS), whereas the α3β4 subtype is the predominant subtype expressed in the autonomic ganglia (Lindstrom, 2000; Alexander et al., 2009).
The nAChRs in the CNS are physiologically relevant in a wide range of neuronal functions (such as cognition, motor control, analgesia, and reward), and may be involved in pathological states such as Alzheimer’s and Parkinson’s diseases (Grady et al., 2007; Quik et al., 2007; Drever et al., 2010). Chronic nicotine exposure, including smoking, is known to increase the number of nAChRs in the CNS of rodents and humans, although there is a region-specific variation in the upregulation (Marks et al., 1992; Sanderson et al., 1993; Davil-Garcia et al., 2003; Govind et al., 2009). Thus, smoking seems to have certain therapeutic effects for Alzheimer’s disease, although this conclusion is controversial (Hellstrom-Lindahl et al., 2004; Connelly and Prentice, 2005; Picciotto and Zoli, 2008). In addition, nAChRs have recently attracted attention as a novel target of general anesthetics and antidepressants (McMorn et al., 1993; Shytle et al., 2002; Tassonyi et al., 2002; Picciotto et al., 2008).
Identifying individual nAChR subtypes in the CNS has been a difficult task. The “high-affinity agonist binding” α4β2 subtype (labeled by [3H]-acetylcholine, [3H]-cytisine, (−)-[3H]-nicotine) and the predominantly or entirely α7 subtype (labeled by [125I]-?α-bungarotoxin) have been extensively characterized (Whiting and Lindstrom, 1987; Schoepfer et al., 1990; Flores et al., 1992; Picciotto et al., 1995; Orr-Urtreger et al., 1997; Marubio et al., 1999). Subsequently, the discovery of epibatidine (an azabicycloheptane alkaloid isolated from the skin of the Ecuadorian frog Epipedobates tricolor) that binds to multiple nAChR subtypes with high-affinity provided a tool to identify novel nAChR subtypes (Houghtling et al., 1995; Perry and Kellar, 1995; Marks et al., 1998; Whiteaker et al., 2000; Grady et al., 2007). In the CNS of rodents, the majority of high-affinity [3H]-epibatidine binding sites represent the α4β2 subtype, but the minority of the sites were distinguished by their relatively low-affinity for cytisine or dihydro-β-erythroidine in small nuclei dispersed across the brain (Grady et al., 2007).
In most studies of nAChRs and other receptors in the CNS, brain tissue was homogenized, and the resulting membrane or particulate preparations have been exclusively used in the radioligand binding assay (Bylund and Toews, 1993; Houghtling et al., 1995; Yang et al., 1998; Marubio et al., 1999; Whiteaker et al., 2000; Schneider and Michel, 2010). Although such a grind-and-bind approach has been successfully used to identify receptor pharmacology for more than two decades, tissue homogenization may cause alterations in receptor environments, resulting in different properties from the inherent profile in some receptors (Hiraizumi-Hiraoka et al., 2004; Anisuzzaman et al., 2008, 2011; Morishima et al., 2008). These recent studies suggest that the in situ environment significantly contributes to some receptor properties. To avoid artificial modification of the receptor environment as much as possible, a binding assay for use with intact tissue segments and with physiological solution (such as Krebs solution) was recently developed and has been demonstrated to be a powerful method for detecting the inherent profiles of receptors in a native manner (Tanaka et al., 2004; Muramatsu et al., 2005; Yoshiki et al., 2009; Anisuzzaman et al., 2011). In contrast to the conventional membrane binding assay, the tissue-segment binding assay is less prone to have a yield loss of receptors and to have a modification of the natural environment/conformation of receptors, which may be caused during tissue homogenization and membrane fractionation.
Previously, we detected a difference in [3H]-epibatidine binding in the rat cerebral cortex between the tissue-segment binding assay and the conventional membrane binding assay (Muramatsu et al., 2005). In this study, we extensively evaluated [3H]-epibatidine binding to nAChRs in the intact segments of rat cerebral cortex and cerebellum, and compared the pharmacological profiles with those in the homogenized preparations. Furthermore, the effects on nAChRs of chronic nicotine administration were examined by the tissue-segment binding assay.
Male Wistar rats weighing 250–350 g (SLC, Shizuoka, Japan) were used. The present study was performed according to the Guidelines for Animal Experiments, University of Fukui (which is accredited by the Ministry of Education, Culture, Sports, Science, and Technology, Japan).
Rats were anesthetized with pentobarbital and killed by cervical dislocation. The brain was rapidly isolated and immersed in modified Krebs–Henseleit solution containing: NaCl, 120.7 mM; KCl, 5.9 mM; MgCl2, 1.2 mM; CaCl2, 2.0 mM; NaH2PO4, 1.2 mM; NaHCO3, 25.5 mM; and D-(+)-glucose, 11.5 mM (pH = 7.4). This solution had been oxygenated with a mixture of 95% O2 and 5% CO2 and was kept at 0°C. The brain was cleaned from the pia mater and substantia alba, and then the cerebral cortex and cerebellum were cut into small pieces (approximately 2 mm × 2 mm × 1 mm). The tissue-segment binding assay was performed at 4°C for 26–30 h, according to the method previously described (Muramatsu et al., 2005; Anisuzzaman et al., 2011). Each segment of the cortex or cerebellum was incubated in 0.5 ml of the modified Krebs–Henseleit solution.
In saturation binding experiments, [3H]-epibatidine was used at concentrations ranging from 30 to 2000 pM. In competition experiments, 400 pM [3H]-epibatidine competed against various concentrations of tested drugs. After incubation at 4°C, tissue segments were quickly moved into a plastic tube containing 1.5 ml of ice-cold (4°C) modified Krebs–Henseleit solution and carefully washed by vortexing for 1 min. By this procedure, most of the unbound radioligand was released from the segments into the buffer medium and adsorbed to the plastic tube. The segments were then solubilized in 0.3 M NaOH solution to estimate the bound radioactivity and protein content. Non-specific binding was determined in the presence of 100 μM nicotine. The bound radioactivity was measured using a liquid scintillation counter (Aloka, Tokyo, Japan). Protein concentration in each tissue segment was measured by the Bio-Rad commercial protein assay. Experiments were done in duplicate at each concentration of [3H]-epibatidine for binding saturation experiments or at each concentration of competing ligand for competition binding experiments.
As reported previously (Anisuzzaman et al., 2011), the rat cerebral cortex and cerebellum were homogenized in 40 volumes (v/w; for cortex) or 20 volumes (v/w; for cerebellum) of the modified Krebs–Henseleit solution using a Polytron homogenizer at 4°C. The resulting homogenates were incubated with [3H]-epibatidine in a final volume of 0.5 ml modified Krebs–Henseleit solution for 5 h at 4°C. In competition experiments, 200 pM (for cortex) and 300 pM (for cerebellum) of [3H]-epibatidine were used. Non-specific binding of [3H]-epibatidine was determined with 100 μM nicotine. The assay was terminated by rapid filtration over Whatman GF/C filters presoaked with 0.3% polyethyleneimine using a Brandel cell harvester and filters were rapidly washed with 5-ml aliquots of ice-cold modified Krebs–Henseleit solution. The filters were then dried and the radioactivity retained on the filter paper was measured by liquid scintillation counting. The protein contents of the homogenates were determined by the Bio-Rad commercial protein assay.
Nicotine was administered for 3 weeks in drinking water. The concentration of nicotine in the drinking water was 50 μg/ml. The control rats drank tap water during the experimental period. The plasma concentration of nicotine after 3 weeks of oral nicotine exposure was measured with high performance liquid chromatography; the plasma level was 18 ± 3 ng/ml (n = 5), which was similar to the nicotine concentrations (10–50 ng/ml) reported in smokers (Benowitz et al., 1982; Pekonen et al., 1993).
After nicotine administration for 3 weeks, rats were killed and the cortex or cerebellum segments were used in the binding experiments, as described above.
Binding data were analyzed using PRISM software (Version 5.01, Graph Pad Software, La Jolla, CA, USA), as previously described (Muramatsu et al., 2005; Yoshiki et al., 2009). Briefly, the data from saturation binding studies were fitted by a one-site saturation binding isotherm and the Kd values and the binding capacity were then calculated. The abundance of the nAChRs is indicated as the maximum binding capacity per milligram of total tissue protein (Bmax). For the competition studies, the data were analyzed using the Binding-Competitive Equation of the PRISM software. A two-site model was adopted only when the residual sums of squares were significantly less (P < 0.05) for a two-site fit to the data than for a one-site by F test comparison.
Data are represented as the mean ± SEM with the number of experiments (n). Results were considered to be significant where P values were less than 0.05 (Student’s t-test).
The following drugs were used in the present study: [5,6-bicycloheptyl-3H]-epibatidine (specific activity, 2065 GBq/mmol; PerkinElmer Life and Analytical Sciences, Boston, MA, USA); 1-quinuclidinyl-[phenyl-4-3H]-benzilate ([3H]-QNB; specific activity, 1.81 TBq/mmol; GE Healthcare, Buckinghamshire, UK); nicotine, nicotine tartrate dihydrate, and hexamethonium chloride (Nacalai Tesque, Kyoto, Japan); dihydro-β-erythroidine, cytisine, and mecamylamine hydrochloride (Tocris Cookson Ltd., Bristol, UK); and α-bungarotoxin (Peptide Institute, Osaka, Japan). For chronic administration of nicotine, aqueous nicotine was used.
The representative time courses of [3H]-epibatidine binding to the intact segments (Figure 1A) and homogenate preparations (Figure 1B) of rat cerebral cortex and to the intact segments (Figure 2A) and homogenate preparations (Figure 2B) of rat cerebellum are shown. At 4°C, the binding to the segments was extremely slow compared with that in the homogenates; therefore, different incubation periods were applied in the segment binding assay (26–30 h) and the homogenate binding assay (5 h). In the saturation experiments, the specific binding of [3H]-epibatidine to both the intact segments (Figure 1C) and homogenate preparations (Figure 1D) of rat cortex was saturable at the concentrations tested and fitted a single-site model. However, the slopes of the saturation curves were apparently different; the slope was steeper for the homogenates than for the segments. Thus, the dissociation constant (Kd) for [3H]-epibatidine was estimated to be four times higher in the segment binding assay than in the homogenate binding assay (Table 1). Furthermore, the maximum binding capacity (Bmax) of [3H]-epibatidine was also approximately three times higher in the segments than in the homogenates of rat cortex (Table 1). Differences in the time course, the density, and the dissociation constant for [3H]-epibatidine binding were also observed between the segments and homogenates of rat cerebellum (Figure 2 and Table 1). The affinity for [3H]-QNB (a muscarinic receptor ligand that was used as control) was also lower in the cortex segments than in the homogenates, but the binding capacity was not significantly different between the segments and the homogenates (Table 1).
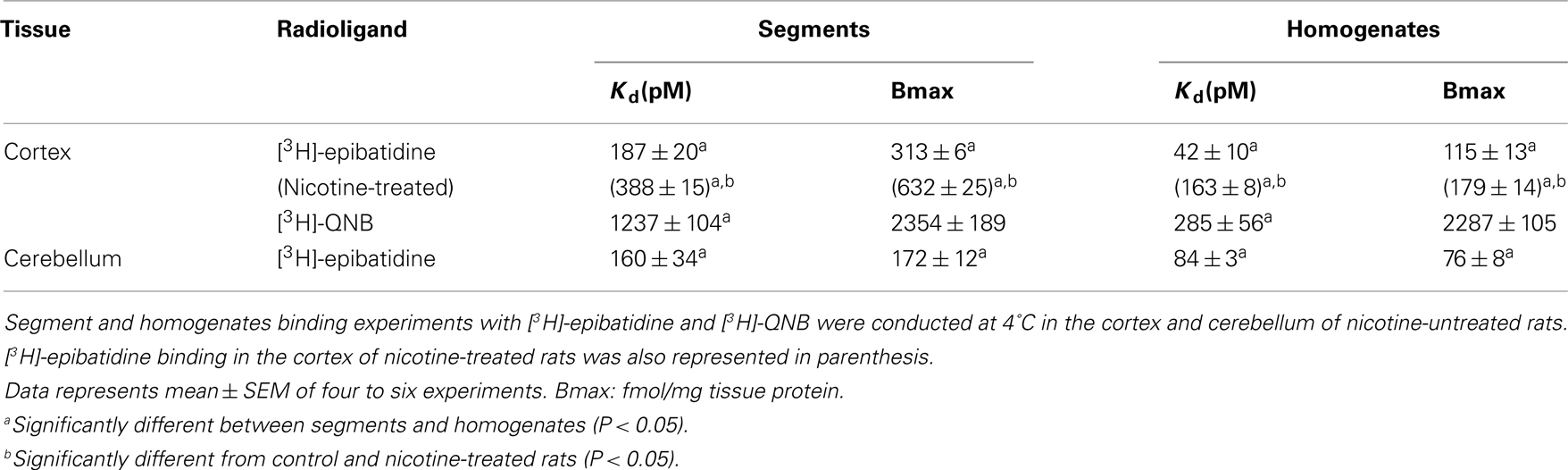
Table 1. Binding parameters of [3H]-epibatidine and [3H]-QNB in rat cortex and cerebellum.
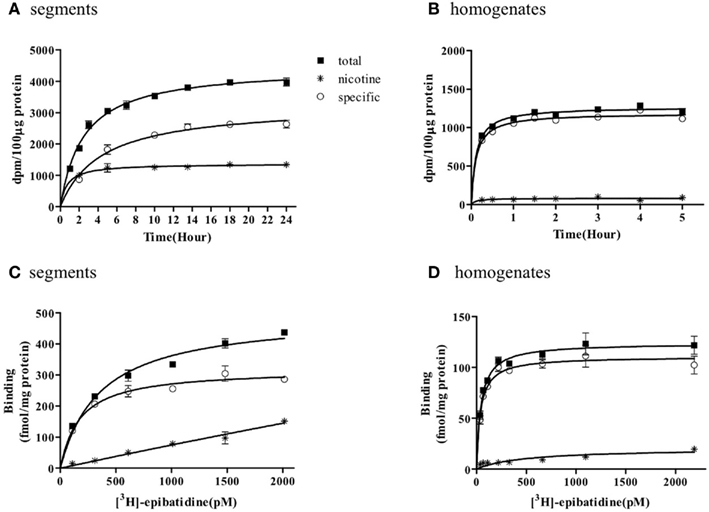
Figure 1. Time course and saturation isotherm of [3H]-epibatidine binding in the intact segments (A,C) and homogenates (B,D) of rat cerebral cortex. Time course (A,B): The intact segments were incubated with 1000 pM [3H]-epibatidine at 4°C for 1–24 h and the homogenates were incubated with 500 pM [3H]-epibatidine at 4°C for 15 min to 5 h. Saturation binding (C,D): The intact segments and homogenates were incubated with [3H]-epibatidine (up to 2000 pM) at 4°C for 28 h (segments) or 5 h (homogenates). Specific binding (open circles) was determined by subtracting the amount bound in the presence of 100 μM nicotine (asterisks) from total binding (closed squares). Each point represents the mean of duplicate determinations. Each figure represents four to five experiments.
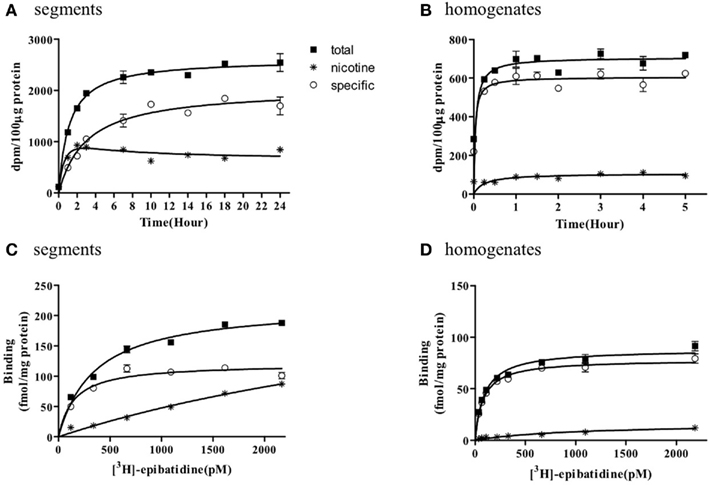
Figure 2. Time course and saturation isotherm of [3H]-epibatidine binding in the intact segments (A,C) and homogenates (B,D) of rat cerebellum. Time course (A,B): The intact segments were incubated with 1000 pM [3H]-epibatidine at 4°C for 1–24 h and the homogenates were incubated with 500 pM [3H]-epibatidine at 4°C for 15 min to 5 h. Saturation binding (C,D): The intact segments and homogenates were incubated with [3H]-epibatidine (up to 2000 pM) at 4°C for 28 h (segments) or 5 h (homogenates). Specific binding (open circles) was determined by subtracting the amount bound in the presence of 100 μM nicotine (asterisks) from total binding (closed squares). Each point represents the mean of duplicate determinations. Each figure represents four to five experiments.
Figures 1 and 2 further demonstrate that non-specific binding of [3H]-epibatidine was significantly higher in the segments than in the homogenates. The non-specific binding in the segments was rapidly equilibrated compared with specific binding (Figures 1A and 2A) and increased lineally with increasing concentrations of [3H]-epibatidine (Figures 1C and 2C). Because the non-specific binding was insensitive to not only nicotine but also the other tested ligands (see the competition experiments), it was considered that there was a significant amount of non-nAChR sites in the segments that were undetectable after homogenization.
Because the binding capacity and dissociation constant for [3H]-epibatidine were significantly different between tissue segments and homogenates, the pharmacological profiles of [3H]-epibatidine binding sites in both preparations of rat cortex were compared in competition binding experiments using various drugs. Nicotine, dihydro-β-erythroidine, and cytisine showed shallow competition curves in the segments of cortex and approximately 70% of the specific binding sites were identified as high-affinity sites for these ligands (Figure 3A). In the homogenates, dihydro-β-erythroidine and cytisine also showed shallow curves, but the competition curve for nicotine better fitted a one-site model with a slightly higher pKi value than that in the segments (Figure 3B and Table 2). Hexamethonium at concentrations higher than 10 μM were competitive, but mecamylamine (1 mM) and α-bungarotoxin (1–30 nM) failed to compete in the [3H]-epibatidine binding assay with both segment and homogenate preparations (Table 2). In general, the estimated ligand affinities were higher in the homogenates compared with the segments.
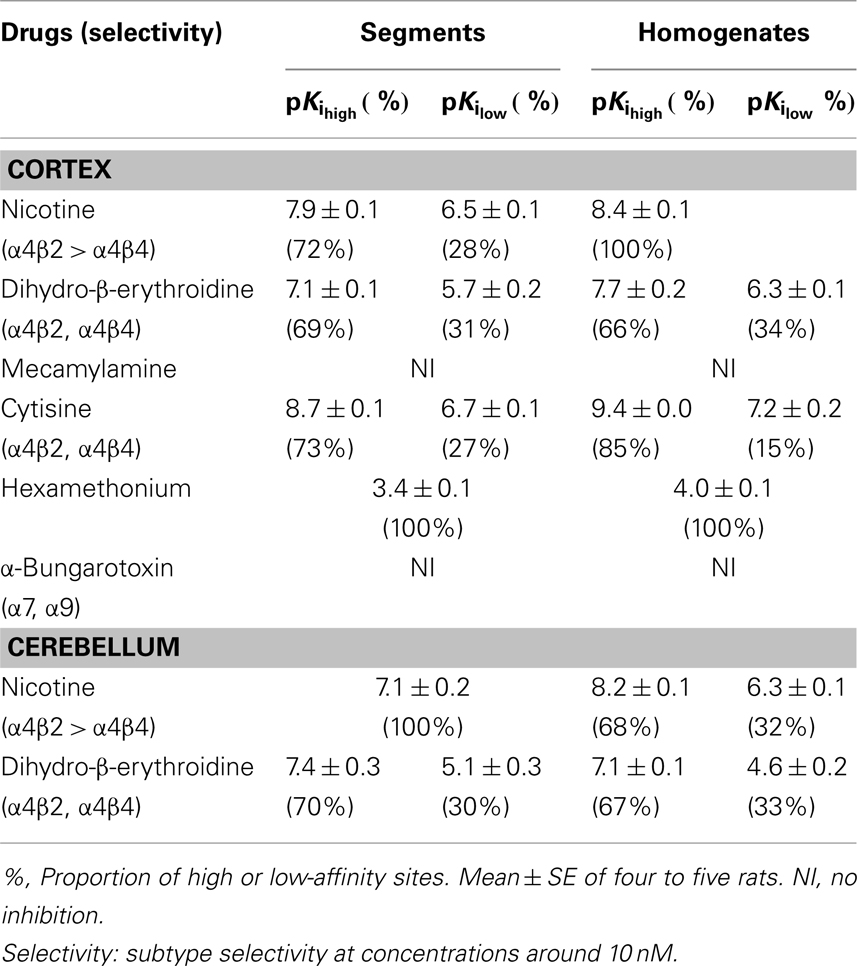
Table 2. Pharmacological characteristics of [3H]-epibatidine binding sites in rat cortex and cerebellum.
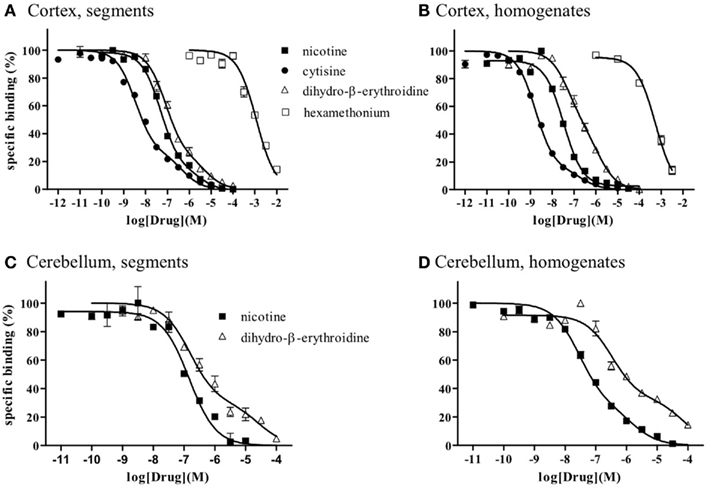
Figure 3. Competition curves for various dugs at [3H]-epibatidine binding sites in intact segments and homogenates of rat cerebral cortex (A,B) and rat cerebellum (C,D). Intact segments (A,C) were incubated with 400 pM [3H]-epibatidine for 28 h at 4°C. Homogenates were incubated with 200 pM [(B): cortex] or 300 pM [(D): cerebellum] [3H]-epibatidine for 5 h at 4°C. Each point represents the mean of duplicate determinations. Each figure represents four to five experiments.
Because of the tissue limitation, the pharmacological profile at [3H]-epibatidine binding sites in the cerebellum was examined with nicotine and dihydro-β-erythroidine. Figure 3 shows the competition curves in the segments (Figure 3C) and the homogenates (Figure 3D) of rat cerebellum. Nicotine showed biphasic competition in the homogenates (pKi = 8.2 and 6.3), but competed for binding to the segments monotonically with an intermediate affinity (pKi = 7.1). These results are summarized in Table 2.
Chronic administration of nicotine is known to cause an increase in nAChRs in the brain, which is called upregulation. Because the number of [3H]-epibatidine binding sites was underestimated in homogenates as mentioned above, the nicotine-induced upregulation was re-evaluated by the tissue-segment binding approach. Administration of nicotine for 3 weeks increased the binding density of [3H]-epibatidine with a slight decrease in affinity in the rat cortex segments (Table 1). A similar tendency was observed in the cortex homogenates, but the density and affinity were also significantly different from the values estimated in the cortex segments of chronically nicotine-treated rats (Table 1). The binding affinities for nicotine in nicotine-treated rat cortex (pKihigh and pKilow = 7.8 ± 0.2 and 6.3 ± 0.2 in the segments and pKi = 8.1 ± 0.1 in the homogenates) were not significantly different from those in rats not treated with nicotine (Table 2). However, the proportion of nicotine-high-affinity sites in the cortex segments increased from 72% in control rats to 86% in nicotine-treated rats (Figure 4). On the other hand, no upregulation was caused by chronic nicotine administration in the cerebellum.
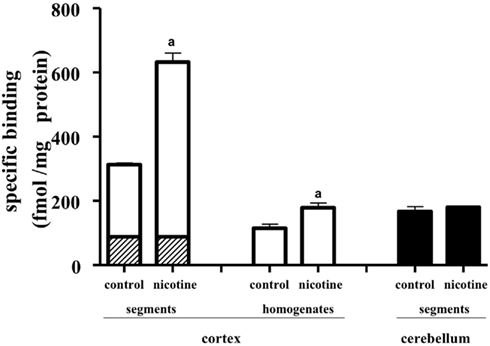
Figure 4. Effects of chronic administration of nicotine for 3 weeks. The tissue segments or homogenates of the cerebral cortex and cerebellum in nicotine-treated and untreated rats were incubated with [3H]-epibatidine for 28 h (segments) or 5 h (homogenates) at 4°C. The total density of [3H]-epibatidine binding sites was estimated from saturation experiments in the cortex and from 2 nM [3H]-epibatidine binding in the cerebellum. From the competition curves for nicotine, high-affinity (open column), and low-affinity sites (hatched column) for nicotine were calculated in the cerebral cortex. The binding sites in the cerebellum were not characterized by nicotine and were represented as black columns. Control, rats not treated with nicotine; nicotine, nicotine-treated rats. Mean ± SEM of four experiments. a: Significantly different from those of the control rats.
In the binding study with [3H]-epibatidine, the nAChRs in the rat cerebral cortex and cerebellum were evaluated and compared between the segments and the homogenates. Two major differences were observed in the present study. The first is different affinities for [3H]-epibatidine and other nicotinic ligands between the two preparations. Previous reports with particulate preparations of rat cortex showed an extremely high affinity for [3H]-epibatidine (Kd = approximately 20 pM; Houghtling et al., 1995; Marks et al., 1998). In the present study as well, such a high affinity for [3H]-epibatidine was estimated in the cortex homogenates (Kd = 42 pM), whereas the affinity in the segments was significantly low (Kd = 187 pM). Consistent with this difference in affinity, the binding time course of [3H]-epibatidine was faster in the homogenates than in the segments.
It has been classically considered that antagonist affinities are constant for a given receptor subtype, regardless of the cell/tissue background in which the receptor is expressed; this has been called “antagonist assumption.” However, this traditional view has now been challenged by the observation of different pharmacological antagonist profiles for the same gene product in different cells/tissues (Kenakin, 2003; Baker and Hill, 2007; Nelson and Challiss, 2007; Muramatsu et al., 2008; Nishimune et al., 2011). The present results also suggest that the pharmacological properties of nAChRs are not necessarily constant, but rather may be easily modified by assay conditions and probably by distinct receptor environments.
In general, there is a tendency for antagonist affinity to become higher after homogenization, even though the other experimental conditions were the same. The tendency was observed in the binding of [3H]-epibatidine to nAChRs (present study) and [3H]-QNB or [3H]-N-methylscopolamine to muscarinic AChRs (present results and the results of Anisuzzaman et al., 2011). Although the precise mechanisms underlying the change in pharmacological profile by homogenization remain to be solved, intact tissue can demonstrate distinct submembrane effector interactions in the presence of constrained membrane architecture, which may be easily destroyed by homogenization. In general, the relatively universal high-affinity obtained in the classical grind-and-bind approach seems to be one of the bases of “antagonist assumption.”
The second difference observed in the present study is distinct densities of nAChRs estimated between segments and homogenates. This is unique for nAChRs ([3H]-epibatidine binding sites), because the abundance of muscarinic AChRs ([3H]-QNB binding sites) did not change after homogenization (Table 1). However, it has been generally reported that homogenization causes a yield loss of receptors (Colucci et al., 1981; Kwan et al., 1981; Faber et al., 2001; Tanaka et al., 2004; Su et al., 2008). The density of [3H]-epibatidine binding sites estimated in the present segment study with rat cortex (313 fmol/mg tissue protein) is greater than the estimates reported previously in conventional membrane binding assays with rat cortex (5–83 fmol/mg of membrane protein; Houghtling et al., 1995; Flores et al., 1997; Davil-Garcia et al., 2003) and with mouse cortex (54.9 fmol/mg of membrane protein; Marks et al., 1998). Previously, we showed that the protein yield of crude membrane preparations in rat cortex after simple centrifugation or fractionation was approximately 50% of the total tissue protein (Morishima et al., 2008). Therefore, it seems that the density of nAChRs estimated previously in the concentrated membrane preparations may be much lower, if the values were adjusted at the same denominator level (that is, per mg of “total tissue protein”).
As mentioned above, the present study reveals that intact tissue conditions or the native physiological environment are very important in the quantitative and qualitative evaluation of nAChRs. However, we are not neglecting the classical grind-and-bind approach and recognize its successful and significant contribution to receptor pharmacology over the past two decades. Indeed, the present competition study with segments and homogenates clearly shows high- and low-affinity sites for dihydro-β-erythroidine and cytisine in the [3H]-epibatidine binding sites, which is consistent with the previous results obtained in membrane binding studies (Whiteaker et al., 2000; Grady et al., 2007). The high-affinity sites in the cortex are major in proportion and seem to correspond to α4β2 and/or α4β4, as reported previously.
In the segments, the [3H]-epibatidine binding sites were composed of high- and low-affinity sites for nicotine in the cortex but a single affinity site in the cerebellum. It is interesting to note that the high affinity for nicotine in the cortex (pKi = 7.9) was slightly higher than the affinity in the cerebellum (pKi = 7.1). Because plasma concentrations of nicotine are reported to be 10–50 ng/ml in nicotine-treated rats and in smokers (present study; Benowitz et al., 1982; Pekonen et al., 1993), the high-affinity sites for nicotine in the cortex may mainly contribute to the upregulation, as shown in Figure 4. The extent of upregulation estimated in the cortex segments (nearly 100%) was greater than those reported in previous membrane binding studies and in the present data of homogenate binding (approximately 50%). On the other hand, no upregulation was observed in the cerebellum in the present segment binding assay as well as previous membrane binding assays (Marks et al., 1992; Flores et al., 1997; Pietila et al., 1998), confirming regional variation of the nicotine-induced upregulation.
In summary, the present comparative binding study with tissue segments and homogenates shows that the native conformation and inherent properties of nAChRs are strongly modified by the receptor environment and suggests that keeping intact tissue conditions in the assays is important for determining the properties of nAChRs and probably other receptors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by a Grant-in Aid for Scientific Research from the Japan Society of the Promotion of Science (JSPS) and a grant from the Smoking Research Foundation of Japan.
Alexander, S. P., Mathie, A., and Peters, J. A. (2009). Guide to receptors and channels (GRAC), 4th edition. Br. J. Pharmacol. 158(Suppl. 1), S1–S254.
Anisuzzaman, A. S. M., Morishima, S., Suzuki, F., Tanaka, T., Yoshiki, H., and Muramatsu, I. (2008). Identification of M1 muscarinic receptor subtype in rat stomach using a tissue segment binding method, and the effects of immobilization stress on the muscarinic receptors. Eur. J. Pharmacol. 599, 146–151.
Anisuzzaman, A. S. M., Nishimune, A., Yoshiki, H., Uwada, J., and Muramatsu, I. (2011). Influence of tissue integrity on pharmacological phenotypes of muscarinic acetylcholine receptors in the rat cerebral cortex. J. Pharmacol. Exp. Ther. 339, 168–193.
Baker, J. G., and Hill, S. J. (2007). Multiple GPCR conformations and signalling pathways: implications for antagonist affinity estimates. Trends Pharmacol. Sci. 28, 374–381.
Benowitz, N. L., Jacob, P., Jones, R. T., and Rosenberg, J. (1982). Interindividual variability in the metabolism and cardiovascular effects of nicotine in man. J. Pharmacol. Exp. Ther. 221, 368–372.
Bylund, D. B., and Toews, M. L. (1993). Radioligand binding methods: practical guide and tips. Am. J. Physiol. 265, L421–L429.
Colucci, W. S., Gimbrone, J. R., and Alexander, R. W. (1981). Regulation of the postsynaptic alpha-adrenergic receptor in rat mesenteric artery. Effects of chemical sympathectomy and epinephrine treatment. Circ. Res. 48, 104–111.
Connelly, P. J., and Prentice, N. P. (2005). Current smoking and response to cholinesterase inhibitor therapy in Alzheimer’s disease. Dement. Geiatr. Cogn. Disord. 19, 11–14.
Davil-Garcia, M. I., Musachio, J. L., and Keller, K. J. (2003). Chronic nicotine administration does not increase nicotinic receptors labeled by [125]epibatidine in adrenal gland, superior cervical ganglia, pineal or retina. J. Neurochem. 85, 1237–1246.
Drever, B. D., Riedel, G., and Platt, B. (2010). The cholinergic system and hippocampal plasticity. Behav. Brain Res. 221, 505–514.
Faber, J. E., Yang, N., and Xin, X. H. (2001). Expression of α-adrenoceptor subtypes by smooth muscle cell and adventitial fibroblast in rat aorta and in culture. J. Pharmacol. Exp. Ther. 298, 441–452.
Flores, C. M., Davila-Gracia, M. I., Ulrich, Y. M., and Keller, K. J. (1997). Differential regulation of neuronal nicotinic receptor binding sites following chronic nicotine administration. J. Neurochem. 69, 2216–2219.
Flores, C. M., Rogers, S. W., Pabreza, L. A., Wolfe, B. B., and Kellar, K. J. (1992). A subtype of nicotinic cholinergic receptor in the brain is composed of α4 and β2 subunits and is upregulated by chronic nicotine treatment. Mol. Pharmacol. 41, 31–37.
Govind, A. P., Vezina, P., and Green, W. N. (2009). Nicotine-induced upregulation of nicotinic receptors: underlying mechanisms and relevance to nicotine addiction. Biochem. Pharmacol. 78, 756–765.
Grady, S. R., Salminen, O., Laverty, D. C., Whiteaker, P., McIntosh, J. M., Collins, A. C., and Marks, M. J. (2007). The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem. Pharmacol. 74, 1235–1246.
Hellstrom-Lindahl, E., Mousavi, M., Ravid, R., and Nordberg, A. (2004). Reduced levels of Abeta 40 and Abeta 42 in brains of smoking controls and Alzheimer’s patients. Neurobiol. Dis. 15, 351–360.
Hiraizumi-Hiraoka, Y., Tanaka, T., Yamamoto, H., Suzuki, F., and Muramatsu, I. (2004). Identification of alpha-1L adrenoceptor in rabbit ear artery. J. Pharmacol. Exp. Ther. 310, 995–1002.
Houghtling, R. A., Davila-Garcia, M. I., Hurt, S. D., and Kellar, K. J. (1995). Characterization of [3H]epibatidine binding to nicotinic cholinergic receptors in rat and human brain. Mol. Pharmacol. 48, 280–287.
Kenakin, T. (2003). Predicting therapeutic value in the lead optimization phase of drug discovery. Nat. Rev. Drug Discov. 2, 429–438.
Kwan, C. Y., Lee, R. M. K. W., and Daniel, E. E. (1981). Isolation of plasma membranes from rat mesenteric veins; a comparison of their physical and biochemical properties with arterial membranes. Blood Vessels 18, 171–186.
Lindstrom, J. (2000). “The structure of neuronal nicotinic receptors,” in Handbook of Experimental Pharmacology, Vol. 144, eds F. Clementi, D. Fornasari and C. Gotti (Berlin: Springer-Verlag), 101–162.
Lindstrom, J., Anand, R., Gerzanich, V., Peng, X., Wang, F., and Wells, G. (1996). Structure and function of neuronal nicotinic acetylcholine receptors. Prog. Brain Res. 109, 125–137.
Marks, M. J., Pauly, J. R., Gross, S. D., Deneris, E. S., Hermans, B. I., Heinemann, S. F., and Collins, A. C. (1992). Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J. Neurosci. 12, 2765–2784.
Marks, M. J., Smith, K. W., and Collins, A. C. (1998). Differential agonist inhibition identifies multiple epibatidine binding sites in mouse brain. J. Pharmacol. Exp. Ther. 285, 377–386.
Marubio, L. M., Arroyo-Jimenez, M. M., Cordero-Erausquin, A. G., Lina, C., Lenovere, N., Exaerde, A. K., Huchet, M., Damaj, M. I., and Changeux, J.-P. (1999). Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature 398, 805–810.
McMorn, R. B., Reeve, H. L., Wyatt, C. N., Vaughan, P. F., and Peers, C. (1993). Inhibition of neuronal nicotinic acetylcholine receptors by imipramine and desipramine. Eur. J. Pharmacol. 250, 247–251.
Morishima, S., Suzuki, F., Yoshiki, H., Anisuzzaman, A. S. M., Sathi, Z. S., Tanaka, T., and Muramatsu, I. (2008). Identification of alpha-1L adrenoceptor in rat cerebral cortex and possible relationship between alpha-1L and alpha-1A adrenoceptors. Br. J. Pharmacol. 153, 1485–1494.
Muramatsu, I., Morishima, S., Suzuki, F., Yoshiki, H., Anisuzzaman, A. S., Tanaka, T., Rodrigo, M. C., Myagmar, B. E., and Simpson, P. C. (2008). Identification of alpha 1L-adrenoceptor in mice and its abolition by alpha 1A-adrenoceptor gene knockout. Br. J. Pharmacol. 155, 1224–1234.
Muramatsu, I., Tanaka, T., Suzuki, F., Li, Z., Hiraizumi-Hiraoka, Y., Anisuzzaman, A. S., Yamamoto, H., Horinouchi, T., and Morishima, S. (2005). Quantifying receptor properties: the tissue segment binding method – a powerful tool for the pharmacome analysis of native receptors. J. Pharmacol. Sci. 98, 331–339.
Nelson, C. P., and Challiss, R. A. (2007) “Phenotypic” pharmacology: the influence of cellular environment on G protein-coupled receptor antagonist and inverse agonist pharmacology. Biochem. Pharmacol. 73, 737–751.
Nishimune, A., Yoshiki, H., Uwada, J., Anisuzzaman, A. S. M., Umada, H., and Muramatsu, I. (2011). Phenotype pharmacology of α1-adrenoceptors in lower urinary tract. Br. J. Pharmacol. doi:10.1111/j.1476-5381.2011.01591.x (in press).
Orr-Urtreger, A., Göldner, F. M., Saeki, M., Lorenzo, I., Goldberg, L., De Biasi, M., Dani, J. A., Patrick, J. W., and Beaudet, A. L. (1997). Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J. Neurosci. 17, 9165–9171.
Pekonen, K., Karlsson, C., Laakso, I., and Ahtee, L. (1993). Plasma nicotine and cotinine concentrations in mice after chronic oral nicotine administration and challenge doses. Eur. J. Pharmacol. 1, 13–18.
Perry, D. C., and Kellar, K. J. (1995). [3H]epibatidine labels nicotinic receptors in rat brain: an autoradiographic study. J. Pharmacol. Exp. Ther. 275, 1030–1034.
Picciotto, M. R., Addy, N. A., Mineur, Y. S., and Brunzell, D. H. (2008). It’s not “either/or”: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog. Neurobiol. 84, 329–342.
Picciotto, M. R., and Zoli, M. (2008). Neuroprotective via nAChRs: the role of nAChRs in neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease. Front. Biosci. 1, 492–504.
Picciotto, M. R., Zoli, M., Léna, C., Bessis, A., Lallemand, Y., Le Novère, N., Vincent, P., Pich, E. M., Brûlet, P., and Changeux, J. P. (1995). Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature 374, 65–67.
Pietila, K., Lahde, T., Attila, M., Ahtee, L., and Nordberg, A. (1998). Regulation of nicotinic receptors in the brain of mice withdrawn from chronic nicotine treatment. Naunyn Schmeideberg’s Arch. Pharmacol. 357, 176–182.
Quik, M., O’neil, M., and Perez, X. A. (2007). Nicotine neuroprotection against nigrostriatal damage: importance of the animal model. Trends Pharmacol. Sci. 28, 229–235.
Sanderson, E. M., Drasdo, A. L., McCrea, K., and Wonnacott, S. (1993). Upregulation of nicotinic receptors following continuous infusion of nicotine is brain-region-specific. Brain Res. 617, 349–352.
Schneider, T., and Michel, M. C. (2010). Can [I]-iodocyanopindolol label β(3)-adrenoceptors in rat urinary bladder? Front. Pharmacol. 1:128. doi:10.3389/fphar.2010.00128
Schoepfer, R., Conroy, W. G., Whiting, P., Gore, M., and Lindstrom, J. (1990). Brain alpha-bungarotoxin binding protein cDNAs and MAbs reveal subtypes of this branch of the ligand-gated ion channel gene superfamily. Neuron 5, 35–48.
Shytle, R. D., Silver, A. A., Lukas, R. J., Newman, M. B., Sheehan, D. V., and Sanberg, P. R. (2002). Nicotinic acetylcholine receptors as targets for antidepressants. Mol. Psychiatry 7, 525–535.
Su, T. H., Morishima, S., Suzuki, F., Yoshiki, H., Anisuzzaman, A. S., Tanaka, T., Cheng, J. T., and Muramatsu, I. (2008). Native profiles of α1A-adrenoceptor phenotypes in rabbit prostate. Br. J. Pharmacol. 155, 906–912.
Tanaka, T., Zang, L., Suzuki, F., and Muramatsu, I. (2004). Alpha-1 adrenoceptors: evaluation of receptor subtype-binding kinetics in intact arterial tissues and comparison with membrane binding. Br. J. Pharmacol. 141, 468–476.
Tassonyi, E., Charpantier, E., Muller, D., Dumont, L., and Bertrand, D. (2002). The role of nicotinic acetylcholine receptors in the mechanisms of anesthesia. Brain Res. Bull. 57, 133–150.
Whiteaker, P., Jimenez, M., McIntosh, J. M., Collins, A. C., and Marks, M. J. (2000). Identification of a novel nicotinic binding site in mouse brain using [125I]-epibatidine. Br. J. Pharmacol. 131, 729–739.
Whiting, P., and Lindstrom, J. (1987). Purification and characterization of a nicotinic acetylcholine receptor from rat brain. Proc. Natl. Acad. Sci. U.S.A. 84, 595–599.
Yang, M., Reese, J., Cotecchia, S., and Michel, M. C. (1998). Murine alpha1-adrenoceptor subtypes. I. Radioligand binding studies. J. Pharmacol. Exp. Ther. 286, 841–847.
Keywords: nicotinic receptor, tissue-segment binding, upregulation
Citation: Wang M-H, Yoshiki H, Anisuzzaman ASM, Uwada J, Nishimune A, Lee K-S, Taniguchi T and Muramatsu I (2011) Re-evaluation of nicotinic acetylcholine receptors in rat brain by a tissue-segment binding assay. Front. Pharmacol. 2:65. doi: 10.3389/fphar.2011.00065
Received: 28 July 2011; Accepted: 05 October 2011;
Published online: 19 October 2011.
Edited by:
Valentina Echeverria Moran, Bay Pines VA Medical Center, USAReviewed by:
David M. Lovinger, National Institutes of Health, USACopyright: © 2011 Wang, Yoshiki, Anisuzzaman, Uwada, Nishimune, Lee, Taniguchi and Muramatsu. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Ikunobu Muramatsu, Division of Pharmacology, Department of Biochemistry and Bioinformative Sciences, School of Medicine, University of Fukui, 23-3 Matsuoka-Shimoaizuki, Eiheiji, Fukui 910-1193, Japan. e-mail: muramatu@u-fukui. ac.jp
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.