


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Pharmacol. , 22 February 2011
Sec. Experimental Pharmacology and Drug Discovery
volume 2 - 2011 | https://doi.org/10.3389/fphar.2011.00009
Receptors for sphingosine-1-phosphate (S1P) have been identified only recently. Their medicinal chemistry is therefore still in its infancy, and few selective agonists or antagonists are available. Furthermore, the selectivity of S1P receptor agonists or antagonists is not well established. JTE-013 and BML-241 (also known as CAY10444), used extensively as specific S1P2 and S1P3 receptors antagonists respectively, are cases in point. When analyzing S1P-induced vasoconstriction in mouse basilar artery, we observed that JTE-013 inhibited not only the effect of S1P, but also the effect of U46619, endothelin-1 or high KCl; JTE-013 strongly inhibited responses to S1P in S1P2 receptor knockout mice. Similarly, BML-241 has been shown to inhibit increases in intracellular Ca2+ concentration via P2 receptor or α1A-adrenoceptor stimulation and α1A-adrenoceptor-mediated contraction of rat mesenteric artery, while it did not affect S1P3-mediated decrease of forskolin-induced cyclic AMP accumulation. Another putative S1P1/3 receptor antagonist, VPC23019, does not inhibit S1P3-mediated vasoconstriction. With these examples in mind, we discuss caveats about relying on available pharmacological tools to characterize receptor subtypes.
Receptors for sphingosine-1-phosphate (S1P) were discovered and characterized in the late nineties. Ten years ago, there was no selective S1P receptor agonist or antagonist. Suramin, an old anti-protozoal drug, was used as S1P3 receptor antagonist (Ancellin and Hla, 1999; Salomone et al., 2003), but its usefulness was limited because it lacked specificity (Voogd et al., 1993). Recently, a number of ligands for S1P receptors have been screened as agonists and antagonist (Im, 2010). Most of these newly developed agents are commercially available and are increasingly being used to characterize the S1P receptor subtypes involved in specific biological mechanisms and functions. However, they have often not been screened for specificity against a wide array of targets, nor have they been systematically studied in vivo, as is required for drugs intended for human use.
In contrast, more detailed pharmacological data is available for compounds that show significant therapeutic potential. Among these, particularly worthy of mention is fingolimod (FTY720), an FDA- and EMA-approved drug, that, once phosphorylated, binds all S1P receptors but S1P2 in vitro. In vivo, phospho-fingolimod down-modulates lymphocytic S1P1 to inhibit S1P-S1P1-signaling and lymphocyte egress (Matloubian et al., 2004; Martin et al., 2010), which induces remission in patients with multiple sclerosis (Kappos et al., 2010). Fingolimod has been screened against a large number of other targets (including about 100 receptors) to insure S1P receptor specificity (V. Brinkmann, Novartis Institutes for BioMedical Research, personal communication).
At variance with most other available S1P receptor agents, FTY720 has been used in a large number of publications (nearly 1000 PubMed-indexed publications as of January 2011). Possibly because it has been the object of such an intense scrutiny, other, non-S1P receptor-mediated effects of FTY720 have been documented. For instance, FTY720 has been shown to inhibit ceramide synthase (Lahiri et al., 2009), cytosolic phospholipase A2 (Payne et al., 2007), S1P lyase (Bandhuvula et al., 2005), sphingosine kinase (SPK) 1 (Vessey et al., 2007; Tonelli et al., 2010), and acid sphingomyelinase (Dawson and Qin, 2011), stimulate 27-hydroxycholesterol production (Blom et al., 2010), bind to 14-3-3 proteins and inhibit their pro-survival signaling (Woodcock et al., 2010). Because most of these effects were observed at relatively high FTY720 concentration, they may not be relevant to the in vivo effects of the drug. But investigators should be cautious when interpreting the results of in vitro studies, particularly when these studies use high FTY720 concentrations. Incidentally, the fact that fingolimod does not discriminate between S1P1, S1P3, S1P4, and S1P5 receptors may not matter in vivo. Because blood levels of FTY720 are low (<3 nM), the drug does not occupy S1P3 receptors to a significant extent. However, after down-regulation of S1P1 receptors, homeostatic activation of S1P2 and S1P3 by endogenous plasma S1P (>200 nM) becomes predominant, and the S1P signaling balance is shifted from S1P1 to S1P2/S1P3, and thus from Gi to G12/13/Rho/Rho kinase activation. The observed functional consequence of FTY720 administration may therefore be a sum of drug- and endogenous S1P-mediated effects, and be very similar with S1P1 selective agents and non-subtype selective S1P modulators.
We chose to focus this Perspective on JTE-013 and BML-241 because they are commonly used despite reports of their lack of specificity. We also discuss VPC23019, for which evidence of S1P3 receptor antagonism activity seems weak. JTE-013 was developed by Central Pharmaceutical Research Institute, Japan Tobacco Inc. Its patent (Patent WO 01/98301; December 27, 2001) stated that JTE-013 inhibited the specific binding of radiolabeled S1P to membranes of CHO cells transfected with human and rat S1P2 receptors, with IC50 values of 17 ± 6 and 22 ± 9 nM, and did not affect S1P binding to S1P3 and S1P1, at concentrations up to 10 μM (Osada et al., 2002; Ohmori et al., 2003). Based on these data, JTE-013 has been considered since then a specific S1P2 receptor ligand and, following further experimental data, a S1P2 antagonist. Because JTE-013 had been used to characterize the S1P receptor mediating canine coronary artery contraction (Ohmori et al., 2003), we used it in rodent isolated vessels to assess the role of S1P2 receptors in S1P-induced vasoconstriction of basilar arteries (Salomone et al., 2008). We knew that S1P-induced vasoconstriction was abolished in 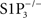 mice and were therefore surprised to see that JTE-013 inhibited vasoconstriction to S1P in arteries from wild type mice, because this suggested that S1P2 instead of, or in addition to, S1P3 was mediating S1P effect. Investigating further, however, we realized that JTE-013 inhibited vasoconstriction not only to S1P, but also to the prostanoid analog U46619, endothelin-1 and high KCl (Salomone et al., 2008; of note, vasoconstriction induced by high KCl is not receptor-mediated but related to L-type Ca2+ channels). We then performed a critical genetic control experiment, and found that JTE-013 inhibited S1P-induced vasoconstriction in
mice and were therefore surprised to see that JTE-013 inhibited vasoconstriction to S1P in arteries from wild type mice, because this suggested that S1P2 instead of, or in addition to, S1P3 was mediating S1P effect. Investigating further, however, we realized that JTE-013 inhibited vasoconstriction not only to S1P, but also to the prostanoid analog U46619, endothelin-1 and high KCl (Salomone et al., 2008; of note, vasoconstriction induced by high KCl is not receptor-mediated but related to L-type Ca2+ channels). We then performed a critical genetic control experiment, and found that JTE-013 inhibited S1P-induced vasoconstriction in 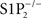 mice, demonstrating that this effect was not related to S1P2 receptors. Yet, JTE-013 is still widely used to characterize S1P2-dependent effects (Table 1). Of note, most studies that did validate S1P2-blocking effects of JTE-013 used concentrations equal to or lower than 1 μM, while we found evidence of non-specificity at 10 μM. It is therefore possible that JTE-013 retains sufficient selectivity and remains a useful antagonist in the submicromolar range, but S1P2 receptor involvement should be validated by other means.
mice, demonstrating that this effect was not related to S1P2 receptors. Yet, JTE-013 is still widely used to characterize S1P2-dependent effects (Table 1). Of note, most studies that did validate S1P2-blocking effects of JTE-013 used concentrations equal to or lower than 1 μM, while we found evidence of non-specificity at 10 μM. It is therefore possible that JTE-013 retains sufficient selectivity and remains a useful antagonist in the submicromolar range, but S1P2 receptor involvement should be validated by other means.
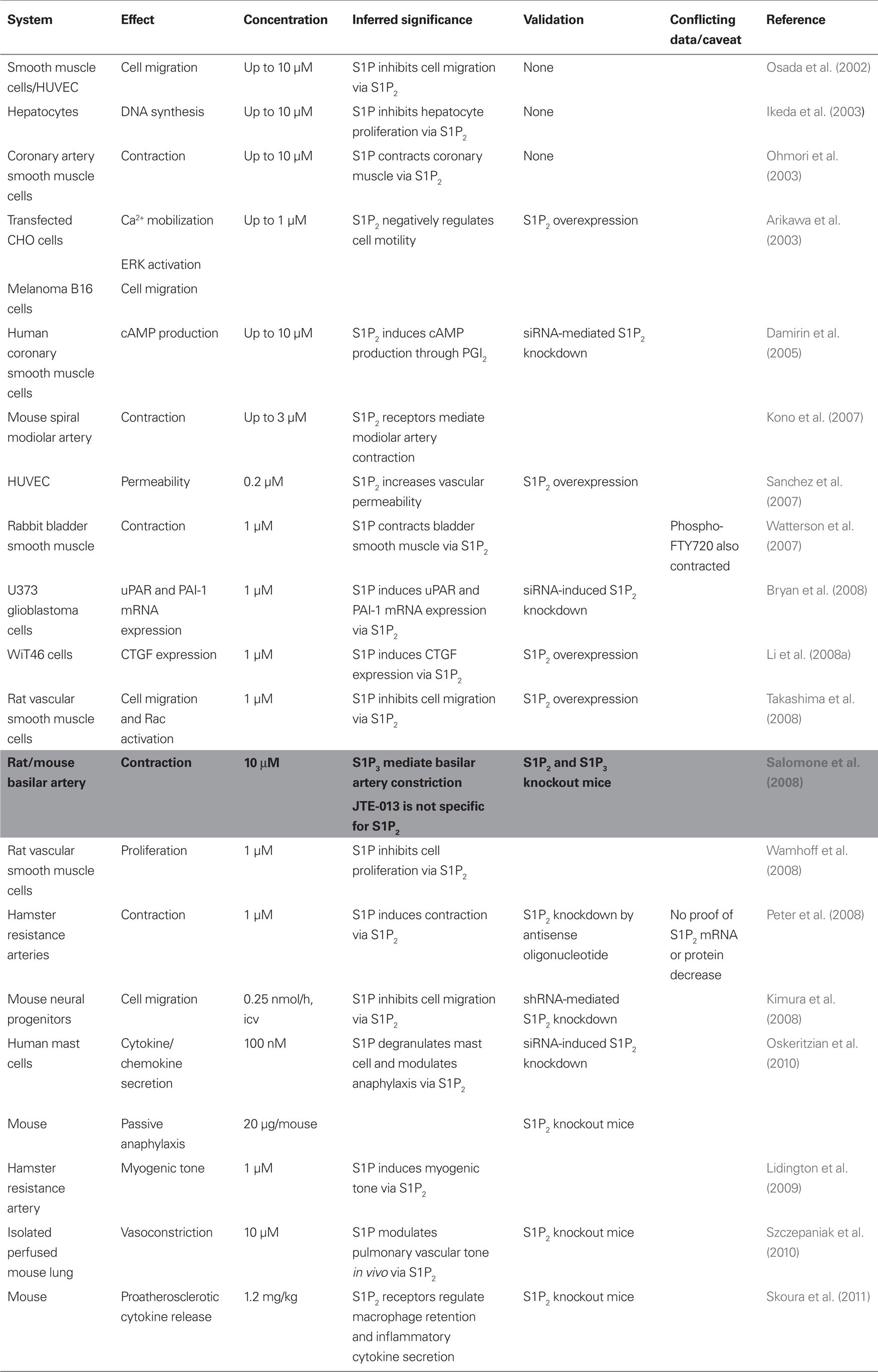
Table 1. Some published data obtained with JTE-013 (among more than 50 PubMed-indexed citations).
BML-241 was developed at the same time as JTE-013, by rational drug design using the structure of S1P to interrogate a three-dimensional database. Two novel compounds were identified that showed antagonist activity. When tested at 10 μM, “Compound 2” inhibited by 37% S1P-induced increases in [Ca2+]i in HeLa cells expressing S1P3 receptors and by about 7% [Ca2+]i increases in cells expressing S1P1 receptors (Koide et al., 2002). Strikingly, this study was based on measurements with a single BML-241 concentration and the comparison of only two potential targets (S1P1 and S1P3 receptors) in one assay; furthermore, an inhibition by less than 40% by a 10-μM antagonist concentration in cell culture might be considered less than impressive. Nevertheless, at least 20 studies (Table 2) were published using BML-241 (also known as CAY10444), most of them after the publication of an article showing that BML-241 inhibits [Ca2+]i increases via purinergic P2 receptor or α1A-adrenoceptor stimulation and α1A-adrenoceptor-mediated contraction, while not affecting the S1P3-mediated decrease of forskolin-induced cAMP accumulation (Jongsma et al., 2006). In a third of these studies, BML-241/CAY10444, tested at 1 or 10 μM, had no effect, leading the authors to conclude on a lack of involvement of S1P3 receptors in their paradigm. It is however possible that the BML-241/CAY10444 concentration used in these studies was too low to block S1P3 receptors to a significant extent; indeed, using a β-arrestin recruitment assay, Wetter et al. (2009) showed that their S1P3 cell line response to S1P was inhibited to 78% of the receptor response by 100 μM BML-241/CAY10444, with an IC50 of 4.6 μM. A less likely, but possible explanation for negative studies with BML-241 is that a non-specific action of the drug may have counteracted the partial blockade of S1P3 receptors. In light of the low affinity of BML-241/CAY10444 for S1P3 receptors, it does not appear that the few studies that documented an effect of the antagonist at 1 or 5 μM were justified in implicating S1P3 receptors. Two studies that used higher BML-241/CAY10444 concentrations (50 and 100 μM) did observe an antagonist effect. In one of these studies, the S1P1 agonist SEW2871 had no effect, while the S1P1/S1P3 receptor antagonist VPC23019 reproduced the effect of BML-241/CAY10444 (Lichte et al., 2008). In the other study, B-cell migration was promoted by a mixed S1P1/S1P3 receptor agonist (VPC24191), but not by the S1P1 agonist SEW2871. Furthermore, B-cells from 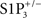 mice migrated to S1P, whereas cells from S1P3 knockout mice were unable to migrate to S1P at all concentrations of S1P tested (Donovan et al., 2010). Interestingly, in this study B-cell migration was only slightly, and not significantly, inhibited by BML-241/CAY10444, tested at concentration as high as 100 μM.
mice migrated to S1P, whereas cells from S1P3 knockout mice were unable to migrate to S1P at all concentrations of S1P tested (Donovan et al., 2010). Interestingly, in this study B-cell migration was only slightly, and not significantly, inhibited by BML-241/CAY10444, tested at concentration as high as 100 μM.
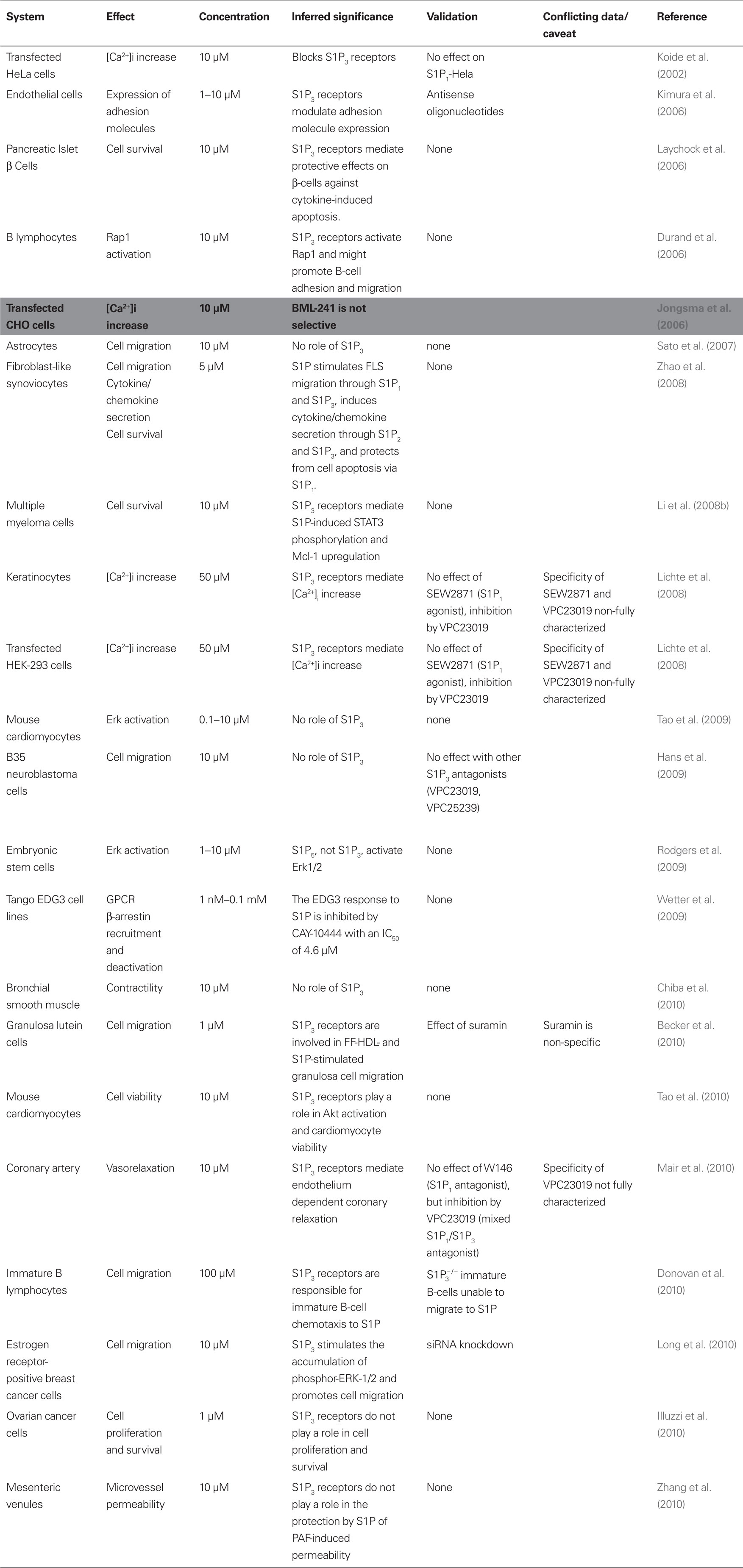
Table 2. Published data obtained with BML-241/CAY10444.
It is increasingly recognized that S1P, generated intracellularly by SPK, can be released into the extracellular space and thereby stimulate membrane S1P receptors, establishing an autocrine loop (Kim et al., 2009; Takabe et al., 2010). When this occurs, even a specific S1P receptor antagonist might inhibit the response to an agonist other than S1P. Autocrine loops might account for the fact that JTE-013 and BML-241 inhibit responses to U46619, endothelin-1 and high KCl, and to purinergic P2 receptor or α1A-adrenoceptor stimulation, respectively. However, autocrine loops cannot explain why an agonist such as JTE-013 has any action in mice lacking S1P2 receptors. Potential S1P autocrine loops should be taken into account when screening S1P receptor antagonists in complex systems (cell or organism level), particularly when both SPK inhibitors and S1P receptor blockade (pharmacologically or by preventing receptor expression) attenuate the response (Peter et al., 2008; Schnitzer et al., 2009; Salomone et al., 2010).
VPC23019 was initially described as an S1P1/3 receptor antagonist, with pKB values of 7.5 and 6.0 for the S1P1 and S1P3 receptors, respectively (Davis et al., 2005). Since then it has been mainly used for characterizing S1P1-receptor mediated responses. In a few studies, however, it has been also used as an S1P3 receptor antagonist. For example, as mentioned above, Lichte et al. (2008), reported that S1P-induced calcium signaling in human keratinocytes is mainly mediated by S1P3, because it can be blocked by the putative S1P3 antagonists BML-241, and VPC23019. Recently, Jongsma et al. (2009), using three different assays, have shown that several compounds of the VPC series, including VPC23019, behave as full or partial agonists at S1P3 receptors. Although obtained in vitro, in a system expressing high S1P3 receptor density, these data suggest that VPC23019 is a less than ideal tool to characterize S1P3-mediated responses. When studying the effects of S1P on vascular tone, we found that VPC23019 potentiated S1P-induced contractile response in both rat and mouse basilar arteries with intact endothelium, while it failed to do so in preparations without endothelium (Salomone et al., 2008). We interpreted this finding as a result of S1P1 antagonism exerted by VPC23019, because S1P1 receptors located on vascular endothelium are known to stimulate nitric oxide release and induce vasodilatation (and may therefore counteract vasoconstriction induced by S1P through S1P3 receptors). However, we were surprised not to observe inhibition of S1P-induced vasoconstriction by VPC23019, at concentrations as high as 10 μM, when solid evidence (S1P3 knockout mice) indicated that S1P-induced vasoconstriction was mediated by S1P3 receptors. Indeed, Murakami et al. (2010) showed that the novel potent and selective S1P3 antagonist TY-52156 restores coronary blood flow reduced by S1P, but VPC23019 was inactive in this system. Interestingly, the same study also showed that in isolated coronary smooth muscle cells, TY-52156 inhibited both Rho activation and Ca2+ signal, whereas VPC23019 only inhibited Ca2+ signal. Taken together with our data, this study suggests that results obtained with VPC23019 should be interpreted with caution.
While we are emphasizing in this Perspective issues related to three particular receptor antagonists, our purpose is to make a broader methodological point on the distinction between selectivity and specificity. The term “selectivity” should refer to the ability of a drug to discriminate between related targets (e.g., receptors or enzymes), showing a higher binding affinity for one subtype or isoform. Selectivity should be assessed by screening in pure systems (e.g., cell lines transfected with one receptor subtype at a time) and eventually in complex systems, including in vivo wild type and genetically altered models. The term “specificity” should refer not only to the ability of a drug to identify a receptor of interest, but also to its potential for discriminating between negative and positive interactions; i.e., the drug should bind the receptor with appropriate affinity, the drug should have low/no cross-reactivity with other receptors. In order for “specificity” to be assessed, the drug should therefore be screened for its interaction with the target of interest and for its interaction with as many unrelated biological targets as possible. For receptors, ligand binding studies may be considered adequate to quantitatively assess affinity for the binding sites under examination, but require further assessment of function, in the presence and absence of the reference agonist, to categorize a given ligand as agonist or antagonist.
Until the end of 1980s, pharmacological studies were mainly based on the use of agonists and antagonists (Salomone, 2010). More recently, investigators have also manipulated the expression levels of receptors using molecular techniques and genetic alterations. This approach represents, under critical evaluation, the most specific pharmacologic strategy available today. When using a drug as an agonist or antagonist, one has to consider the probability of off-target effects; instead, when using a receptor knock out animal, one can exclude, with great degree of confidence, that an effect is mediated by the receptor product of the deleted gene. We therefore believe that such a molecular genetic control, whenever available, should be considered the best validation in analytical and experimental pharmacology. Unfortunately, investigational pharmacological agents, agonists or antagonists, are increasingly being used to come to pharmacologic conclusions on receptor function, even though data on their selectivity is seldom sufficient and little information on their specificity is available. As this Perspective demonstrates, these conclusions are sometimes drawn despite conflicting evidence (see shaded rows in Tables 1 and 2), such as that coming from gene-deletion studies.
In conclusion, experimental ligands not used in humans (for which extensive pre-clinical characterization has uncovered potential off-target actions) and for which only limited information from simple systems (in vitro, transfected cell lines) is available should be used with caution and pharmacological data obtained with them should be considered as weak evidence, unless supported by consistent stronger evidence (for example genetic knockdown or knockout data, or concurrent pharmacological evidence obtained with other chemically unrelated ligands). Furthermore, negative and/or conflicting data obtained with experimental ligands should be considered and quoted; in other words, negative and/or conflicting data should be used for retrospective analysis and to interpret data already published with these compounds. Finally, potential off-target effects, including non-receptor-mediated effects, should always be considered and might sometimes be suspected by looking at the reversibility (many receptor-mediated effects are reversible, toxic non-specific effects are often irreversible), concentration–response or dose–response relationship, or at the kinetics of the response (some effects are too slow or too fast to be compatible with a given receptor function).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ancellin, N., and Hla, T. (1999). Differential pharmacological properties and signal transduction of the sphingosine 1-phosphate receptors EDG-1, EDG-3, and EDG-5. J. Biol. Chem. 274, 18997–19002.
Arikawa, K., Takuwa, N., Yamaguchi, H., Sugimoto, N., Kitayama, J., Nagawa, H., Takehara, K., and Takuwa, Y. (2003). Ligand-dependent inhibition of B16 melanoma cell migration and invasion via endogenous S1P2 G protein-coupled receptor. Requirement of inhibition of cellular RAC activity. J. Biol. Chem. 278, 32841–32851.
Bandhuvula, P., Tam, Y. Y., Oskouian, B., and Saba, J. D. (2005). The immune modulator FTY720 inhibits sphingosine-1-phosphate lyase activity. J. Biol. Chem. 280, 33697–33700.
Becker, S., von Otte, S., Robenek, H., Diedrich, K., and Nofer, J. R. (2010). Follicular fluid high-density lipoprotein (FF-HDL)-associated sphingosine 1-phosphate (S1P) promotes human granulosa lutein cell migration via S1P receptor type 3 (S1PR3) and small G protein RAC1. Biol. Reprod. doi: 10.1095/biolreprod.110.084152
Blom, T., Back, N., Mutka, A. L., Bittman, R., Li, Z., de Lera, A., Kovanen, P. T., Diczfalusy, U., and Ikonen, E. (2010). FTY720 stimulates 27-hydroxycholesterol production and confers atheroprotective effects in human primary macrophages. Circ. Res. 106, 720–729.
Bryan, L., Paugh, B. S., Kapitonov, D., Wilczynska, K. M., Alvarez, S. M., Singh, S. K., Milstien, S., Spiegel, S., and Kordula, T. (2008). Sphingosine-1-phosphate and interleukin-1 independently regulate plasminogen activator inhibitor-1 and urokinase-type plasminogen activator receptor expression in glioblastoma cells: implications for invasiveness. Mol. Cancer Res. 6, 1469–1477.
Chiba, Y., Suzuki, K., Uechi, M., Kurihara, E., Goto, K., Sakai, H., and Misawa, M. (2010). Downregulation of sphingosine-1-phosphate receptors in bronchial smooth muscle of mouse experimental asthma. Pharmacol. Res. 62, 357–363.
Damirin, A., Tomura, H., Komachi, M., Tobo, M., Sato, K., Mogi, C., Nochi, H., Tamoto, K., and Okajima, F. (2005). Sphingosine 1-phosphate receptors mediate the lipid-induced cAMP accumulation through cyclooxygenase-2/prostaglandin I2 pathway in human coronary artery smooth muscle cells. Mol. Pharmacol. 67, 1177–1185.
Davis, M. D., Clemens, J. J., Macdonald, T. L., and Lynch, K. R. (2005). Sphingosine 1-phosphate analogs as receptor antagonists. J. Biol. Chem. 280, 9833–9841.
Dawson, G., and Qin, J. (2011). Gilenya (FTY720) inhibits acid sphingomyelinase by a mechanism similar to tricyclic antidepressants. Biochem. Biophys. Res. Commun. 404, 321–323.
Donovan, E. E., Pelanda, R., and Torres, R. M. (2010). S1P3 confers differential S1P-induced migration by autoreactive and non-autoreactive immature B cells and is required for normal B-cell development. Eur. J. Immunol. 40, 688–698.
Durand, C. A., Westendorf, J., Tse, K. W., and Gold, M. R. (2006). The Rap GTPases mediate CXCL13- and sphingosine1-phosphate-induced chemotaxis, adhesion, and Pyk2 tyrosine phosphorylation in B lymphocytes. Eur. J. Immunol. 36, 2235–2249.
Hans, M., Pusch, A., Dai, L., Racke, K., Swandulla, D., Gieselmann, V., and Kappler, J. (2009). Lysosulfatide regulates the motility of a neural precursor cell line via calcium-mediated process collapse. Neurochem. Res. 34, 508–517.
Ikeda, H., Satoh, H., Yanase, M., Inoue, Y., Tomiya, T., Arai, M., Tejima, K., Nagashima, K., Maekawa, H., Yahagi, N., Yatomi, Y., Sakurada, S., Takuwa, Y., Ogata, I., Kimura, S., and Fujiwara, K. (2003). Antiproliferative property of sphingosine 1-phosphate in rat hepatocytes involves activation of Rho via Edg-5. Gastroenterology 124, 459–469.
Illuzzi, G., Bernacchioni, C., Aureli, M., Prioni, S., Frera, G., Donati, C., Valsecchi, M., Chigorno, V., Bruni, P., Sonnino, S., and Prinetti, A. (2010). Sphingosine kinase mediates resistance to the synthetic retinoid N-(4-hydroxyphenyl)retinamide in human ovarian cancer cells. J. Biol. Chem. 285, 18594–18602.
Im, D. S. (2010). Pharmacological tools for lysophospholipid GPCRs: development of agonists and antagonists for LPA and S1P receptors. Acta Pharmacol. Sin. 31, 1213–1222.
Jongsma, M., Hendriks-Balk, M. C., Michel, M. C., Peters, S. L., and Alewijnse, A. E. (2006). BML-241 fails to display selective antagonism at the sphingosine-1-phosphate receptor, S1P(3). Br. J. Pharmacol. 149, 277–282.
Jongsma, M., van Unen, J., van Loenen, P. B., Michel, M. C., Peters, S. L., and Alewijnse, A. E. (2009). Different response patterns of several ligands at the sphingosine-1-phosphate receptor subtype 3 (S1P3). Br. J. Pharmacol. 156, 1305–1311.
Kappos, L., Radue, E. W., O’Connor, P., Polman, C., Hohlfeld, R., Calabresi, P., Selmaj, K., Agoropoulou, C., Leyk, M., Zhang-Auberson, L., and Burtin, P. (2010). A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 362, 387–401.
Kim, R. H., Takabe, K., Milstien, S., and Spiegel, S. (2009). Export and functions of sphingosine-1-phosphate. Biochim. Biophys. Acta 1791, 692–696.
Kimura, A., Ohmori, T., Kashiwakura, Y., Ohkawa, R., Madoiwa, S., Mimuro, J., Shimazaki, K., Hoshino, Y., Yatomi, Y., and Sakata, Y. (2008). Antagonism of sphingosine 1-phosphate receptor-2 enhances migration of neural progenitor cells toward an area of brain. Stroke 39, 3411–3417.
Kimura, T., Tomura, H., Mogi, C., Kuwabara, A., Ishiwara, M., Shibasawa, K., Sato, K., Ohwada, S., Im, D. S., Kurose, H., Ishizuka, T., Murakami, M., and Okajima, F. (2006). Sphingosine 1-phosphate receptors mediate stimulatory and inhibitory signalings for expression of adhesion molecules in endothelial cells. Cell. Signal. 18, 841–850.
Koide, Y., Hasegawa, T., Takahashi, A., Endo, A., Mochizuki, N., Nakagawa, M., and Nishida, A. (2002). Development of novel EDG3 antagonists using a 3D database search and their structure-activity relationships. J. Med. Chem. 45, 4629–4638.
Kono, M., Belyantseva, I. A., Skoura, A., Frolenkov, G. I., Starost, M. F., Dreier, J. L., Lidington, D., Bolz, S. S., Friedman, T. B., Hla, T., and Proia, R. L. (2007). Deafness and stria vascularis defects in S1P2 receptor-null mice. J. Biol. Chem. 282, 10690–10696.
Lahiri, S., Park, H., Laviad, E. L., Lu, X., Bittman, R., and Futerman, A. H. (2009). Ceramide synthesis is modulated by the sphingosine analog FTY720 via a mixture of uncompetitive and noncompetitive inhibition in an Acyl-CoA chain length-dependent manner. J. Biol. Chem. 284, 16090–16098.
Laychock, S. G., Sessanna, S. M., Lin, M. H., and Mastrandrea, L. D. (2006). Sphingosine 1-phosphate affects cytokine-induced apoptosis in rat pancreatic islet beta-cells. Endocrinology 147, 4705–4712.
Li, M. H., Sanchez, T., Pappalardo, A., Lynch, K. R., Hla, T., and Ferrer, F. (2008a). Induction of antiproliferative connective tissue growth factor expression in Wilms’ tumor cells by sphingosine-1-phosphate receptor 2. Mol. Cancer Res. 6, 1649–1656.
Li, Q. F., Wu, C. T., Guo, Q., Wang, H., and Wang, L. S. (2008b). Sphingosine 1-phosphate induces Mcl-1 upregulation and protects multiple myeloma cells against apoptosis. Biochem. Biophys. Res. Commun. 371, 159–162.
Lichte, K., Rossi, R., Danneberg, K., ter Braak, M., Kurschner, U., Jakobs, K. H., Kleuser, B., and Meyer zu Heringdorf, D. (2008). Lysophospholipid receptor-mediated calcium signaling in human keratinocytes. J. Invest. Dermatol. 128, 1487–1498.
Lidington, D., Peter, B. F., Meissner, A., Kroetsch, J. T., Pitson, S. M., Pohl, U., and Bolz, S. S. (2009). The phosphorylation motif at serine 225 governs the localization and function of sphingosine kinase 1 in resistance arteries. Arterioscler. Thromb. Vasc. Biol. 29, 1916–1922.
Long, J. S., Edwards, J., Watson, C., Tovey, S., Mair, K. M., Schiff, R., Natarajan, V., Pyne, N. J., and Pyne, S. (2010). Sphingosine kinase 1 induces tolerance to human epidermal growth factor receptor 2 and prevents formation of a migratory phenotype in response to sphingosine 1-phosphate in estrogen receptor-positive breast cancer cells. Mol. Cell. Biol. 30, 3827–3841.
Mair, K. M., Robinson, E., Kane, K. A., Pyne, S., Brett, R. R., Pyne, N. J., and Kennedy, S. (2010). Interaction between anandamide and sphingosine-(1)-phosphate in mediating vasorelaxation in rat coronary artery. Br. J. Pharmacol. 161, 176–192.
Martin, M., Mory, C., Prescher, A., Wittekind, C., Fiedler, M., and Uhlmann, D. (2010). Protective effects of early CD4(+) T cell reduction in hepatic ischemia/reperfusion injury. J. Gastrointest. Surg. 14, 511–519.
Matloubian, M., Lo, C. G., Cinamon, G., Lesneski, M. J., Xu, Y., Brinkmann, V., Allende, M. L., Proia, R. L., and Cyster, J. G. (2004). Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427, 355–360.
Murakami, A., Takasugi, H., Ohnuma, S., Koide, Y., Sakurai, A., Takeda, S., Hasegawa, T., Sasamori, J., Konno, T., Hayashi, K., Watanabe, Y., Mori, K., Sato, Y., Takahashi, A., Mochizuki, N., and Takakura, N. (2010). Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: investigation based on a new S1P3 receptor antagonist. Mol. Pharmacol. 77, 704–713.
Ohmori, T., Yatomi, Y., Osada, M., Kazama, F., Takafuta, T., Ikeda, H., and Ozaki, Y. (2003). Sphingosine 1-phosphate induces contraction of coronary artery smooth muscle cells via S1P2. Cardiovasc. Res. 58, 170–177.
Osada, M., Yatomi, Y., Ohmori, T., Ikeda, H., and Ozaki, Y. (2002). Enhancement of sphingosine 1-phosphate-induced migration of vascular endothelial cells and smooth muscle cells by an EDG-5 antagonist. Biochem. Biophys. Res. Commun. 299, 483–487.
Oskeritzian, C. A., Price, M. M., Hait, N. C., Kapitonov, D., Falanga, Y. T., Morales, J. K., Ryan, J. J., Milstien, S., and Spiegel, S. (2010). Essential roles of sphingosine-1-phosphate receptor 2 in human mast cell activation, anaphylaxis, and pulmonary edema. J. Exp. Med. 207, 465–474.
Payne, S. G., Oskeritzian, C. A., Griffiths, R., Subramanian, P., Barbour, S. E., Chalfant, C. E., Milstien, S., and Spiegel, S. (2007). The immunosuppressant drug FTY720 inhibits cytosolic phospholipase A2 independently of sphingosine-1-phosphate receptors. Blood 109, 1077–1085.
Peter, B. F., Lidington, D., Harada, A., Bolz, H. J., Vogel, L., Heximer, S., Spiegel, S., Pohl, U., and Bolz, S. S. (2008). Role of sphingosine-1-phosphate phosphohydrolase 1 in the regulation of resistance artery tone. Circ. Res. 103, 315–324.
Rodgers, A., Mormeneo, D., Long, J. S., Delgado, A., Pyne, N. J., and Pyne, S. (2009). Sphingosine 1-phosphate regulation of extracellular signal-regulated kinase-1/2 in embryonic stem cells. Stem Cells Dev. 18, 1319–1330.
Salomone, S. (2010). Analytical and experimental pharmacology, challenges ahead. Front. Pharmacol. 1:119. doi: 10.3389/fphar.2010.00119
Salomone, S., Potts, E. M., Tyndall, S., Ip, P. C., Chun, J., Brinkmann, V., and Waeber, C. (2008). Analysis of sphingosine 1-phosphate receptors involved in constriction of isolated cerebral arteries with receptor null mice and pharmacological tools. Br. J. Pharmacol. 153, 140–147.
Salomone, S., Soydan, G., Ip, P. C., Hopson, K. M., and Waeber, C. (2010). Vessel-specific role of sphingosine kinase 1 in the vasoconstriction of isolated basilar arteries. Pharmacol. Res. 62, 465–474.
Salomone, S., Yoshimura, S., Reuter, U., Foley, M., Thomas, S. S., Moskowitz, M. A., and Waeber, C. (2003). S1P(3) receptors mediate the potent constriction of cerebral arteries by sphingosine-1-phosphate. Eur. J. Pharmacol. 469, 125–134.
Sanchez, T., Skoura, A., Wu, M. T., Casserly, B., Harrington, E. O., and Hla, T. (2007). Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler. Thromb. Vasc. Biol. 27, 1312–1318.
Sato, K., Malchinkhuu, E., Horiuchi, Y., Mogi, C., Tomura, H., Tosaka, M., Yoshimoto, Y., Kuwabara, A., and Okajima, F. (2007). HDL-like lipoproteins in cerebrospinal fluid affect neural cell activity through lipoprotein-associated sphingosine 1-phosphate. Biochem. Biophys. Res. Commun. 359, 649–654.
Schnitzer, S. E., Weigert, A., Zhou, J., and Brune, B. (2009). Hypoxia enhances sphingosine kinase 2 activity and provokes sphingosine-1-phosphate-mediated chemoresistance in A549 lung cancer cells. Mol. Cancer Res. 7, 393–401.
Skoura, A., Michaud, J., Im, D. S., Thangada, S., Xiong, Y., Smith, J. D., and Hla, T. (2011). Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 31, 81–85.
Szczepaniak, W. S., Pitt, B. R., and McVerry, B. J. (2010). S1P2 receptor-dependent Rho-kinase activation mediates vasoconstriction in the murine pulmonary circulation induced by sphingosine 1-phosphate. Am. J. Physiol. Lung Cell Mol. Physiol. 299, L137–L145.
Takabe, K., Kim, R. H., Allegood, J. C., Mitra, P., Ramachandran, S., Nagahashi, M., Harikumar, K. B., Hait, N. C., Milstien, S., and Spiegel, S. (2010). Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J. Biol. Chem. 285, 10477–10486.
Takashima, S., Sugimoto, N., Takuwa, N., Okamoto, Y., Yoshioka, K., Takamura, M., Takata, S., Kaneko, S., and Takuwa, Y. (2008). G12/13 and Gq mediate S1P2-induced inhibition of Rac and migration in vascular smooth muscle in a manner dependent on Rho but not Rho kinase. Cardiovasc. Res. 79, 689–697.
Tao, R., Hoover, H. E., Honbo, N., Kalinowski, M., Alano, C. C., Karliner, J. S., and Raffai, R. (2010). High-density lipoprotein determines adult mouse cardiomyocyte fate after hypoxia-reoxygenation through lipoprotein-associated sphingosine 1-phosphate. Am. J. Physiol. Heart Circ. Physiol. 298, H1022–H1028.
Tao, R., Hoover, H. E., Zhang, J., Honbo, N., Alano, C. C., and Karliner, J. S. (2009). Cardiomyocyte S1P1 receptor-mediated extracellular signal-related kinase signaling and desensitization. J. Cardiovasc. Pharmacol. 53, 486–494.
Tonelli, F., Lim, K. G., Loveridge, C., Long, J., Pitson, S. M., Tigyi, G., Bittman, R., Pyne, S., and Pyne, N. J. (2010). FTY720 and (S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1 and promote its proteasomal degradation in human pulmonary artery smooth muscle, breast cancer and androgen-independent prostate cancer cells. Cell. Signal. 22, 1536–1542.
Vessey, D. A., Kelley, M., Zhang, J., Li, L., Tao, R., and Karliner, J. S. (2007). Dimethylsphingosine and FTY720 inhibit the SK1 form but activate the SK2 form of sphingosine kinase from rat heart. J. Biochem. Mol. Toxicol. 21, 273–279.
Voogd, T. E., Vansterkenburg, E. L., Wilting, J., and Janssen, L. H. (1993). Recent research on the biological activity of suramin. Pharmacol. Rev. 45, 177–203.
Wamhoff, B. R., Lynch, K. R., Macdonald, T. L., and Owens, G. K. (2008). Sphingosine-1-phosphate receptor subtypes differentially regulate smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 28, 1454–1461.
Watterson, K. R., Berg, K. M., Kapitonov, D., Payne, S. G., Miner, A. S., Bittman, R., Milstien, S., Ratz, P. H., and Spiegel, S. (2007). Sphingosine-1-phosphate and the immunosuppressant, FTY720-phosphate, regulate detrusor muscle tone. FASEB J. 21, 2818–2828.
Wetter, J. A., Revankar, C., and Hanson, B. J. (2009). Utilization of the Tango beta-arrestin recruitment technology for cell-based EDG receptor assay development and interrogation. J. Biomol. Screen 14, 1134–1141.
Woodcock, J. M., Ma, Y., Coolen, C., Pham, D., Jones, C., Lopez, A. F., and Pitson, S. M. (2010). Sphingosine and FTY720 directly bind pro-survival 14-3-3 proteins to regulate their function. Cell. Signal. 22, 1291–1299.
Zhang, G., Xu, S., Qian, Y., and He, P. (2010). Sphingosine-1-phosphate prevents permeability increases via activation of endothelial sphingosine-1-phosphate receptor 1 in rat venules. Am. J. Physiol. Heart Circ. Physiol. 299, H1494–H1504.
Keywords: JTE-013, BML-241, CAY10444, VPC23019, antagonist
Citation: Salomone S and Waeber C (2011) Selectivity and specificity of sphingosine-1-phosphate receptor ligands: caveats and critical thinking in characterizing receptor-mediated effects. Front. Pharmacol. 2:9. doi: 10.3389/fphar.2011.00009
Received: 17 December 2010;
Accepted: 09 February 2011;
Published online: 22 February 2011.
Edited by:
Martin C. Michel, Academic Medical Center, NetherlandsReviewed by:
Dagmar Meyer Zu Heringdorf, Goethe University, GermanyCopyright: © 2011 Salomone and Waeber. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Christian Waeber, Massachusetts General Hospital, CNY149 Room 6403, 149 13th Street, Charlestown, MA 02129, USA. e-mail:d2FlYmVyQGhlbGl4Lm1naC5oYXJ2YXJkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.