

- Department of Pharmacology and Toxicology, Stark Neurosciences Research Institute, Indiana University School of Medicine, Indianapolis, IN, USA
Chronic and neuropathic pain constitute significant health problems affecting millions of individuals each year. Pain sensations typically originate in sensory neurons of the peripheral nervous system which relay information to the central nervous system (CNS). Pathological pain sensations can arise as result of changes in excitability of these peripheral sensory neurons. Voltage-gated sodium channels are key determinants regulating action potential generation and propagation; thus, changes in sodium channel function can have profound effects on neuronal excitability and pain signaling. At present, most of the clinically available sodium channel blockers used to treat pain are non-selective across sodium channel isoforms and can contribute to cardio-toxicity, motor impairments, and CNS side effects. Numerous strides have been made over the last decade in an effort to develop more selective and efficacious sodium channel blockers to treat pain. The purpose of this review is to highlight some of the more recent developments put forth by research universities and pharmaceutical companies alike in the pursuit of developing more targeted sodium channel therapies for the treatment of a variety of neuropathic pain conditions.
Introduction
The International Association for the Study of Pain (ISAP) defines pain as “an unpleasant sensory and emotional experience associated with actual or potential tissue damage.” The ability to experience painful stimuli is essential for an organism’s survival by alerting the individual to engage in protective behaviors to prevent further tissue damage or to seek appropriate actions to ameliorate the painful condition. Pain sensations typically originate in nociceptors, peripheral nerve fibers which transduce noxious thermal, mechanical, or chemical stimuli into electrical impulses that are then encoded via central pathways. The cell bodies of these sensory nociceptors are located in the dorsal root ganglia (DRG) with afferent projections to the dorsal horn of the spinal cord. Persistent or chronic pain may linger beyond the initial acute pain-producing stimulus and can become debilitating, severely affecting an individual’s quality of life. It is estimated that in the general population one out of five suffer from moderate or severe chronic pain. Whereas certain types of persistent inflammatory pain can be seen as an extension of the normal healing process, neuropathic pain serves no known protective or healing purpose. The ISAP defines neuropathic pain as “pain caused by a lesion or disease of the somatosensory nervous system,” which can be subdivided into central and peripheral neuropathic pain. Common types of neuropathic pain for which many people seek treatment include post-herpetic neuralgia, painful diabetic neuropathy, phantom limb pain, and spinal cord injury pain.
Neuropathic pain is typically characterized by allodynia (pain produced by otherwise non-painful stimuli) and/or hyperalgesia (exacerbated responses to painful stimuli). Neuropathic pain can arise following an increase in intrinsic nerve excitability, generally manifested in impulses generated ectopically or with minimal stimulation. DRG neurons express a wide variety of voltage-gated sodium channels which regulate the excitability of these neurons (Rush et al., 2007). Nerve injury can result in changes in sodium channel trafficking, gene expression, and/or channel kinetics, all of which contribute to neuronal membrane remodeling and hyperexcitability associated with neuropathic pain (Devor, 2006). As such, voltage-gated sodium channels are attractive targets for the development of novel pain therapeutics. Currently used medications for the treatment of neuropathic pain which have demonstrable actions against sodium channels include tricyclic antidepressants (TCAs: amitriptyline and nortriptyline), local anesthetics (lidocaine, mexiletine), and anticonvulsants (carbamazepine, lamotrigine, phenytoin). However, most of the sodium channel blockers that are currently available are often associated with cardio-toxicity and central nervous system (CNS) side effects (Mulroy, 2002; Walia et al., 2004).
Several new strategies are emerging in the pursuit of providing effective pain relief in patients exhibiting neuropathic pain while minimizing adverse side effects typical of many currently available medications. In this review, we will highlight (1) the development of sodium channel blockers targeted at isoforms preferentially expressed in peripheral sensory neurons involved in the initiation and transduction of pain sensations, (2) techniques for limiting the action of sodium channel blockers to the periphery, and (3) the development of sodium channel modulators that target specific patterns of sodium channel activity associated with problematic pain.
Overview of Voltage-Gated Sodium Channels
Voltage-gated sodium channels mediate the inward sodium current of excitable cells and are thus key determinants regulating action potential generation and propagation (Hodgkin and Huxley, 1952). Voltage-gated sodium channels can also influence the resting potential of neurons and play critical roles in setting the threshold for generation of action potentials (Rush et al., 2007). Therefore, alterations in sodium channel function or expression can have profound effects on normal cell excitability. Sodium channels are dynamic transmembrane proteins consisting of a pore-forming α-subunit (220–260 kDa) and auxiliary β-subunits (32–36 kDa; Catterall, 2000). The α-subunit consists of four homologous domains (I–IV), each consisting of six transmembrane α-helices (S1–S6). Additional loops join S5–S6 segments of each domain to form the outer mouth of the channel pore, with residues of the α-helical S6 segment forming the inner mouth of the pore. The S1–S4 segments of each domain serve as the voltage-sensor, with translocation of the positively charged residues in the S4 segment initiating channel activation in response to changes in the membrane potential. At resting membrane potentials, the majority of the sodium channels are in the closed configuration, prohibiting sodium influx. Upon depolarization, the channels proceed through multiple activation states with translocation of all four S4 segments into the outward configuration allowing sodium influx. Within milliseconds, the channel inactivates via a hinged-lid mechanism whereby a short cytoplasmic loop (which includes an isoleucine, phenylalanine and methionine, IFM, motif) connecting domains III and IV folds into the channel structure, occluding the pore (Vassilev et al., 1988; West et al., 1992). Channel inactivation persists throughout the depolarizing pulse, thus underlying the action potential refractory period. Following hyperpolarization of the membrane potential, the channel recovers from inactivation by returning to the closed, resting state and is re-primed and available again for activation. Perturbations in channel activation and inactivation properties can modulate the onset, duration and frequency of action potentials, thus affecting physiological neuronal signaling.
To date, nine α-subunits (Nav1.1–1.9) and four β-subunits (β1–4) have been identified in mammals (Goldin et al., 2000). Voltage-gated sodium channel isoforms exhibit differential distribution (Felts et al., 1997) as well as distinguishing electrophysiological (Catterall et al., 2005; Rush et al., 2007) and pharmacological properties (England and De Groot, 2009). Seven of the nine isoforms are neuronal, with Nav1.4 and Nav1.5 expressed in skeletal and cardiac muscle, respectively. Nav1.7, Nav1.8, and Nav1.9 are expressed almost exclusively in the peripheral nervous system (PNS). Most of the sodium channel isoforms are sensitive to nanomolar concentrations of the puffer fish toxin, tetrodotoxin (TTX), while the Nav1.5, Nav1.8, and Nav1.9 are resistant to TTX up to millimolar concentrations. Table 1 summarizes some of the properties of these channels.

Table 1. Summary of mammalian voltage-gated sodium channels.
Nav1.3, Nav1.7, Nav1.8, and Nav1.9 have been identified as possible targets for analgesics. Nav1.3 and Nav1.7 are very similar in structure to other neuronal sodium channels (Nav1.1, Nav1.2, and Nav1.6) and the skeletal muscle sodium channel (Nav1.4), but there are some interesting functional differences. By contrast, Nav1.8 and Nav1.9 exhibit pronounced differences in their sequences from other sodium channels (D:Goldin:2000]) and striking functional differences. The next section provides additional information on these four isoforms.
Sodium Channel Isoforms Implicated in Acquired and Inherited Disorders of Neuropathic Pain
Nav1.7
The Nav1.7 isoform (originally PN1, hNE) encoded by the SCN9A gene is expressed in the PNS, in both sensory and sympathetic neurons as well as in Schwann cells and neuroendocrine cells (Klugbauer et al., 1995; Felts et al., 1997; Sangameswaran et al., 1997; Toledo-Aral et al., 1997). Nav1.7 is TTX-sensitive (TTX-S) and like other TTX-S isoforms, it displays rapid activation and inactivation kinetics. However, Nav1.7 does exhibit several distinguishing characteristics with important functional effects including a slow recovery from inactivation and a slow onset of closed-state inactivation, resulting in the generation of prominent ramp currents – inward currents generated during a slow, depolarizing voltage ramp (Cummins et al., 1998; Herzog et al., 2003). As such, neurons expressing Nav1.7 are capable of amplifying slowly developing sub-threshold depolarizing inputs – such as generator potentials arising in peripheral nociceptor terminals – thereby directly modulating action potential threshold and ultimately, neuronal excitability.
Recent clinical and experimental studies have implicated Nav1.7 in playing a crucial role in inherited neuropathic pain mechanisms. Specifically, mutations in SCN9A have been linked to inherited pain syndromes (Figure 1). Mutations associated with congenital insensitivity to pain (CIP) result in truncated, non-functional Nav1.7 channels, and individuals unable to experience pain (Cox et al., 2006). In contrast, inherited erythromelalgia (IEM) and paroxysmal extreme pain disorder (PEPD) are distinct severe pain syndromes associated with gain-of-function mutations in SCN9A. IEM is characterized by episodes of burning pain, erythema, and mild swelling in the hands and feet (Waxman and Dib-Hajj, 2005). PEPD is characterized by severe rectal, ocular, and mandibular pain (Fertleman et al., 2007). Sensory neurons expressing either IEM or PEPD mutant channels are hyperexcitable, however, via distinct mechanisms (Rush et al., 2006; Dib-Hajj et al., 2008). Whereas IEM mutations hyperpolarize the voltage-dependence of activation and slow the rate of deactivation (Cummins et al., 2004; Dib-Hajj et al., 2005; Choi et al., 2006; Theile et al., 2011), PEPD mutations destabilize fast inactivation via a depolarizing shift in steady-state fast inactivation, slowed rate of open-channel fast inactivation and produce persistent currents (Fertleman et al., 2006; Dib-Hajj et al., 2008; Jarecki et al., 2008; Theile et al., 2011). PEPD mutant channels, likely due to destabilized inactivation, also enhance resurgent sodium currents (Jarecki et al., 2010; Theile et al., 2011), which have been demonstrated to facilitate high-frequency firing (Raman and Bean, 1997; Khaliq et al., 2003; Castelli et al., 2007) and as such, may contribute to increased neuronal excitability and extreme pain sensations associated with PEPD (Jarecki et al., 2010). Interestingly, a mutation in Nav1.7 was described in a patient with clinical characteristics of both IEM and PEPD. This mutation has biophysical characteristics common to both IEM and PEPD mutations, suggesting that these disorders may be part of a physiological continuum (Estacion et al., 2008). Furthermore, a single nucleotide polymorphism in the SCN9A (rs6746030 in the Single Nucleotide Polymorphisms database), is associated with increased pain perception in patients with osteoarthritis, sciatica, and phantom limb pain (Reimann et al., 2010), although the mechanism by which this polymorphism might increase pain sensations is not clear. Overall these studies indicate that alterations in Nav1.7 properties can profoundly impact pain sensitivity. Based on these findings, it has been proposed that Nav1.7 is also likely to play important roles in the more common acquired neuropathic pain syndromes.
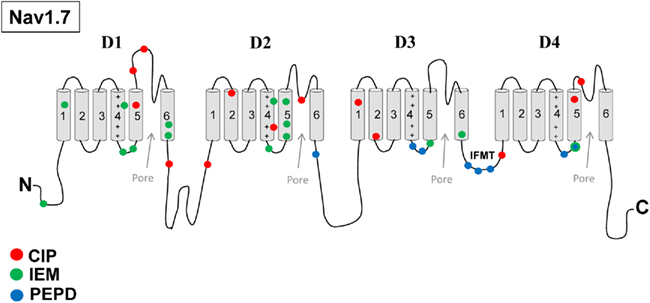
Figure 1. Nav1.7 mutations associated with inherited pain syndromes. A linear representation of the Nav1.7 α-subunit showing the approximate mutation sites for CIP, congenital insensitivity to pain; IEM, inherited erythromelalgia; PEPD, paroxysmal extreme pain disorder.
Nav1.8
The Nav1.8 isoform (originally PN3, SNS), encoded by the SCN10A gene, is expressed in nociceptive trigeminal and DRG neurons (Akopian et al., 1996; Djouhri et al., 2003; Ho and O’leary, 2011). Nav1.8 is TTX-resistant (TTX-R), with an IC50 > 50 μM when expressed in Xenopus oocytes (Akopian et al., 1996; Sangameswaran et al., 1996). Early pharmacological characterization of Nav1.8 was limited due to its poor expression in several mammalian heterologous expression systems, although functional expression has been demonstrated in an immortalized rat DRG/mouse neuroblastoma hybridoma cell line, ND7–23 (John et al., 2004). DRG neurons express a second TTX-R isoform, Nav1.9 (Cummins et al., 1999). Due to differences in voltage-dependent properties and kinetics of Nav1.8 and Nav1.9, implementation of specific voltage-clamp protocols can allow for near complete isolation of either current in DRG neurons (Dib-Hajj et al., 1999; Priest et al., 2005; Sheets et al., 2008). In contrast to the fast and rapidly inactivating TTX-S channels, Nav1.8 channels exhibit ∼10-fold slower kinetics with a depolarized voltage-dependence of activation and inactivation (Akopian et al., 1996; Cummins and Waxman, 1997). Reported values for kinetics and voltage-dependent properties of Nav1.8 vary depending on a number of factors including: (1) cell background (John et al., 2004), (2) channel ortholog (Browne et al., 2009), splice variants (Kerr et al., 2004), and (3) possible contaminating expression of Nav1.9 currents. All of these factors can have an important impact on drug discovery, and must be taken into careful consideration.
Nav1.8 channel-mediated currents are important determinants in sensory neuron excitability and pain signaling. Studies using Nav1.8-null mice have demonstrated that Nav1.8 carries the majority of current underlying the upstroke of the action potential in nociceptive neurons (Renganathan et al., 2001). The use of Nav1.8 knock-out mice and antisense oligodeoxynucleotide Nav1.8 knockdown have demonstrated a role for Nav1.8 in visceral and inflammatory pain (Akopian et al., 1999; Joshi et al., 2006). However, whether Nav1.8 plays any role in neuropathic pain is still a matter for debate (Kerr et al., 2001; Decosterd et al., 2002; Nassar et al., 2005; Siqueira et al., 2009; Leo et al., 2010). There are several lines of evidence that suggest some role of Nav1.8 in certain models of neuropathic pain. Increased levels of TNF-α in the cerebrospinal fluid and in DRG tissue following L5 ventral root transection results in the upregulation of Nav1.8 channel mRNA and protein levels (He et al., 2010). TNF-α mediated increase in Nav1.8 current density increases neuronal excitability, likely contributing to increased mechanical allodynia following motor nerve injury (Chen et al., 2011). Following sciatic nerve entrapment (SNE), a peripheral nerve injury model similar to that of the chronic constriction injury (CCI) model, upregulation of Nav1.8 mRNA is observed in sciatic nerve axons which may underlie the observed increase excitability as recorded from both A- and C-fibers (Thakor et al., 2009). However, in the SNE injury model, a concomitant decrease is observed in Nav1.8 in L4–L5 DRG neuron cell bodies, suggesting that the increased levels of mRNA may have occurred as a result of translocation from the cell bodies. Similar changes in Nav1.8 protein have been observed following CCI (Novakovic et al., 1998). As will be discussed below, several Nav1.8-selective inhibitors have been shown to be analgesic in several neuropathic pain animal models.
Nav1.3
The TTX-S isoform Nav1.3 is the predominant sodium channel isoform expressed in the CNS and PNS during embryogenesis, with very low levels in the PNS in adults (Waxman et al., 1994). However, Nav1.3 levels increase in the periphery following nerve injury and inflammation, suggesting that this channel could play a role in pain (Black et al., 2004). Nav1.3 levels are also increased in the dorsal horn following SCI (Lampert et al., 2006) and in DRG neurons following motor fiber injury (He et al., 2010). In addition, patients with trigeminal neuralgia, a disorder characterized by ectopic action potentials in trigeminal neurons, exhibit increased expression of Nav1.3 in gingival tissue (Siqueira et al., 2009). Nav1.3 channels exhibit fast kinetics with rapid recovery from inactivation, poising these channels to contribute to pathological high-frequency firing following nerve injury (Cummins and Waxman, 1997), although Nav1.3 knock-out animals still exhibit normal neuropathic pain behavior and ectopic discharges from damaged nerves (Nassar et al., 2006).
Nav1.9
Nav1.9-mediated TTX-R sodium currents are extraordinarily slow persistent currents with a relatively hyperpolarized voltage-dependency compared to the other isoforms (Cummins et al., 1999). Due to the slow, persistent nature of Nav1.9 currents these channels likely do not contribute to the upstroke of the action potential, though they may amplify sub-threshold depolarizations and lower the threshold for action potential induction (Cummins et al., 1999; Herzog et al., 2001; Baker et al., 2003). Studies using Nav1.9 KO mice suggest that this peripheral isoform plays a predominant role in inflammatory pain but not neuropathic pain (Priest et al., 2005; Amaya et al., 2006). However, even the role in inflammatory pain is somewhat unclear as antisense oligodeoxynucleotide-mediated knockdown of Nav1.9 does not reduce thermal hypersensitivity associated with complete Freund’s adjuvant (CFA)-induced inflammatory pain (Yu et al., 2011). Although Nav1.9 currents are downregulated following nerve injury (Cummins and Waxman, 1997; Decosterd et al., 2002), in an L5 spinal nerve ligation model of neuropathy reduction of Nav1.9 mRNA expression is minimal in animals that exhibited the most pain and maximal in animals with minimal pain (Persson et al., 2009), suggesting that Nav1.9 may play a role in determining pain threshold. As Nav1.9 expression is restricted to the PNS, Nav1.9 is an attractive target for modulating pain sensitivity.
Development of Drugs for Nociceptor-Selective Blockade
Nav1.7-selective Inhibitors
Given the role of Nav1.7 in pain and its expression limited to the periphery, it has been proposed that drugs selectively targeting Nav1.7 may be ideal analgesics. Despite the uncertain role of Nav1.7 in acquired neuropathic pain disorders (see above), recently developed Nav1.7-selective blockers have been shown to be efficacious in rat models of neuropathic pain. Researchers at Merck, using benzodiazepine as a molecular building block, developed a series of structurally novel benzazepinone-based state-dependent selective hNav1.7 blockers that displayed nearly complete inhibition of spontaneous neuronal firing in vivo in a rat peripheral axotomy model and reversed tactile allodynia in a rat model of spinal nerve ligation (SNL; Hoyt et al., 2007a). Following up on that study, two additional related compounds were developed with improved oral bioavailability and were highly efficacious in a rat model of SNL, with comparable or increased efficacy compared to the clinical standard, mexiletine (Hoyt et al., 2007b). However, these benzazepinone-based Nav1.7 blockers exhibited less than ideal pharmacokinetics (PK) with high clearance rates, thus Merck Research Laboratories developed a series of imidazopyridine-based blockers with improved PK and excellent efficacy in a rat SNL model with an associated reversal of inflammatory pain (London et al., 2008). Additional Nav1.7-selective compounds developed by Merck include the biphenyl thiazole carboxamides (Tyagarajan et al., 2010a) and biphenyl pyrazoles (Tyagarajan et al., 2010b,c). ProTx-II from the tarantula Thrixopelma prurient has also been reported to inhibit Nav1.7 by inducing a depolarizing shift in the voltage-dependence of activation and displays about 100-fold selectivity over the other Nav isoforms (Schmalhofer et al., 2008). Interestingly, ProTx-II seems to target the voltage-sensors of Nav isoforms (Schmalhofer et al., 2008; Xiao et al., 2010), suggesting that drugs targeting specific voltage-sensors of Nav1.7 may exhibit enhanced isoform selectivity. However, the therapeutic utility of ProTx-II in vivo may be limited by its nanomolar affinity to the other Nav isoforms and the ineffectiveness in reducing short-term or inflammatory pain following intravenous or intrathecal delivery, possibly related to its inability to cross the blood–nerve barrier (Schmalhofer et al., 2008). Amgen, Inc., has recently reported on a state-dependent Nav1.7 inhibitor which displays efficacy in a formalin model of pain in rodents, although this compound displays only modest selectivity for Nav1.7 compared to Nav1.5 and Nav1.8 (Bregman et al., 2011). Interestingly, the binding site of this compound does not appear to overlap the local anesthetic binding site, as displacement of bound tritiated batrachotoxin (3H-BTX) is not observed. Additionally, Xenon Pharmaceuticals currently has a Nav1.7-selective compound (XEN402) in the form of a topical ointment for the treatment of post-herpetic neuralgia in Phase II clinical trials.
Nav1.8-Selective Inhibitors
Although early studies using Nav1.8 knock-out mice suggested a lack of a role of Nav1.8 in the development of neuropathic pain (Akopian et al., 1999; Nassar et al., 2005), the discovery of the μO-conotoxin MrVIB and the small molecule A-803476, both potent and selective Nav1.8 inhibitors demonstrating efficacy in attenuating neuropathic pain in rats, fueled interest in the search for selective Nav1.8 inhibitors for the treatment of neuropathic pain. MrVIB inhibition of sodium channels is not voltage-dependent and has only 10-fold higher selectivity for Nav1.8 over TTX-S currents in DRG neurons (Ekberg et al., 2006). While exhibiting 100-fold selectivity of Nav1.8 over Nav1.9, MrVIB inhibits Nav1.4 channels almost equally well as Nav1.8, potentially limiting the therapeutic effectiveness of MrVIB. In contrast, A-803467 reportedly exhibits >100-fold selectivity for both resting and inactivated hNav1.8 channels over hNav1.2, hNav1.3, hNav1.5, and hNav1.7 channels expressed in HEK293 cells (Jarvis et al., 2007). A-803467 has demonstrated 10-fold greater selectivity for inactivated Nav1.8 compared to the anticonvulsant compound V102862 and 100- to 1000-fold greater selectivity compared to the local anesthetic tetracaine, the α-amino amide derivative ralfinamide, and the lamotrigine derivative 227c89 (Browne et al., 2009). Systemic administration of A-803467 demonstrated acute antinociceptive activity as measured as a reduction in mechanical allodynia in several models of inflammatory and neuropathic pain in rats (Jarvis et al., 2007). Additionally, systemic and intraspinal delivery of A-803467 attenuates both evoked and spontaneous firing of wide dynamic range neurons in rats with spinal nerve ligations (Mcgaraughty et al., 2008). Following the discovery of A-803467, a series of structurally related compounds have been developed also exhibiting low nanomolar IC50 for Nav1.8 (Kort et al., 2008, 2010; Scanio et al., 2010).
More recently, scientists at Abbott Laboratories and Icagen, Inc., collaborated on the development of an additional Nav1.8-selective blocker, A-887826 (Zhang et al., 2010). A-887826 is structurally distinct from A-803467, and displays enhanced potency toward Nav1.8. At a holding potential of −40 mV, A-887826 blocks TTX-R currents in small diameter DRG neurons with an IC50 of 8 nM, compared to A-803467 with an IC50 of 140 nM (Jarvis et al., 2007). Although A-887826 displays greater potency for Nav1.8 compared to A-803467, its selectivity to other channels is reduced (3-fold, 30-fold, and 28-fold more potent for Nav1.8 compared to Nav1.2, Nav1.5, and Nav1.7 respectively). Oral administration of A-887826 dose-dependently attenuates tactile allodynia in a spinal nerve ligation model of neuropathic pain (Zhang et al., 2010). Both compounds attenuated evoked action potential firing in neurons clamped at −40 mV and suppressed spontaneous firing induced 2 days following CFA-induced inflammation. Interestingly, unlike most sodium channel blockers, both compounds demonstrate high nanomolar affinity for block of resting channels and neither compound displays frequency-dependent inhibition of Nav1.8, thus suggesting that the mechanism of alleviating mechanical allodynia likely does not involve selective inhibition of high-frequency firing neurons due to progressive block of Nav1.8, as is the case of typical anticonvulsant sodium channel blockers.
For both A-803467 and A-887826, there were considerable differences in IC50 values for different TTX-R currents, with hNav1.8 < rNav1.8 < native TTX-R currents in DRG neurons, suggesting that subtle differences in either channel sequence or in cell background can affect the drug/channel interaction. Pursuant to this, hNav1.8 and rNav1.8 channels display significant differences in the voltage-dependence of steady-state fast inactivation when both channels are expressed in ND7–23 cells (Browne et al., 2009). Furthermore, in that report, the half-inactivation for human (∼−80 mV) and rat (∼−64 mV) Nav1.8 channels expressed in ND7–23 cells are significantly hyperpolarized compared to other reports for Nav1.8 in Xenopus oocytes (∼−30 mV; Akopian et al., 1999), DRG neurons (∼−30 mV; Cummins and Waxman, 1997; Sheets et al., 2008), and HEK293 cells (∼−50 mV; Jarvis et al., 2007). Collectively, these reports underscore the importance of considering both the cell background and channel ortholog used in the drug development process. Additionally, due to the state-dependent binding characteristics of many of the small molecule Nav1.8 sodium channel blockers, careful consideration must be used in the design of voltage-protocols, as the membrane holding potential can influence both the half-inactivation potential as well as the relative drug binding affinity (John et al., 2004; Browne et al., 2009). Despite initial excitement regarding the ability of A-803467 to modulate pain sensations (Rush and Cummins, 2007), its usefulness as a research tool has been limited by these and other factors.
Ambroxol, a secretolytic used predominantly in the treatment of respiratory disorders and as a cough suppressant, inhibits both TTX-S and TTX-R sodium currents, although with a slightly higher potency for TTX-R channels (Weiser and Wilson, 2002). Ambroxol shares an overlapping binding site with local anesthetics and exhibits preferential use-dependent block, but not tonic block, for Nav1.8 (Leffler et al., 2010). Although weakly effective in acute pain models, ambroxol reduced mechanical allodynia in a partial nerve ligation (PNL) model and reduced thermal hyperalgesia, cold allodynia, and mechanical hyperalgesia in a CCI model (Gaida et al., 2005). Oral administration of ambroxol reversed below-injury level tactile and noxious heat hypersensitivity in SCI rats (Hama et al., 2010).
Nav1.3 and Nav1.9-Selective Inhibitors
Although Nav1.3 channels have been implicated in pain mechanisms (Hains et al., 2003; Siqueira et al., 2009), Nav1.3 specific blockers have not been identified. Nav1.3 is highly homologous to other sodium channels. For example, Nav1.2 and Nav1.3 are more than 85% identical. This likely limits the probability that Nav1.3 selective blockers can be developed (Catterall et al., 2005). In contrast, Nav1.9 is only about 45% identical to the other voltage-gated sodium channel isoforms. However, it has been difficult to identify chemicals that modify Nav1.9 currents because Nav1.9 has proven very difficult to express in heterologous systems and typically exhibits rapid run-down in DRG neurons (Leffler et al., 2005). Recently a clever approach has been employed to help identify toxins that interact with Nav1.9 channels (Bosmans et al., 2011). In this study, individual voltage-sensor paddles of Nav1.9 were transplanted into potassium channel constructs and these chimeric constructs were used to screen for toxins that might target full-length Nav1.9 channels. Although this study indicated that Nav1.9 channels have distinctive pharmacological sensitivities and that the voltage-sensor paddles of Nav1.9 might be excellent targets, it is unclear to what extent the chimeric channels faithfully reproduce the pharmacological properties of the full-length Nav1.9 channels.
Techniques for Limiting the Action of Sodium Channel Blockers to the Periphery
Combinational Therapy: TRPV1 Agonists and Local Anesthetics
Local anesthetics such as lidocaine block nociceptive nerve transmission via inhibition of sodium channels and are thus clinically useful for the treatment of pain. Local anesthetics are lipophilic in their uncharged form and gain access to the intracellular sodium channel pore binding site by diffusion across cell membranes. However, local anesthetics are non-selective across sodium channel isoforms causing block of cardiac sodium channels as well as non-nociceptive sensory, sympathetic and motor fibers leading to adverse and potentially toxic side effects. Thus, development of localized anesthesia via selective inhibition of nociceptive fibers would greatly enhance the clinical utility of local anesthetics in treating pain.
A novel strategy for such an approach was recently developed that exploited the relatively selective expression of the transient receptor potential vanilloid 1 (TRPV1) receptor to nociceptive sensory neurons (Binshtok et al., 2007). TRP channels are key regulators of sensory transduction (Patapoutian et al., 2009). Specifically, TRPV1 channels are polymodal nocitransducers activated by noxious stimuli including high heat (>43°C), low pH (<6.0) and the pungent ingredient of chili peppers, capsaicin (Caterina et al., 1997; Tominaga et al., 1998). TRPV1 channels have large pores capable of passing small charged molecules, such as QX-314, a permanently charged quaternary derivative of lidocaine. Normally, QX-314 has no effect on neuronal sodium channels when applied extracellularly but does induce block when applied intracellularly (Figure 2). Following activation of TRPV1 via capsaicin, Woolf and colleagues utilized the large pore of TRPV1 as a conduit by which to deliver QX-314 into the cell interior in order to block Na+ currents in DRG neurons (Binshtok et al., 2007). Furthermore, because TRPV1 channels are restricted primarily to nociceptive neurons, co-injection of capsaicin and QX-314 into the rat hindpaw or near the sciatic nerve produced a long-lasting decrease in pain sensitivity without any motor or tactile deficits. However, the use of capsaicin as a TRPV1 agonist presents an obvious downside for pain relief in that capsaicin itself produces acute pain upon application prior to the slowly developing QX-314 mediated block of nociceptive transmission. Interestingly, pre-treating with QX-314 prior to capsaicin injection imparted little or no response to capsaicin, possibly due to the immediate entry of QX-314 already in the extracellular space following activation of TRPV1, or inhibition of TRPV1 channels by low concentrations of QX-314 (Rivera-Acevedo et al., 2011). Although the use of a non-pungent TRPV1 agonist, such as eugenol (Yang et al., 2003) would circumvent the irritating aspect of capsaicin, formulation issues have so far prevented successful co-application of eugenol and QX-314 in vivo (Roberson et al., 2011). It should be noted that intrathecal administration of QX-314 in mice can be lethal, suggesting that QX-314 should be avoided for spinal anesthesia in humans (Schwarz et al., 2010).
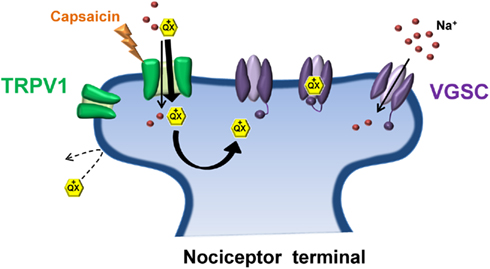
Figure 2. Transient receptor potential vanilloid 1 and QX-314: No pain, no gain. Due to its positive charge, the lidocaine derivative QX-314 is unable to pass through the plasma membrane to gain access to its binding site on the intracellular face of the sodium channel pore. Activation of TRPV1 by capsaicin (the pungent ingredient in chili peppers) allows QX-314 to pass through the relatively large pore of TRPV1, shuttling QX-314 into the cytosol where it can bind and inhibit the sodium channel. TRPV1 receptors are preferentially expressed on nociceptive terminals thus allowing selective inhibition of pain-transmitting nerve fibers.
Interestingly, lidocaine itself is an activator of TRPV1 channels, although only at a concentration sufficient to produce appreciable block of sodium channels (Leffler et al., 2008). Nonetheless, co-application of lidocaine and QX-314 produces a long-lasting differential nerve block (with nociceptive block far outlasting motor block) better than lidocaine alone (Binshtok et al., 2009), without the initial irritation induced by capsaicin administration. Following pre-treatment with an amphipathic quaternary ammonium sodium channel blocker (N-methyl amitriptyline) and tertiary amine sodium channel blockers (amitriptyline, bupivacaine, and lidocaine), injection of capsaicin near the rat sciatic nerve results in long-lasting differential nerve block with the more hydrophobic drugs producing the largest differential block (Gerner et al., 2008). In an effort to determine the optimal concentration and ratio of lidocaine and QX-314 resulting in long-lasting duration of nociceptive-selective differential nerve block, Woolf and colleagues recently underwent a systematic study exploring multiple combinations of drug doses in a rat sciatic nerve model (Roberson et al., 2011). The authors determined that the combination of 0.5% QX-314 and 2% lidocaine administered near the sciatic nerve produced 1 h of non-selective sensory and motor block followed by >9 h of pain selective block. Though initially counter-intuitive (causing pain in order to alleviate pain), the use of TRPV1 as a drug delivery portal represents an ingenious new platform for future researchers to build upon in the possible development of membrane-impermeable, subtype-selective sodium channel blockers that can be delivered to pain sensing terminals.
Development of Sodium Channel Blockers that Do not Cross the Blood Brain Barrier
Due to the restricted expression of Nav1.7, Nav1.8, and Nav1.9 to the periphery, development of poorly brain-penetrant Nav selective compounds may increase tolerability due to reduced actions on CNS Nav isoforms. Indeed, a small molecule Nav1.7 inhibitor developed by Merck, N-[(R)-1-((R)-7-chloro-1-isopropyl-2-oxo-2,3,4,5-tetrahydro-1H-benzo[b]aze pin-3-ylcarbamoyl)-2-(2-flu-orophenyl)-ethyl]-4-fluoro-2-trifluoromethyl-benzamide (BZP), produces substantial reversal of inflammatory hyperalgesia and mechanical allodynia comparable to that of standard clinical drugs, but due to its poor brain-penetrance, BZP conferred fewer sedative, and motor-coordination impairments (Mcgowan et al., 2009). Although sodium channel blockers that do not penetrate the BBB may help reduce CNS associated side effects, actions against cardiac sodium channels could still be problematic, as could the use of such compounds in conditions which compromise the BBB (e.g., certain spinal cord injuries) or the blood-spinal cord barrier (Beggs et al., 2010).
Targeting Sodium Channel Activity Rather than the Isoform
As detailed above, isoform-specific sodium channel blockers that are targeted at isoforms preferentially expressed in the peripheral sensory neurons (Nav1.7, Nav1.8, and Nav1.9) may be very effective at treating pain while minimizing significant side effects. An alternative approach is to identify sodium channel modulators that target specific patterns of sodium channel activity that are associated with problematic pain. Below we highlight several properties of sodium channel function which have shown to be promising pain targets.
Slow-Inactivation
Within milliseconds following activation sodium channels enter a non-conducting fast-inactivated state; accounting for the action potential refractory period until the membrane potential has been sufficiently repolarized allowing the channels to become available again for activation. However, under conditions of prolonged (within seconds to minutes) depolarizations, sodium channels can also enter a slow-inactivated state (Figure 3). Whereas the mechanism of fast inactivation entails occlusion of the pore by translocation of the IFM motif of the DIII–DIV intracellular linker, the mechanism of slow-inactivation is believed to be due to structural rearrangement of the pore (Goldin, 2003). In neuropathic pain, sensory neurons become hyperexcitable, eliciting repetitive action potentials resulting in a progressively depolarized membrane potential. These sustained depolarizations reduce sodium channel availability as a result of accumulation into the slow-inactivated state, dampening neuronal excitability. As typical anticonvulsants curtail pathological high-frequency firing by preferentially binding open/inactivated channels, small molecules capable of stabilizing channels into the slow-inactivated state may represent an additional therapeutic approach for selectively targeting hyperexcitable neurons while sparing normal, low-frequency spiking activity.
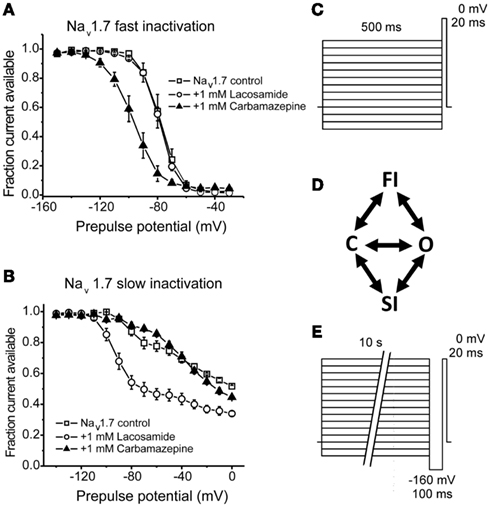
Figure 3. Lacosamide enhances sodium channel slow-inactivation without altering fast inactivation. (A) Carbamazepine, but not lacosamide, enhances Nav1.7 fast inactivation as evident by a hyperpolarizing shift in the voltage-dependence of steady-state fast inactivation. (B) Lacosamide, but not carbamazepine, causes a significant shift in the voltage-dependence of Nav1.7 slow-inactivation. (C,E) Pulse protocols for determining the voltage-dependence of fast and slow-inactivation, respectively. (D) A simplified diagram showing the different channel occupancy states, where C indicates the closed-state, O the open state, FI the fast-inactivated state and SI the slow-inactivated state. (A,B) adapted from Sheets et al. (2008 with permission from The Journal of Pharmacology and Experimental Therapeutics.
Lacosamide is a functionalized amino acid that was synthesized during the development of anticonvulsant drug candidates and has displayed antinociceptive properties in inflammatory and neuropathic pain (Beyreuther et al., 2006; Hao et al., 2006; Stohr et al., 2006; Bee and Dickenson, 2009; Wymer et al., 2009; Ziegler et al., 2010). Lacosamide displays a unique mechanism of action in that it seemingly selectively stabilizes channels into the slow-inactivated state (Errington et al., 2008). In that study, lacosamide had no apparent effect on fast inactivation of sodium channels from neocortical neurons, nor did it exhibit rapid use-dependent block which is typical of the prototypical anticonvulsants. In a similar study, lacosamide inhibited currents from Nav1.3, Nav1.7, and Nav1.8, but only after prolonged depolarizations, consistent with an enhancement in slow-inactivation with no effect on fast inactivation (Sheets et al., 2008). Furthermore, Sheets et al. (2008) observed that lacosamide was better able to discriminate between resting and inactivated channels compared to lidocaine or carbamazepine (Figure 3), thus likely allowing for improved selectivity over neurons with a depolarized membrane potential, with little tonic block.
Recently, several other compounds have been identified that also seem to selectively stabilize the slow-inactivated state (Table 2). Structurally novel derivatives of lacosamide have been developed that exhibit enhanced binding to the slow-inactivated state compared to lacosamide (Wang et al., 2011). Brilliant Blue G, also known as Coomassie blue, was recently demonstrated to be a potent inhibitor of sodium channels with a much higher affinity for the slow-inactivated state compared to the fast-inactivated state (Jo and Bean, 2011). Zalicus Pharmaceuticals recently developed a small, organic compound, Z123212, which selectively stabilizes slow-inactivation of recombinant Nav1.7, Nav1.8, and Cav3.2 T-type currents, as well as TTX-S and TTX-R currents from lamina I/II spinal cord neurons, with no effect on the voltage-dependence of fast inactivation for sodium or Cav3.2 currents (Hildebrand et al., 2011). In that study, Z123212 is considerably more potent (IC50 = 480 nM) than lacosamide (IC50 = 150 μM) in inhibiting AP firing elicited in response to a steady current injection. Furthermore, oral administration of Z123212 at doses within the range to enhance slow-inactivation resulted in a significant reversal of tactile allodynia and thermal hyperalgesia in SNL rats with no observable effects on motor-coordination or cardiovascular properties. These studies suggest that targeting slow-inactivation represents a potentially advantageous approach for the targeting of pathologically depolarized neurons associated with neuropathic pain. However, it should be noted that it can be very difficult to distinguish between a compound that targets slow-inactivation and one that simply exhibits very slow binding and unbinding to fast-inactivated channels. This caveat must be kept in mind when trying to definitively determine a mechanism of action with inactivation modifiers.

Table 2. Summary of voltage-gated sodium channel blockers.
Persistent Sodium Currents
Persistent sodium currents can arise when sodium channels fail to fully inactivate after opening, resulting in a very small residual sodium influx, representing only a fraction of the transient current (∼1%). Additionally, persistent currents can arise at voltages in which channels are activated but inactivation is sub-maximal (“window currents”). Persistent currents can play a critical role in modulating both resting membrane potential and action potential threshold, thus modulating neuronal excitability. Increased persistent currents have been observed under pathological conditions of hyperexcitability and may be associated with neuropathic pain. Nav1.7-PEPD mutations exhibited increased persistent currents (Jarecki et al., 2008; Theile et al., 2011), and inhibition of these currents is a proposed mechanism by which carbamazepine is effective in treating pain in PEPD patients (Fertleman et al., 2006). Upregulation of Nav1.3-mediated persistent currents in dorsal horn neurons is observed following contusive SCI in rats, and likely contributes to neuronal hyperexcitability seen in these neurons (Lampert et al., 2006). In rats, chronic constriction of L5 dorsal root ganglion (CCD) results in mechanical allodynia and increased persistent currents in injured A-type fibers, both of which are reduced by the persistent current-selective inhibitor, riluzole (Xie et al., 2011).
Riluzole is a neuroprotective agent mainly used in the treatment of amyotrophic lateral sclerosis (Bensimon et al., 1994), however it also displays anticonvulsant and antiepileptic properties in animals models (Romettino et al., 1991; Zgrajka et al., 2010). The neuroprotective properties of riluzole partially stem from its actions to reduce pre-synaptic glutamate release and/or via post-synaptic inhibition of NMDA receptors (Cheramy et al., 1992; Debono et al., 1993). Riluzole also displays activity against sodium channels, inhibiting TTX-S and TTX-R currents in DRG neurons equally well in a state-dependent manner (Song et al., 1997). In CNS and cardiac sodium channel isoforms, riluzole at low concentrations selectively inhibits persistent sodium currents compared to transient currents (Urbani and Belluzzi, 2000; Weiss et al., 2010). Recently, riluzole has demonstrated antinociceptive properties on below-level cutaneous hypersensitivity in rats with spinal cord injury, although it is not clear if this effect is mediated via sodium channel inhibition or a reduction in glutamatergic transmission (Hama and Sagen, 2011). The authors also observed general antinociceptive effects of riluzole on uninjured rats when administered peripherally but not centrally. Because persistent sodium currents, especially those that occur at or near the threshold for action potential generation, can substantially increase excitability, drugs that selectively target persistent currents in nociceptive neurons could be very useful in treating pain.
The analgesic potential of ranolazine, a drug that is FDA approved for treatment of chronic angina pectoris, has been investigated because ranolazine preferentially inhibits persistent cardiac sodium currents as well as sensory neuronal sodium channels (Rajamani et al., 2008, 2009; Wang et al., 2008). Ranolazine inhibits sensory neuronal excitability and the behavioral signs of inflammatory and neuropathic pain (Gould et al., 2009; Casey et al., 2010; Estacion et al., 2010). Although targeting persistent sodium currents in sensory neurons may be efficacious in treating pain, both riluzole and ranolazine interact with a broad spectrum of sodium channels, including cardiac and CNS channels, which may not be ideal.
Resurgent Sodium Currents
Recently it has been proposed that resurgent sodium currents might also contribute to enhanced pain sensitivity (Jarecki et al., 2010). After opening, Nav channels normally undergo inactivation within milliseconds via occlusion of the pore with the IMF inactivation particle. Channels are refractory following inactivation until the membrane has been hyperpolarized to resting potentials, repriming the channel. Under certain conditions channels can re-open during repolarization to moderately negative potentials allowing a surge of inward current (resurgent current; Figure 4). Resurgent sodium currents were first identified in cerebellar Purkinje neurons (Raman and Bean, 1997) and more recently in DRG neurons (Cummins et al., 2005). These currents are proposed to flow following relief of ultra-fast open-channel block by an endogenous cytosolic blocking particle, proposed to be the C-terminal tail of the auxiliary Navβ4 subunit (Grieco et al., 2005; Bant and Raman, 2010). Resurgent currents facilitate recovery from inactivation and are maximal at potentials near the action potential threshold, and as such, may contribute to high-frequency firing (Raman and Bean, 1997; Khaliq et al., 2003; Castelli et al., 2007). Although Nav1.6 is the predominant carrier of resurgent current in DRG neurons (Cummins et al., 2005), other isoforms can carry this current under pathological conditions of impaired inactivation (Grieco and Raman, 2004; Jarecki et al., 2010). Furthermore, enhanced resurgent currents are associated with the Nav1.7-PEPD mutations (Jarecki et al., 2010; Theile et al., 2011). Thus, resurgent currents potentially represent a novel drug target for the treatment of pain. As resurgent currents arise following transition to a unique channel state (open-channel block), it may be possible to develop small molecules capable of selectively targeting resurgent currents. Indeed, a recent study showed that anandamide is able to inhibit resurgent sodium currents at concentrations that have little impact on peak transient currents (Theile and Cummins, 2011).
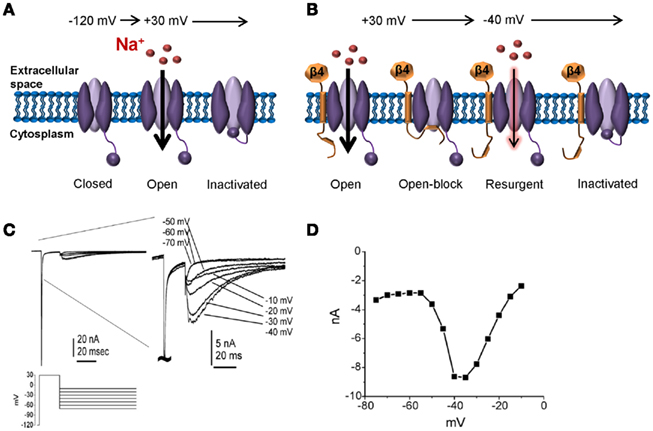
Figure 4. Resurgent sodium currents. (A) Following a strong depolarization, sodium channels transition from the resting closed-state to open, allowing influx of sodium. Within milliseconds, the channel inactivates via a hinged-lid mechanism and remains inactivated until the membrane potential has been sufficiently hyperpolarized. This cycle of events underlies the action potential refractory period. (B) Following a strong depolarization, a blocking particle (likely the C-terminal portion of the auxiliary Navβ4 subunit) can occlude the open-channel before the inactivation gate can bind, thus resulting in open-channel block. Following a hyperpolarization to an intermediate potential, the blocker is expelled resulting in an additional surge in current. (C) Representative resurgent sodium currents recorded from a large Nav1.8-null DRG neuron. The traces are magnified in the right panel to better see the resurgent currents. (D) The voltage-dependence of the resurgent currents is shown by plotting the peak resurgent current amplitude against the repolarization pulse potential. (C,D) adapted from Cummins et al. 2005) with permission from FEBS Letters.
Conclusion
Voltage-gated sodium channels are emerging as exciting targets for the treatment of neuropathic pain, albeit several challenges must be overcome. The currently available sodium channel blockers used for treatment are non-specific among sodium channel subtypes, resulting in undesirable side effects thus limiting their clinical utility. However, as highlighted in this review, researchers have made considerable advances in the pursuit of developing sodium channel blockers with improved efficacy while minimizing off-target effects. Several pharmaceutical companies are engaged in the development of subtype-selective blockers targeted to the peripheral neuronal isoforms Nav1.7 and Nav1.8, which have a demonstrable role in pain mechanisms. In fact, several such blockers have entered into human clinical trials. Other exciting new approaches involve techniques limiting drug delivery to the periphery via the development of brain-impenetrable blockers and a unique approach that exploits the TRPV1 receptor. Additionally, as the widely used anticonvulsants antagonize aberrant sodium channel activity due to their state-dependence, identifying, and developing other state-dependent drugs which target pathological sodium channel activity may circumvent the challenges associated with developing drugs targeting the subtle differences between the highly homologous channel isoforms. Furthermore, resurgent current generation relies on the presence of the accessory Navβ4 protein. As resurgent currents may play a role in pathological pain, drugs targeting Navβ4 or other accessory proteins associated with sodium channels may represent yet another avenue for pain therapy. Overall, recent advances in our knowledge of sodium channel properties and increased understanding of the roles that specific isoforms play in abnormal pain syndromes makes this an exciting time for novel pain therapeutics that target voltage-gated sodium channels.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akopian, A. N., Sivilotti, L., and Wood, J. N. (1996). A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 379, 257–262.
Akopian, A. N., Souslova, V., England, S., Okuse, K., Ogata, N., Ure, J., Smith, A., Kerr, B. J., Mcmahon, S. B., Boyce, S., Hill, R., Stanfa, L. C., Dickenson, A. H., and Wood, J. N. (1999). The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat. Neurosci. 2, 541–548.
Amaya, F., Wang, H., Costigan, M., llchorne, A. J., Hatcher, J. P., Egerton, J., Stean, T., Morisset, V., Grose, D., Gunthorpe, M. J., Chessell, I. P., Tate, S., Green, P. J., and Woolf, C. J. (2006). The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J. Neurosci. 26, 12852–12860.
Baker, M. D., Chandra, S. Y., Ding, Y., Waxman, S. G., and Wood, J. N. (2003). GTP-induced tetrodotoxin-resistant Na+ current regulates excitability in mouse and rat small diameter sensory neurones. J. Physiol. (Lond.) 548, 373–382.
Bant, J. S., and Raman, I. M. (2010). Control of transient, resurgent, and persistent current by open-channel block by Na channel β4 in cultured cerebellar granule neurons. Proc. Natl. Acad. Sci. U.S.A. 107, 12357–12362.
Bee, L. A., and Dickenson, A. H. (2009). Effects of lacosamide, a novel sodium channel modulator, on dorsal horn neuronal responses in a rat model of neuropathy. Neuropharmacology 57, 472–479.
Beggs, S., Liu, X. J., Kwan, C., and Salter, M. W. (2010). Peripheral nerve injury and TRPV1-expressing primary afferent C-fibers cause opening of the blood-brain barrier. Mol. Pain 6, 74.
Bensimon, G., Lacomblez, L., and Meininger, V. (1994). A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 330, 585–591.
Beyreuther, B., Callizot, N., and Stohr, T. (2006). Antinociceptive efficacy of lacosamide in a rat model for painful diabetic neuropathy. Eur. J. Pharmacol. 539, 64–70.
Binshtok, A. M., Bean, B. P., and Woolf, C. J. (2007). Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature 449, 607–610.
Binshtok, A. M., Gerner, P., Oh, S. B., Puopolo, M., Suzuki, S., Roberson, D. P., Herbert, T., Wang, C. F., Kim, D., Chung, G., Mitani, A. A., Wang, G. K., Bean, B. P., and Woolf, C. J. (2009). Coapplication of lidocaine and the permanently charged sodium channel blocker QX-314 produces a long-lasting nociceptive blockade in rodents. Anesthesiology 111, 127–137.
Black, J. A., Liu, S., Tanaka, M., Cummins, T. R., and Waxman, S. G. (2004). Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108, 237–247.
Bosmans, F., Puopolo, M., Martin-Eauclaire, M. F., Bean, B. P., and Swartz, K. J. (2011). Functional properties and toxin pharmacology of a dorsal root ganglion sodium channel viewed through its voltage sensors. J. Gen. Physiol. 138, 59–72.
Bregman, H., Berry, L., Buchanan, J. L., Chen, A., Du, B., Feric, E., Hierl, M., Huang, L., Immke, D., Janosky, B., Johnson, D., Li, X., Ligutti, J., Liu, D., Malmberg, A., Matson, D., Mcdermott, J., Miu, P., Nguyen, H. N., Patel, V. F., Waldon, D., Wilenkin, B., Zheng, X. M., Zou, A., Mcdonough, S. I., and Dimauro, E. F. (2011). Identification of a potent, state-dependent inhibitor of Nav1.7 with oral efficacy in the formalin model of persistent pain. J. Med. Chem. 54, 4427–4445.
Browne, L. E., Clare, J. J., and Wray, D. (2009). Functional and pharmacological properties of human and rat NaV1.8 channels. Neuropharmacology 56, 905–914.
Casey, G. P., Roberts, J. S., Paul, D., Diamond, I., and Gould, H. J. III. (2010). Ranolazine attenuation of CFA-induced mechanical hyperalgesia. Pain Med. 11, 119–126.
Castelli, L., Biella, G., Toselli, M., and Magistretti, J. (2007). Resurgent Na+ current in pyramidal neurones of rat perirhinal cortex: axonal location of channels and contribution to depolarizing drive during repetitive firing. J. Physiol. (Lond.) 582, 1179–1193.
Caterina, M. J., Schumacher, M. A., Tominaga, M., Rosen, T. A., Levine, J. D., and Julius, D. (1997). The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824.
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25.
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2005). International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409.
Chen, X., Pang, R. P., Shen, K. F., Zimmermann, M., Xin, W. J., Li, Y. Y., and Liu, X. G. (2011). TNF-alpha enhances the currents of voltage gated sodium channels in uninjured dorsal root ganglion neurons following motor nerve injury. Exp. Neurol. 227, 279–286.
Cheramy, A., Barbeito, L., Godeheu, G., and Glowinski, J. (1992). Riluzole inhibits the release of glutamate in the caudate nucleus of the cat in vivo. Neurosci. Lett. 147, 209–212.
Choi, J. S., Dib-Hajj, S. D., and Waxman, S. G. (2006). Inherited erythermalgia: limb pain from an S4 charge-neutral Na channelopathy. Neurology 67, 1563–1567.
Cox, J. J., Reimann, F., Nicholas, A. K., Thornton, G., Roberts, E., Springell, K., Karbani, G., Jafri, H., Mannan, J., Raashid, Y., Al-Gazali, L., Hamamy, H., Valente, E. M., Gorman, S., Williams, R., Mchale, D. P., Wood, J. N., Gribble, F. M., and Woods, C. G. (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898.
Cummins, T. R., Dib-Hajj, S. D., Black, J. A., Akopian, A. N., Wood, J. N., and Waxman, S. G. (1999). A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J. Neurosci. 19, RC43.
Cummins, T. R., Dib-Hajj, S. D., Herzog, R. I., and Waxman, S. G. (2005). Nav1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett. 579, 2166–2170.
Cummins, T. R., Dib-Hajj, S. D., and Waxman, S. G. (2004). Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J. Neurosci. 24, 8232–8236.
Cummins, T. R., Howe, J. R., and Waxman, S. G. (1998). Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. 18, 9607–9619.
Cummins, T. R., and Waxman, S. G. (1997). Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J. Neurosci. 17, 3503–3514.
Debono, M. W., Le Guern, J., Canton, T., Doble, A., and Pradier, L. (1993). Inhibition by riluzole of electrophysiological responses mediated by rat kainate and NMDA receptors expressed in Xenopus oocytes. Eur. J. Pharmacol. 235, 283–289.
Decosterd, I., Ji, R. R., Abdi, S., Tate, S., and Woolf, C. J. (2002). The pattern of expression of the voltage-gated sodium channels Na(v)1.8 and Na(v)1.9 does not change in uninjured primary sensory neurons in experimental neuropathic pain models. Pain 96, 269–277.
Dib-Hajj, S. D., Estacion, M., Jarecki, B. W., Tyrrell, L., Fischer, T. Z., Lawden, M., Cummins, T. R., and Waxman, S. G. (2008). Paroxysmal extreme pain disorder M1627K mutation in human Nav1.7 renders DRG neurons hyperexcitable. Mol. Pain 4, 37.
Dib-Hajj, S. D., Rush, A. M., Cummins, T. R., Hisama, F. M., Novella, S., Tyrrell, L., Marshall, L., and Waxman, S. G. (2005). Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 128, 1847–1854.
Dib-Hajj, S. D., Tyrrell, L., Cummins, T. R., Black, J. A., Wood, P. M., and Waxman, S. G. (1999). Two tetrodotoxin-resistant sodium channels in human dorsal root ganglion neurons. FEBS Lett. 462, 117–120.
Djouhri, L., Fang, X., Okuse, K., Wood, J. N., Berry, C. M., and Lawson, S. N. (2003). The TTX-resistant sodium channel Nav1.8 (SNS/PN3): expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J. Physiol. 550, 739–752.
Ekberg, J., Jayamanne, A., Vaughan, C. W., Aslan, S., Thomas, L., Mould, J., Drinkwater, R., Baker, M. D., Abrahamsen, B., Wood, J. N., Adams, D. J., Christie, M. J., and Lewis, R. J. (2006). muO-conotoxin MrVIB selectively blocks Nav1.8 sensory neuron specific sodium channels and chronic pain behavior without motor deficits. Proc. Natl. Acad. Sci. U.S.A. 103, 17030–17035.
England, S., and De Groot, M. J. (2009). Subtype-selective targeting of voltage-gated sodium channels. Br. J. Pharmacol. 158, 1413–1425.
Errington, A. C., Stohr, T., Heers, C., and Lees, G. (2008). The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol. Pharmacol. 73, 157–169.
Estacion, M., Dib-Hajj, S. D., Benke, P. J., Te Morsche, R. H., Eastman, E. M., Macala, L. J., Drenth, J. P., and Waxman, S. G. (2008). NaV1.7 gain-of-function mutations as a continuum: A1632E displays physiological changes associated with erythromelalgia and paroxysmal extreme pain disorder mutations and produces symptoms of both disorders. J. Neurosci. 28, 11079–11088.
Estacion, M., Waxman, S. G., and Dib-Hajj, S. D. (2010). Effects of ranolazine on wild-type and mutant hNav1.7 channels and on DRG neuron excitability. Mol. Pain 6, 35.
Felts, P. A., Yokoyama, S., Dib-Hajj, S., Black, J. A., and Waxman, S. G. (1997). Sodium channel alpha-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res. Mol. Brain Res. 45, 71–82.
Fertleman, C. R., Baker, M. D., Parker, K. A., Moffatt, S., Elmslie, F. V., Abrahamsen, B., Ostman, J., Klugbauer, N., Wood, J. N., Gardiner, R. M., and Rees, M. (2006). SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 52, 767–774.
Fertleman, C. R., Ferrie, C. D., Aicardi, J., Bednarek, N. A., Eeg-Olofsson, O., Elmslie, F. V., Griesemer, D. A., Goutieres, F., Kirkpatrick, M., Malmros, I. N., Pollitzer, M., Rossiter, M., Roulet-Perez, E., Schubert, R., Smith, V. V., Testard, H., Wong, V., and Stephenson, J. B. (2007). Paroxysmal extreme pain disorder (previously familial rectal pain syndrome). Neurology 69, 586–595.
Gaida, W., Klinder, K., Arndt, K., and Weiser, T. (2005). Ambroxol, a Nav1.8-preferring Na(+) channel blocker, effectively suppresses pain symptoms in animal models of chronic, neuropathic and inflammatory pain. Neuropharmacology 49, 1220–1227.
Gerner, P., Binshtok, A. M., Wang, C. F., Hevelone, N. D., Bean, B. P., Woolf, C. J., and Wang, G. K. (2008). Capsaicin combined with local anesthetics preferentially prolongs sensory/nociceptive block in rat sciatic nerve. Anesthesiology 109, 872–878.
Goldin, A. L. (2003). Mechanisms of sodium channel inactivation. Curr. Opin. Neurobiol. 13, 284–290.
Goldin, A. L., Barchi, R. L., Caldwell, J. H., Hofmann, F., Howe, J. R., Hunter, J. C., Kallen, R. G., Mandel, G., Meisler, M. H., Netter, Y. B., Noda, M., Tamkun, M. M., Waxman, S. G., Wood, J. N., and Catterall, W. A. (2000). Nomenclature of voltage-gated sodium channels. Neuron 28, 365–368.
Gould, H. J. III, Garrett, C., Donahue, R. R., Paul, D., Diamond, I., and Taylor, B. K. (2009). Ranolazine attenuates behavioral signs of neuropathic pain. Behav. Pharmacol. 20, 755–758.
Grieco, T. M., Malhotra, J. D., Chen, C., Isom, L. L., and Raman, I. M. (2005). Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron 45, 233–244.
Grieco, T. M., and Raman, I. M. (2004). Production of resurgent current in NaV1.6-null Purkinje neurons by slowing sodium channel inactivation with beta-pompilidotoxin. J. Neurosci. 24, 35–42.
Hains, B. C., Klein, J. P., Saab, C. Y., Craner, M. J., Black, J. A., and Waxman, S. G. (2003). Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J. Neurosci. 23, 8881–8892.
Hama, A., and Sagen, J. (2011). Antinociceptive effect of riluzole in rats with neuropathic spinal cord injury pain. J. Neurotrauma 28, 127–134.
Hama, A. T., Plum, A. W., and Sagen, J. (2010). Antinociceptive effect of ambroxol in rats with neuropathic spinal cord injury pain. Pharmacol. Biochem. Behav. 97, 249–255.
Hao, J. X., Stohr, T., Selve, N., Wiesenfeld-Hallin, Z., and Xu, X. J. (2006). Lacosamide, a new anti-epileptic, alleviates neuropathic pain-like behaviors in rat models of spinal cord or trigeminal nerve injury. Eur. J. Pharmacol. 553, 135–140.
He, X. H., Zang, Y., Chen, X., Pang, R. P., Xu, J. T., Zhou, X., Wei, X. H., Li, Y. Y., Xin, W. J., Qin, Z. H., and Liu, X. G. (2010). TNF-alpha contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury. Pain 151, 266–279.
Herzog, R. I., Cummins, T. R., Ghassemi, F., Dib-Hajj, S. D., and Waxman, S. G. (2003). Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J. Physiol. 551, 741–750.
Herzog, R. I., Cummins, T. R., and Waxman, S. G. (2001). Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J. Neurophysiol. 86, 1351–1364.
Hildebrand, M. E., Smith, P. L., Bladen, C., Eduljee, C., Xie, J. Y., Chen, L., Fee-Maki, M., Doering, C. J., Mezeyova, J., Zhu, Y., Belardetti, F., Pajouhesh, H., Parker, D., Arneric, S. P., Parmar, M., Porreca, F., Tringham, E., Zamponi, G. W., and Snutch, T. P. (2011). A novel slow-inactivation-specific ion channel modulator attenuates neuropathic pain. Pain 152, 833–843.
Ho, C., and O’leary, M. E. (2011). Single-cell analysis of sodium channel expression in dorsal root ganglion neurons. Mol. Cell. Neurosci. 46, 159–166.
Hodgkin, A. L., and Huxley, A. F. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. (Lond.) 117, 500–544.
Hoyt, S. B., London, C., Gorin, D., Wyvratt, M. J., Fisher, M. H., Abbadie, C., Felix, J. P., Garcia, M. L., Li, X., Lyons, K. A., Mcgowan, E., Macintyre, D. E., Martin, W. J., Priest, B. T., Ritter, A., Smith, M. M., Warren, V. A., Williams, B. S., Kaczorowski, G. J., and Parsons, W. H. (2007a). Discovery of a novel class of benzazepinone Na(v)1.7 blockers: potential treatments for neuropathic pain. Bioorg. Med. Chem. Lett. 17, 4630–4634.
Hoyt, S. B., London, C., Ok, H., Gonzalez, E., Duffy, J. L., Abbadie, C., Dean, B., Felix, J. P., Garcia, M. L., Jochnowitz, N., Karanam, B. V., Li, X., Lyons, K. A., Mcgowan, E., Macintyre, D. E., Martin, W. J., Priest, B. T., Smith, M. M., Tschirret-Guth, R., Warren, V. A., Williams, B. S., Kaczorowski, G. J., and Parsons, W. H. (2007b). Benzazepinone Nav1.7 blockers: potential treatments for neuropathic pain. Bioorg. Med. Chem. Lett. 17, 6172–6177.
Jarecki, B. W., Piekarz, A. D., Jackson, J. O. II, and Cummins, T. R. (2010). Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J. Clin. Invest. 120, 369–378.
Jarecki, B. W., Sheets, P. L., Jackson, J. O. II, and Cummins, T. R. (2008). Paroxysmal extreme pain disorder mutations within the D3/S4-S5 linker of Nav1.7 cause moderate destabilization of fast inactivation. J. Physiol. 586, 4137–4153.
Jarvis, M. F., Honore, P., Shieh, C. C., Chapman, M., Joshi, S., Zhang, X. F., Kort, M., Carroll, W., Marron, B., Atkinson, R., Thomas, J., Liu, D., Krambis, M., Liu, Y., Mcgaraughty, S., Chu, K., Roeloffs, R., Zhong, C., Mikusa, J. P., Hernandez, G., Gauvin, D., Wade, C., Zhu, C., Pai, M., Scanio, M., Shi, L., Drizin, I., Gregg, R., Matulenko, M., Hakeem, A., Gross, M., Johnson, M., Marsh, K., Wagoner, P. K., Sullivan, J. P., Faltynek, C. R., and Krafte, D. S. (2007). A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. U.S.A. 104, 8520–8525.
Jo, S., and Bean, B. P. (2011). Inhibition of neuronal voltage-gated sodium channels by brilliant blue G. Mol. Pharmacol. 80, 247–257.
John, V. H., Main, M. J., Powell, A. J., Gladwell, Z. M., Hick, C., Sidhu, H. S., Clare, J. J., Tate, S., and Trezise, D. J. (2004). Heterologous expression and functional analysis of rat Nav1.8 (SNS) voltage-gated sodium channels in the dorsal root ganglion neuroblastoma cell line ND7-23. Neuropharmacology 46, 425–438.
Joshi, S. K., Mikusa, J. P., Hernandez, G., Baker, S., Shieh, C. C., Neelands, T., Zhang, X. F., Niforatos, W., Kage, K., Han, P., Krafte, D., Faltynek, C., Sullivan, J. P., Jarvis, M. F., and Honore, P. (2006). Involvement of the TTX-resistant sodium channel Nav 1.8 in inflammatory and neuropathic, but not post-operative, pain states. Pain 123, 75–82.
Kerr, B. J., Souslova, V., Mcmahon, S. B., and Wood, J. N. (2001). A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport 12, 3077–3080.
Kerr, N. C., Holmes, F. E., and Wynick, D. (2004). Novel isoforms of the sodium channels Nav1.8 and Nav1.5 are produced by a conserved mechanism in mouse and rat. J. Biol. Chem. 279, 24826–24833.
Khaliq, Z. M., Gouwens, N. W., and Raman, I. M. (2003). The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J. Neurosci. 23, 4899–4912.
Klugbauer, N., Lacinova, L., Flockerzi, V., and Hofmann, F. (1995). Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 14, 1084–1090.
Kort, M. E., Atkinson, R. N., Thomas, J. B., Drizin, I., Johnson, M. S., Secrest, M. A., Gregg, R. J., Scanio, M. J., Shi, L., Hakeem, A. H., Matulenko, M. A., Chapman, M. L., Krambis, M. J., Liu, D., Shieh, C. C., Zhang, X., Simler, G., Mikusa, J. P., Zhong, C., Joshi, S., Honore, P., Roeloffs, R., Werness, S., Antonio, B., Marsh, K. C., Faltynek, C. R., Krafte, D. S., Jarvis, M. F., and Marron, B. E. (2010). Subtype-selective Na(v)1.8 sodium channel blockers: identification of potent, orally active nicotinamide derivatives. Bioorg. Med. Chem. Lett. 20, 6812–6815.
Kort, M. E., Drizin, I., Gregg, R. J., Scanio, M. J., Shi, L., Gross, M. F., Atkinson, R. N., Johnson, M. S., Pacofsky, G. J., Thomas, J. B., Carroll, W. A., Krambis, M. J., Liu, D., Shieh, C. C., Zhang, X., Hernandez, G., Mikusa, J. P., Zhong, C., Joshi, S., Honore, P., Roeloffs, R., Marsh, K. C., Murray, B. P., Liu, J., Werness, S., Faltynek, C. R., Krafte, D. S., Jarvis, M. F., Chapman, M. L., and Marron, B. E. (2008). Discovery and biological evaluation of 5-aryl-2-furfuramides, potent and selective blockers of the Nav1.8 sodium channel with efficacy in models of neuropathic and inflammatory pain. J. Med. Chem. 51, 407–416.
Lampert, A., Hains, B. C., and Waxman, S. G. (2006). Upregulation of persistent and ramp sodium current in dorsal horn neurons after spinal cord injury. Exp. Brain Res. 174, 660–666.
Leffler, A., Fischer, M. J., Rehner, D., Kienel, S., Kistner, K., Sauer, S. K., Gavva, N. R., Reeh, P. W., and Nau, C. (2008). The vanilloid receptor TRPV1 is activated and sensitized by local anesthetics in rodent sensory neurons. J. Clin. Invest. 118, 763–776.
Leffler, A., Herzog, R. I., Dib-Hajj, S. D., Waxman, S. G., and Cummins, T. R. (2005). Pharmacological properties of neuronal TTX-resistant sodium channels and the role of a critical serine pore residue. Pflugers Arch. 451, 454–463.
Leffler, A., Reckzeh, J., and Nau, C. (2010). Block of sensory neuronal Na+ channels by the secreolytic ambroxol is associated with an interaction with local anesthetic binding sites. Eur. J. Pharmacol. 630, 19–28.
Leo, S., D’hooge, R., and Meert, T. (2010). Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behav. Brain Res. 208, 149–157.
London, C., Hoyt, S. B., Parsons, W. H., Williams, B. S., Warren, V. A., Tschirret-Guth, R., Smith, M. M., Priest, B. T., Mcgowan, E., Martin, W. J., Lyons, K. A., Li, X., Karanam, B. V., Jochnowitz, N., Garcia, M. L., Felix, J. P., Dean, B., Abbadie, C., Kaczorowski, G. J., and Duffy, J. L. (2008). Imidazopyridines: a novel class of hNav1.7 channel blockers. Bioorg. Med. Chem. Lett. 18, 1696–1701.
Mcgaraughty, S., Chu, K. L., Scanio, M. J., Kort, M. E., Faltynek, C. R., and Jarvis, M. F. (2008). A selective Nav1.8 sodium channel blocker, A-803467 [5-(4-chlorophenyl-N-(3,5-dimethoxyphenyl)furan-2-carboxamide], attenuates spinal neuronal activity in neuropathic rats. J. Pharmacol. Exp. Ther. 324, 1204–1211.
Mcgowan, E., Hoyt, S. B., Li, X., Lyons, K. A., and Abbadie, C. (2009). A peripherally acting Na(v)1.7 sodium channel blocker reverses hyperalgesia and allodynia on rat models of inflammatory and neuropathic pain. Anesth. Analg. 109, 951–958.
Mulroy, M. F. (2002). Systemic toxicity and cardiotoxicity from local anesthetics: incidence and preventive measures. Reg. Anesth. Pain Med. 27, 556–561.
Nassar, M. A., Baker, M. D., Levato, A., Ingram, R., Mallucci, G., Mcmahon, S. B., and Wood, J. N. (2006). Nerve injury induces robust allodynia and ectopic discharges in Nav1.3 null mutant mice. Mol. Pain 2, 33.
Nassar, M. A., Levato, A., Stirling, L. C., and Wood, J. N. (2005). Neuropathic pain develops normally in mice lacking both Na(v)1.7 and Na(v)1.8. Mol. Pain 1, 24.
Novakovic, S. D., Tzoumaka, E., Mcgivern, J. G., Haraguchi, M., Sangameswaran, L., Gogas, K. R., Eglen, R. M., and Hunter, J. C. (1998). Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J. Neurosci. 18, 2174–2187.
Patapoutian, A., Tate, S., and Woolf, C. J. (2009). Transient receptor potential channels: targeting pain at the source. Nat. Rev. Drug Discov. 8, 55–68.
Persson, A. K., Thun, J., Xu, X. J., Wiesenfeld-Hallin, Z., Strom, M., Devor, M., Lidman, O., and Fried, K. (2009). Autotomy behavior correlates with the DRG and spinal expression of sodium channels in inbred mouse strains. Brain Res. 1285, 1–13.
Priest, B. T., Murphy, B. A., Lindia, J. A., Diaz, C., Abbadie, C., Ritter, A. M., Liberator, P., Iyer, L. M., Kash, S. F., Kohler, M. G., Kaczorowski, G. J., Macintyre, D. E., and Martin, W. J. (2005). Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc. Natl. Acad. Sci. U.S.A. 102, 9382–9387.
Rajamani, S., El-Bizri, N., Shryock, J. C., Makielski, J. C., and Belardinelli, L. (2009). Use-dependent block of cardiac late Na(+) current by ranolazine. Heart Rhythm 6, 1625–1631.
Rajamani, S., Shryock, J. C., and Belardinelli, L. (2008). Block of tetrodotoxin-sensitive, Na(V)1.7 and tetrodotoxin-resistant, Na(V)1.8, Na+ channels by ranolazine. Channels (Austin) 2, 449–460.
Raman, I. M., and Bean, B. P. (1997). Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci. 17, 4517–4526.
Reimann, F., Cox, J. J., Belfer, I., Diatchenko, L., Zaykin, D. V., Mchale, D. P., Drenth, J. P., Dai, F., Wheeler, J., Sanders, F., Wood, L., Wu, T. X., Karppinen, J., Nikolajsen, L., Mannikko, M., Max, M. B., Kiselycznyk, C., Poddar, M., Te Morsche, R. H., Smith, S., Gibson, D., Kelempisioti, A., Maixner, W., Gribble, F. M., and Woods, C. G. (2010). Pain perception is altered by a nucleotide polymorphism in SCN9A. Proc. Natl. Acad. Sci. U.S.A. 107, 5148–5153.
Renganathan, M., Cummins, T. R., and Waxman, S. G. (2001). Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J. Neurophysiol. 86, 629–640.
Rivera-Acevedo, R. E., Pless, S. A., Ahern, C. A., and Schwarz, S. K. (2011). The quaternary lidocaine derivative, QX-314, exerts biphasic effects on transient receptor potential vanilloid subtype 1 channels in vitro. Anesthesiology 114, 1425–1434.
Roberson, D., Binshtok, A., Blasl, F., Bean, B., and Woolf, C. (2011). Targeting of sodium channel blockers into nociceptors to produce long-duration analgesia: a systematic study and review. Br. J. Pharmacol. 164, 48–58.
Romettino, S., Lazdunski, M., and Gottesmann, C. (1991). Anticonvulsant and sleep-waking influences of riluzole in a rat model of absence epilepsy. Eur. J. Pharmacol. 199, 371–373.
Rush, A. M., and Cummins, T. R. (2007). Painful research: identification of a small-molecule inhibitor that selectively targets Nav1.8 sodium channels. Mol. Interv. 7, 192–195.
Rush, A. M., Cummins, T. R., and Waxman, S. G. (2007). Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. (Lond.) 579, 1–14.
Rush, A. M., Dib-Hajj, S. D., Liu, S., Cummins, T. R., Black, J. A., and Waxman, S. G. (2006). A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 8245–8250.
Sangameswaran, L., Delgado, S. G., Fish, L. M., Koch, B. D., Jakeman, L. B., Stewart, G. R., Sze, P., Hunter, J. C., Eglen, R. M., and Herman, R. C. (1996). Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J. Biol. Chem. 271, 5953–5956.
Sangameswaran, L., Fish, L. M., Koch, B. D., Rabert, D. K., Delgado, S. G., Ilnicka, M., Jakeman, L. B., Novakovic, S., Wong, K., Sze, P., Tzoumaka, E., Stewart, G. R., Herman, R. C., Chan, H., Eglen, R. M., and Hunter, J. C. (1997). A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J. Biol. Chem. 272, 14805–14809.
Scanio, M. J., Shi, L., Drizin, I., Gregg, R. J., Atkinson, R. N., Thomas, J. B., Johnson, M. S., Chapman, M. L., Liu, D., Krambis, M. J., Liu, Y., Shieh, C. C., Zhang, X., Simler, G. H., Joshi, S., Honore, P., Marsh, K. C., Knox, A., Werness, S., Antonio, B., Krafte, D. S., Jarvis, M. F., Faltynek, C. R., Marron, B. E., and Kort, M. E. (2010). Discovery and biological evaluation of potent, selective, orally bioavailable, pyrazine-based blockers of the Na(v)1.8 sodium channel with efficacy in a model of neuropathic pain. Bioorg. Med. Chem. 18, 7816–7825.
Schmalhofer, W. A., Calhoun, J., Burrows, R., Bailey, T., Kohler, M. G., Weinglass, A. B., Kaczorowski, G. J., Garcia, M. L., Koltzenburg, M., and Priest, B. T. (2008). ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol. Pharmacol. 74, 1476–1484.
Schwarz, S. K., Cheung, H. M., Ries, C. R., Lee, S. M., Wang, J. T., and Macleod, B. A. (2010). Lumbar intrathecal administration of the quaternary lidocaine derivative, QX-314, produces irritation and death in mice. Anesthesiology 113, 438–444.
Sheets, P. L., Heers, C., Stoehr, T., and Cummins, T. R. (2008). Differential block of sensory neuronal voltage-gated sodium channels by lacosamide [(2R)-2-(acetylamino)-N-benzyl-3-methoxypropanamide], lidocaine, and carbamazepine. J. Pharmacol. Exp. Ther. 326, 89–99.
Siqueira, S. R., Alves, B., Malpartida, H. M., Teixeira, M. J., and Siqueira, J. T. (2009). Abnormal expression of voltage-gated sodium channels Nav1.7, Nav1.3 and Nav1.8 in trigeminal neuralgia. Neuroscience 164, 573–577.
Song, J. H., Huang, C. S., Nagata, K., Yeh, J. Z., and Narahashi, T. (1997). Differential action of riluzole on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J. Pharmacol. Exp. Ther. 282, 707–714.
Stohr, T., Krause, E., and Selve, N. (2006). Lacosamide displays potent antinociceptive effects in animal models for inflammatory pain. Eur. J. Pain 10, 241–249.
Thakor, D. K., Lin, A., Matsuka, Y., Meyer, E. M., Ruangsri, S., Nishimura, I., and Spigelman, I. (2009). Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painful neuropathy. Mol. Pain 5, 14.
Theile, J. W., and Cummins, T. R. (2011). Inhibition of Nav{beta}4 peptide-mediated resurgent sodium currents in Nav1.7 channels by carbamazepine, riluzole and anandamide. Mol. Pharmacol. 80, 724–734.
Theile, J. W., Jarecki, B. W., Piekarz, A. D., and Cummins, T. R. (2011). Nav1.7 mutations associated with paroxysmal extreme pain disorder, but not erythromelalgia, enhance Nav{beta}4 peptide-mediated resurgent sodium currents. J. Physiol. 589, 597–608.
Toledo-Aral, J. J., Moss, B. L., He, Z. J., Koszowski, A. G., Whisenand, T., Levinson, S. R., Wolf, J. J., Silos-Santiago, I., Halegoua, S., and Mandel, G. (1997). Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc. Natl. Acad. Sci. U.S.A. 94, 1527–1532.
Tominaga, M., Caterina, M. J., Malmberg, A. B., Rosen, T. A., Gilbert, H., Skinner, K., Raumann, B. E., Basbaum, A. I., and Julius, D. (1998). The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21, 531–543.
Tyagarajan, S., Chakravarty, P. K., Zhou, B., Fisher, M. H., Wyvratt, M. J., Lyons, K., Klatt, T., Li, X., Kumar, S., Williams, B., Felix, J., Priest, B. T., Brochu, R. M., Warren, V., Smith, M., Garcia, M., Kaczorowski, G. J., Martin, W. J., Abbadie, C., Mcgowan, E., Jochnowitz, N., and Parsons, W. H. (2010a). Substituted biaryl oxazoles, imidazoles, and thiazoles as sodium channel blockers. Bioorg. Med. Chem. Lett. 20, 5536–5540.
Tyagarajan, S., Chakravarty, P. K., Zhou, B., Taylor, B., Eid, R., Fisher, M. H., Parsons, W. H., Wyvratt, M. J., Lyons, K. A., Klatt, T., Li, X., Kumar, S., Williams, B., Felix, J., Priest, B. T., Brochu, R. M., Warren, V., Smith, M., Garcia, M., Kaczorowski, G. J., Martin, W. J., Abbadie, C., Mcgowan, E., Jochnowitz, N., Weber, A., and Duffy, J. L. (2010b). Discovery of a novel class of biphenyl pyrazole sodium channel blockers for treatment of neuropathic pain. Bioorg. Med. Chem. Lett. 20, 7479–7482.
Tyagarajan, S., Chakravarty, P. K., Zhou, B., Taylor, B., Fisher, M. H., Wyvratt, M. J., Lyons, K., Klatt, T., Li, X., Kumar, S., Williams, B., Felix, J., Priest, B. T., Brochu, R. M., Warren, V., Smith, M., Garcia, M., Kaczorowski, G. J., Martin, W. J., Abbadie, C., Mcgowan, E., Jochnowitz, N., and Parsons, W. H. (2010c). Substituted biaryl pyrazoles as sodium channel blockers. Bioorg. Med. Chem. Lett. 20, 5480–5483.
Urbani, A., and Belluzzi, O. (2000). Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur. J. Neurosci. 12, 3567–3574.
Vassilev, P. M., Scheuer, T., and Catterall, W. A. (1988). Identification of an intracellular peptide segment involved in sodium channel inactivation. Science 241, 1658–1661.
Walia, K. S., Khan, E. A., Ko, D. H., Raza, S. S., and Khan, Y. N. (2004). Side effects of antiepileptics – a review. Pain Pract. 4, 194–203.
Wang, G. K., Calderon, J., and Wang, S. Y. (2008). State- and use-dependent block of muscle Nav1.4 and neuronal Nav1.7 voltage-gated Na+ channel isoforms by ranolazine. Mol. Pharmacol. 73, 940–948.
Wang, Y., Park, K. D., Salome, C., Wilson, S. M., Stables, J. P., Liu, R., Khanna, R., and Kohn, H. (2011). Development and characterization of novel derivatives of the antiepileptic drug lacosamide that exhibit far greater enhancement in slow inactivation of voltage-gated sodium channels. ACS Chem. Neurosci. 2, 90–106.
Waxman, S. G., and Dib-Hajj, S. (2005). Erythermalgia: molecular basis for an inherited pain syndrome. Trends Mol. Med. 11, 555–562.
Waxman, S. G., Kocsis, J. D., and Black, J. A. (1994). Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J. Neurophysiol. 72, 466–470.
Weiser, T., and Wilson, N. (2002). Inhibition of tetrodotoxin (TTX)-resistant and TTX-sensitive neuronal Na(+) channels by the secretolytic ambroxol. Mol. Pharmacol. 62, 433–438.
Weiss, S., Benoist, D., White, E., Teng, W., and Saint, D. A. (2010). Riluzole protects against cardiac ischaemia and reperfusion damage via block of the persistent sodium current. Br. J. Pharmacol. 160, 1072–1082.
West, J. W., Patton, D. E., Scheuer, T., Wang, Y., Goldin, A. L., and Catterall, W. A. (1992). A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc. Natl. Acad. Sci. U.S.A. 89, 10910–10914.
Wymer, J. P., Simpson, J., Sen, D., and Bongardt, S. (2009). Efficacy and safety of lacosamide in diabetic neuropathic pain: an 18-week double-blind placebo-controlled trial of fixed-dose regimens. Clin. J. Pain 25, 376–385.
Xiao, Y., Blumenthal, K., Jackson, J. O. II, Liang, S., and Cummins, T. R. (2010). The tarantula toxins ProTx-II and huwentoxin-IV differentially interact with human Nav1.7 voltage sensors to inhibit channel activation and inactivation. Mol. Pharmacol. 78, 1124–1134.
Xie, R. G., Zheng, D. W., Xing, J. L., Zhang, X. J., Song, Y., Xie, Y. B., Kuang, F., Dong, H., You, S. W., Xu, H., and Hu, S. J. (2011). Blockade of persistent sodium currents contributes to the riluzole-induced inhibition of spontaneous activity and oscillations in injured DRG neurons. PLoS ONE 6, e18681. doi: 10.1371/journal.pone.0018681
Yang, B. H., Piao, Z. G., Kim, Y. B., Lee, C. H., Lee, J. K., Park, K., Kim, J. S., and Oh, S. B. (2003). Activation of vanilloid receptor 1 (VR1) by eugenol. J. Dent. Res. 82, 781–785.
Yu, Y. Q., Zhao, F., Guan, S. M., and Chen, J. (2011). Antisense-mediated knockdown of Na(V)1.8, but not Na(V)1.9, generates inhibitory effects on complete Freund’s adjuvant-induced inflammatory pain in rat. PLoS One 6, e19865. doi: 10.1371/journal.pone.0019865
Zgrajka, W., Nieoczym, D., Czuczwar, M., Kis, J., Brzana, W., Wlaz, P., and Turski, W. A. (2010). Evidences for pharmacokinetic interaction of riluzole and topiramate with pilocarpine in pilocarpine-induced seizures in rats. Epilepsy Res. 88, 269–274.
Zhang, X. F., Shieh, C. C., Chapman, M. L., Matulenko, M. A., Hakeem, A. H., Atkinson, R. N., Kort, M. E., Marron, B. E., Joshi, S., Honore, P., Faltynek, C. R., Krafte, D. S., and Jarvis, M. F. (2010). A-887826 is a structurally novel, potent and voltage-dependent Na(v)1.8 sodium channel blocker that attenuates neuropathic tactile allodynia in rats. Neuropharmacology 59, 201–207.
Ziegler, D., Hidvegi, T., Gurieva, I., Bongardt, S., Freynhagen, R., Sen, D., and Sommerville, K. (2010). Efficacy and safety of lacosamide in painful diabetic neuropathy. Diabetes Care 33, 839–841.
Keywords: voltage-gated sodium channel, neuropathic pain, TRPV1, Nav1.7, Nav1.8, resurgent currents
Citation: Theile JW and Cummins TR (2011) Recent developments regarding voltage-gated sodium channel blockers for the treatment of inherited and acquired neuropathic pain syndromes. Front. Pharmacol. 2:54. doi: 10.3389/fphar.2011.00054
Received: 20 July 2011;
Paper pending published: 10 August 2011;
Accepted: 12 September 2011;
Published online: 04 October 2011.
Edited by:
Mohamed Chahine, Laval University, CanadaReviewed by:
Mohamed Chahine, Laval University, CanadaChris Ahern, University of British Columbia, Canada
Saïd Bendahhou, Cantre National de la Recherche Scientifique, France
Copyright: © 2011 Theile and Cummins. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Theodore R. Cummins, Department of Pharmacology and Toxicology, Stark Neurosciences Research Institute, Indiana University School of Medicine, 950 West Walnut Street, R2-459, Indianapolis, IN 46202, USA. e-mail:dHJjdW1taW5AaXVwdWkuZWR1