



- 1 Novamass Ltd, Medipolis Center, Oulu, Finland
- 2 Department of Chemistry, University of Oulu, Oulu, Finland
- 3 Department of Pharmacology and Toxicology, Institute of Biomedicine, University of Oulu, Oulu, Finland
Liquid chromatography–mass spectrometry (LC–MS) with generic gradient elution for a large number of chemically different compounds is a common approach in drug development, used to acquire a large amount of data in a short time frame for drug candidates. The analysis with non-optimized parameters however may lead to a poor method performance for many compounds, and contains a risk of losing important information. Here, generic electrospray time of flight (ESI-TOF) MS methods in various pH conditions were tested for 55 chemically diverse compounds (10 acids, 25 bases, 17 neutrals, and 3 amphoterics), aiming to find best analytical conditions for each compound, for studies of in vitro metabolic properties in liver preparations. The effect of eluent pH and elution gradient strength on chromatographic performance and electrospray MS ionization efficiency were examined for each compound. The data are evaluated how well the best generic approach could cover the analysis of test compounds and how many compounds would still need completely different analytical conditions after that. Aqueous mobile phase consisting of 0.05% acetic acid and 5 mM ammonium acetate (pH 4.4) showed the best general suitability for the analyses, showing adequate performance for metabolite profiling for 41 out of 55 compounds either in positive or negative ion mode. In positive ion mode, the main limitation of performance in various pH conditions was generally not the lack of ionization, but rather the poor chromatographic performance (inadequate retention or poor peak shape), suggesting that more emphasis should be put in finding conditions providing best chromatographic performance, rather than highest ionization properties. However, a single generic approach for a large number of different compounds is not likely to produce good results for all compounds. Preferably, at least two or three different conditions are needed for the coverage of a larger number of structurally diverse compounds.
Introduction
Liquid chromatography–mass spectrometry (LC–MS) techniques employed in drug metabolism studies of early drug discovery very often utilize the so called generic gradient elution, i.e. only structure group-optimized or even totally non-optimized eluent pH and gradient strength are used for all analytes (Lee and Kerns, 1999; Cheng et al., 2001; Hop et al., 2002; Kostiainen et al., 2003; Castro-Perez et al., 2005: Kamel and Prakash, 2006; Pedraglio et al., 2007). This provides high throughput and fast turn-around time, and is usually considered as the approach giving the highest amount of data with the lowest time consumption. The use of non-optimized test conditions however may lead to sacrificed data quality for many compounds, producing biased results, often in a form of false negatives in metabolite screening.
Despite their common use in industry, performance and data quality obtained with such methods are however rarely studied, at least in format of published data with a high number of compounds with diverse chemical nature. Also not much attention is paid to what kind of eluent conditions in case of LC–MS analysis would lead to highest general applicability, even though it is very well known that with LC–ESI–MS acidic compounds are usually negatively ionized (deprotonated) in basic conditions and basic compounds are on the contrary positively ionized (protonated) in acidic conditions (Kostiainen and Kauppila, 2009). The electrospray ionization process in LC–MS is however a very complex phenomenon having multiple affecting factors, such as volatility, viscosity, surface tension, conductivity, ion strength, and pH of the used solvents and additives in LC mobile phase, and depending on the properties of analyte compounds and mobile phase constituents, sometimes better ionization may occur in the conditions clearly against these rules (Kostiainen and Kauppila, 2009). In addition, the chromatographically co-eluting matrix components may suppress or enhance the ionization process (Mallet et al., 2004; Cappiello et al., 2008). Therefore, mobile phase constituents have very high and sometimes unpredictable effect to the ESI response of the analytes, and solvents and additives used must be carefully chosen and tested to obtain high-quality data from analyses.
Chromatographically valid data of gradient pH conditions considering the generic elution gradients with a very high number of compounds is described in the literature by Law (2004), resulting in a conclusion that gradient elution at neutral pH conditions is highly successful for most compounds, as long as logD values of the analytes are > –2. If the pH is increased above 8, the chromatographic performance for acidic compounds is weakened, whereas the pH below 4 decreases the performance with basic compounds. This study did not examine the effect of pH from the mass spectrometric point of view. Despite the chromatographic performance, most of the generic gradient conditions for drugs or drug-like compounds are at pH conditions 3–4 or even below, as in most cases with LC–MS utilizing electrospray ionization (ESI) the protonation (and thus detection sensitivity) is favored at this pH, especially with basic compounds.
Some papers focusing on development of generic gradients for LC–MS have been published, describing however the use of only minor groups of compounds (2–13 compounds), and thus not showing the real applicability of the methods for compounds of higher chemical diversity (Ayrton et al., 1998; Chu et al., 2002; Seto et al., 2002; Mensch et al., 2007). Ayrton et al. (1998) described the development of generic HPLC–UV gradient elution conditions for 11 compounds, but tested the method only for two compounds with mass spectrometric detection. Seto et al. (2002) in turn described a generic gradient LC–MS method with ion pairing reagents, considering the use of various mobile phase additives, but focusing on analysis of only four acidic compounds. Chu et al. (2002) built up a generic LC–MS–MS method for in vitro blood–brain barrier test assay; showing its applicability with 28 new chemical entities (NCE). They however applied both positive and negative mode ionization polarities with both ESI and APCI ionization modes; and three different chromatographic columns for compounds with different retention behavior, so that the methods used in the end were really not so “generic”. Also Mensch et al. (2007) described a generic UPLC–MS–MS method for analysis of NCEs in drug discovery phase permeability studies, but only six model compounds were used in evaluating the performance of the method. The effect of eluent pH on chromatographic and mass spectrometric performance in generic gradient elution was most comprehensively studied by Delatour, who concluded that with their 11 basic drugs the basic pH provided clearly better chromatography in comparison to acidic conditions, without significant decrease in positive ion mode ESI–MS detection (Delatour and Leclercq, 2005).
Our actual goal was to develop LC–ESI–MS methods to be used in in vitro metabolism studies of 55 chemically diverse compounds in rat and human liver microsome and homogenate incubations, monitoring both substrate depletion and metabolite formation at the same time. The compounds were selected from the list of validation substances of European Centre for the Validation of Alternative Methods (ECVAM), and they have been used to study the comparison of metabolic properties in rat and human liver homogenate and microsomal incubations in vitro. The ECVAM list of validation compounds is not based on chemical classes (acids, bases, neutrals, amphoterics) but rather on the different toxicities of the compounds. For this reason the final set contained a relatively high number of chemically neutral compounds, so that this class of compounds was present in a clearly higher proportion than neutral compounds in drug discovery in general (Law, 2004). However, as the multiresidual techniques are nowadays of high importance in several analytical fields, such as metabolism, environmental and forensic sciences, or plant and human metabolomics, the search of compromise between optimal conditions for chromatographic separation, and detection is of great interest also in the application areas other than drug metabolism studies.
The methods for each compound were developed with one generic method as a starting point, and testing the effect of eluent pH on both chromatographic performance and mass spectrometric ionization efficiency (detection sensitivity). The data acquired during this stage was combined and analyzed here for assessing whether certain conditions would show the best general applicability in this type of studies, and thus evaluating the performance of the most typical gradient elution conditions for LC–ESI–MS used in drug discovery studies. It is worth stressing that our aim was not to develop one set of LC–MS conditions that would enable analysis of samples from all 55 test compounds; therefore the pH and the ion strengths of the LC effluents were not optimized more than by using six of the most typically used LC–MS aqueous mobile phases.
Materials and Methods
Chemicals
All chemicals and solvents were HPLC grade and were purchased from VWR International Ltd (Espoo, Finland). LichroSolv acetonitrile was from Merck KGaA (Darmstadt, Germany), while formic acid, acetic acid, ammonium formate, ammonium acetate, and ammonia were from BDH Laboratories Ltd (Poole, England). Laboratory water was prepared with Direct-Q (Millipore S.A., Molsheim, France) water purification system and was UP grade (ultrapure, 18.2 MΩ).
Sample Preparation
The test samples were prepared to provide similar injection conditions with real incubation samples. Thus, samples were prepared to 0.5 μM concentration in 50% acetonitrile in 150 mM phosphate buffer, so that this corresponds to 1 μM incubation (4% of the initial substrate concentration of 25 μM to be used in the following metabolism studies) where enzymatic reactions are quenched with an equal volume of acetonitrile.
Liquid Chromatography Mass Spectrometry
A Waters Alliance 2695 high performance liquid chromatographic system (Waters Corp., Milford, MA, USA) with an autosampler, a column oven, and a vacuum degasser was used with Waters XBridge Shield RP18 column and Phenomenex Luna C18 (2 × 4 mm) precolumn. In some cases also Waters XBridge C18 or Phenomenex Luna-C18 columns were used if poor peak shape was observed with the first tested column. All columns used had 2.0–2.1 × 50 mm dimensions and 3–3.5 μm particle sizes. The column selection was based on stationary phases able to cover wide pH range (XBridge C18 pH 1–12, XBridgeShieldRP pH 2–11, Luna 1.5–10) and authors’ good experiences about the columns over several years in various chromatographic conditions. Temperature of column oven was 30°C and injection volumes used were 10 μl. The data was acquired with a Micromass LCT time of flight (TOF) mass spectrometer (Micromass Ltd., Manchester, England) equipped with a LockSpray electrospray ionization source, used both in positive and negative ion polarity. Cone voltages between 16 and 28 V were used (roughly optimized for each compound). Capillary voltages of 3.5 and 2.8 kV were used at positive and negative ion mode, respectively. Nitrogen was used as a nebulizer and drying gas with flow rates of 100 and 800 l/h, respectively. The source and desolvation temperatures used were 150 and 350°C, respectively. Leucine encephalin ([M+H]+ m/z 556.2771) and raffinose ([M–H]– m/z 503.1612) were used as lock mass compounds in accurate mass measurement with positive and negative ion modes, respectively, and were delivered into LockSpray probe with syringe pump to obtain about 160 counts/s. The mass spectrometer and HPLC system were operated under MassLynx 4.0 software.
Chromatographic Conditions and Method Development
In method development phase the study compounds were divided to batches of 3–10 compounds, based on their chemical structures or acid/base properties, and LC–MS conditions for each compound were briefly optimized by acquiring test data for all compounds in a single batch at the same time. As a first run conditions a gradient elution with 2%–2%–60%–90%–90% acetonitrile in 0–1–3–5–6 min was applied for each compound (in batches of 3–10 compounds), followed by column equilibration. After this, few runs for optimizing gradient strength (slope of the organic phase proportion) were acquired, followed by runs with various aqueous phase pH conditions and different MS ionization polarities. In all cases, gradient elution was used (no isocratic runs). Acetonitrile was chosen as organic eluent phase over methanol, as it usually provides better chromatographic peak shapes. The eluent flow rate used was 0.4 ml/min. The aqueous eluent phases used were 0.1% formic acid (FA, pH 2.8), 0.1% acetic acid (AA, pH 3.2), 0.05% acetic acid +5 mM ammonium acetate (pH 4.4), 2 mM ammonium formate (NH4OF, pH 6.0), 2 mM ammonium acetate (NH4OAc, pH 6.7), and 10 mM ammonia (NH4OH, pH 9.8). After this the acquired data was used to extract the optimum LC–MS conditions for each single compound; and these methods were used later in in vitro metabolism studies (data to be reported elsewhere). For compounds with poor peak shape, also the effect of different columns (see above) were tested for improving chromatographic peak shapes.
Results and Discussion
Suitability of Generic Gradient Conditions for the Test Set Compounds
The suitability of the tested HPLC eluent conditions for each 55 test compounds are shown in Table 1. Mass spectrometric ionization efficiencies were rated for each compound with symbols to mark detection sensitivity (a) high enough for monitoring formed metabolites “++”, (b) good enough to monitor substrate depletion but not the formed metabolites “+”, or (c) poor sensitivity being not enough even to monitor substrate depletion “−”. The sensitivity limit for monitoring metabolites (metabolite profiling) was considered as signal-to-noise ratio >10 from 1% concentration of the actual initial incubation concentration (25 μM), whereas the limit for monitoring substrate depletion was considered as a detected signal (S/N > 3) from test sample having 4% concentration of the actual initial incubation concentration. When chromatographic problems were observed, in form of poor peak shape or inadequate retention behavior (to separate parent and metabolites) preventing the use of certain eluent conditions for metabolite profiling despite of good ionization, the abovementioned symbol was marked within parentheses “(++)”. Poor peak shape was specified as asymmetric factor A/B < 0.4 (tailing peak) or A/B > 2 (fronting peak), where A = peak width before the peak top and B = peak width after the peak top (both measured from 5% peak height). As the chromatographic separation or peak shape are not so crucial in analysis of metabolic stability, this chromatographic criteria was not applied to “+” − symbols. The criterion for adequate retention was set so that normalized retention time (retention time minus dead time) should be at least ten times the peak width (measured from 5% peak height). In addition, the number of each symbol for each HPLC conditions in Table 1 is summarized to Table 2. In addition, the best conditions for each compound were marked with a “*” symbol.
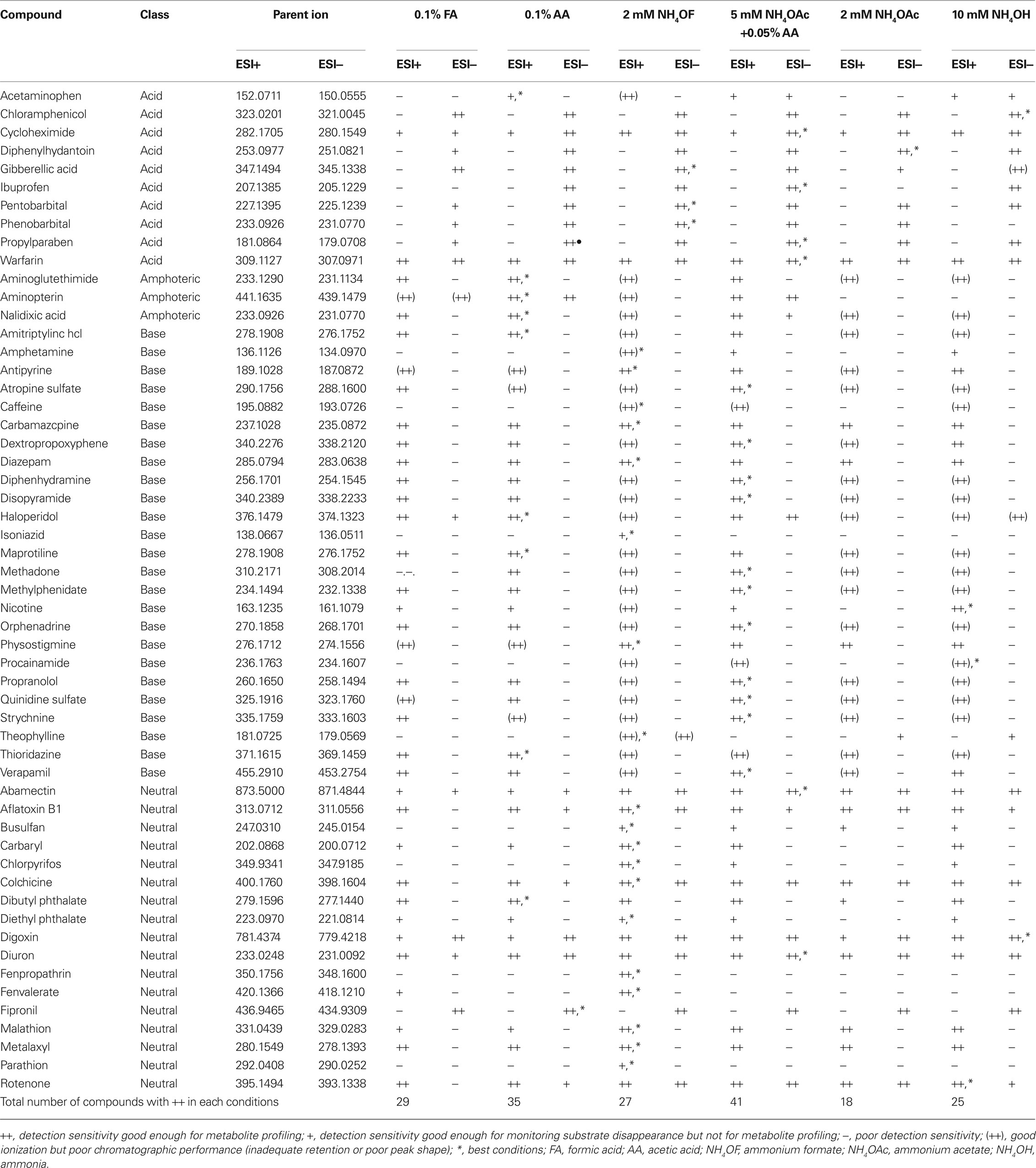
Table 1. Performance obtained for the 55 test compounds in the tested LC–ESI–TOF–MS mobile phase conditions.
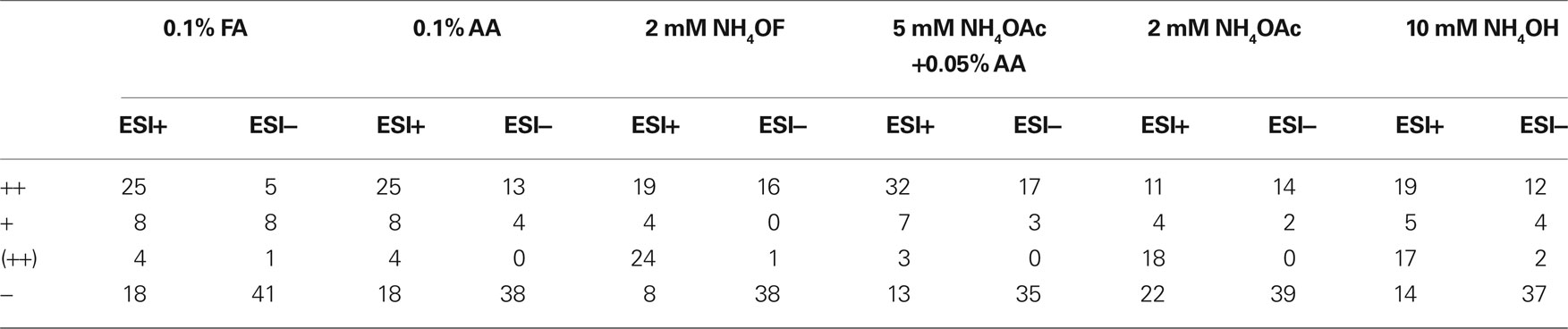
Table 2. Number of different symbols in Table 1 in the tested LC–ESI–TOF–MS mobile phase conditions.
As already mentioned above, the test compound set was not selected primarily for the analytical considerations discussed in the present study, but rather focused to obtain a suitable test set for metabolism studies. For this reason the number of neutral compounds is relatively high with respect to other classes, whereas only few amphoteric compounds are included. Even if the 55 compounds offer structurally quite a diverse selection of compounds, the number is still not very high, as some metabolomics studies may describe methods monitoring several hundreds of compounds at the same time. However, metabolomics studies do not provide similar approach for the assessment of analytical conditions as followed here.
Generally, and not surprisingly, the results showed clearly that none of the six eluent pH conditions was able to provide adequate analytical conditions to cover successful metabolite profiling, or even monitoring the substrate depletion, for all the 55 test compounds. However, suitable conditions for monitoring at least the substrate disappearance were found for all of the 55 test compounds. As expected, the positive ion mode electrospray ionization covered a higher number of test compounds in all conditions, even when working with ammonia in clearly basic conditions at pH 9.8. However, this latter result is somewhat biased by a high number of basic compounds in test group (bases 25/55, acids 10/55). The results are collected to Table 1. It is obvious that negative ion mode was superior in ionization efficiency with acidic compounds in all tested pH conditions, whereas the basic compounds were self-evidently ionized better with positive ion mode. The neutral compounds were generally ionized with positive ion mode, and suitability of negative ion mode increased with increasing pH.
Of the six tested eluent conditions, the one with 0.05% acetic acid and 5 mM ammonium acetate (pH 4.4) showed the highest suitability for the test compounds, as 41 of the 55 compounds showed LC–MS performance filling the criteria set as adequate for metabolite profiling, either with positive or negative ion mode, when using this eluent. More specifically, 31 of the test compounds worked well in positive ion mode and 17 in negative ion mode (Table 2). The actual strength of these conditions seemed to be a good chromatographic performance for most of the compounds, as only three compounds showed inadequate chromatographic retention behavior or poor peak shape. However, 13 of the test compounds showed completely inadequate performance even for monitoring substrate depletion in these conditions due to poor detection sensitivity. Also, even though the mobile phase with 0.05% acetic acid and 5 mM ammonium acetate showed the best general suitability for the test compounds, it was the best aqueous mobile phase only for 17 of the compounds. This again shows that even though this mobile phase is generally highly useful, it still leads to a less than-optimal performance for most of the compounds.
The second highest suitability was obtained with 0.1% acetic acid (pH 3.2) as an aqueous mobile phase, as for 35 out of 55 compounds the criteria set for monitoring both substrate depletion and the forming metabolites were fulfilled either with positive or negative ion mode. More specifically, 25 of the compounds filled the criteria in positive ion mode ionization, and 13 in negative ion mode ionization. The chromatographic performance was found unsuitable for seven compounds. The 0.1% acetic acid as an aqueous mobile phase provided the best performance for ten of the 55 compounds.
In terms of the number of compounds fulfilling the set criteria for metabolite profiling, either with positive or negative ion mode electrospray ionization, the suitability of 0.1% formic acid (pH 2.8), 2 mM ammonium formate (pH 6.0) and 10 mM ammonia (pH 9.8) were generally about equal, all leading to an adequate performance for 25–29 compounds. Clear differences between these three mobile phases were however apparent: 2 mM ammonium formate was the best aqueous mobile phase for 22 of the compounds, while ammonia was the most suitable only for five of the compounds, and formic acid was not the best for any of the 55 compounds. This latter issue is worth stressing, as though several mobile phase conditions gave performance that fulfilled the criteria set for metabolite profiling (marked “**”), large, even over 10-fold, sensitivity differences were observed between them in some cases. The 2 mM ammonium acetate was suggested to have clearly the lowest general suitability for the analysis of the used test compounds, as the metabolite profiling criteria were fulfilled only with 18 of the 55 compounds. The relatively poor performance with ammonium acetate and formic acid as mobile phase additives is somewhat unexpected and worth noticing, as these both are commonly used in LC–ESI–MS.
With 2 mM ammonium formate, 2 mM ammonium acetate, and 10 mM ammonia as an aqueous mobile phase, it was clearly observed that most of the detected problems were introduced by inadequate chromatographic performance (poor peak shape), not by the mass spectrometric ionization properties. The Table 2 shows that when using 2 mM ammonium formate as an aqueous mobile phase, there were 24 compounds for which positive ion mode ESI ionization response high enough for metabolite profiling was observed, but at the same time their chromatographic properties were very poor and non-suitable for the intended study. When using 2 mM ammonium acetate and 10 mM ammonia as an aqueous eluent phase, the corresponding numbers of compounds with good ionization but poor chromatography were 18 and 17, respectively. In this context there was a striking difference between these three aqueous mobile phases and the mixture containing 0.05% acetic acid and 5 mM ammonium acetate (pH 4.4) providing clearly better chromatographic performance for a large set of compounds. This effect was most distinct for basic compounds, as peak shapes for acidic and neutral compounds were generally clearly better. An example of the pH/mobile phase additive effect for peak shapes is shown in Figure 1, where propranolol has very poor tailing peaks when ammonium formate, ammonium acetate and ammonia are used in aqueous mobile phase (traces C, E, and F), but on the contrary very good chromatographic performance when formic acid, acetic acid or mixture of ammonium acetate and acetic acid are used (traces A, B, and D). The ion strengths of the buffers were intentionally kept rather low to provide a good ESI–MS ionization efficiency, which may in some cases be the reason for the observed poor chromatographic peak shapes. Thus, from the chromatographical point of view higher buffer ion strength would improve the quality of the data for certain compounds, but this would also lead to clearly lower ionization response and thus poorer detection sensitivity.
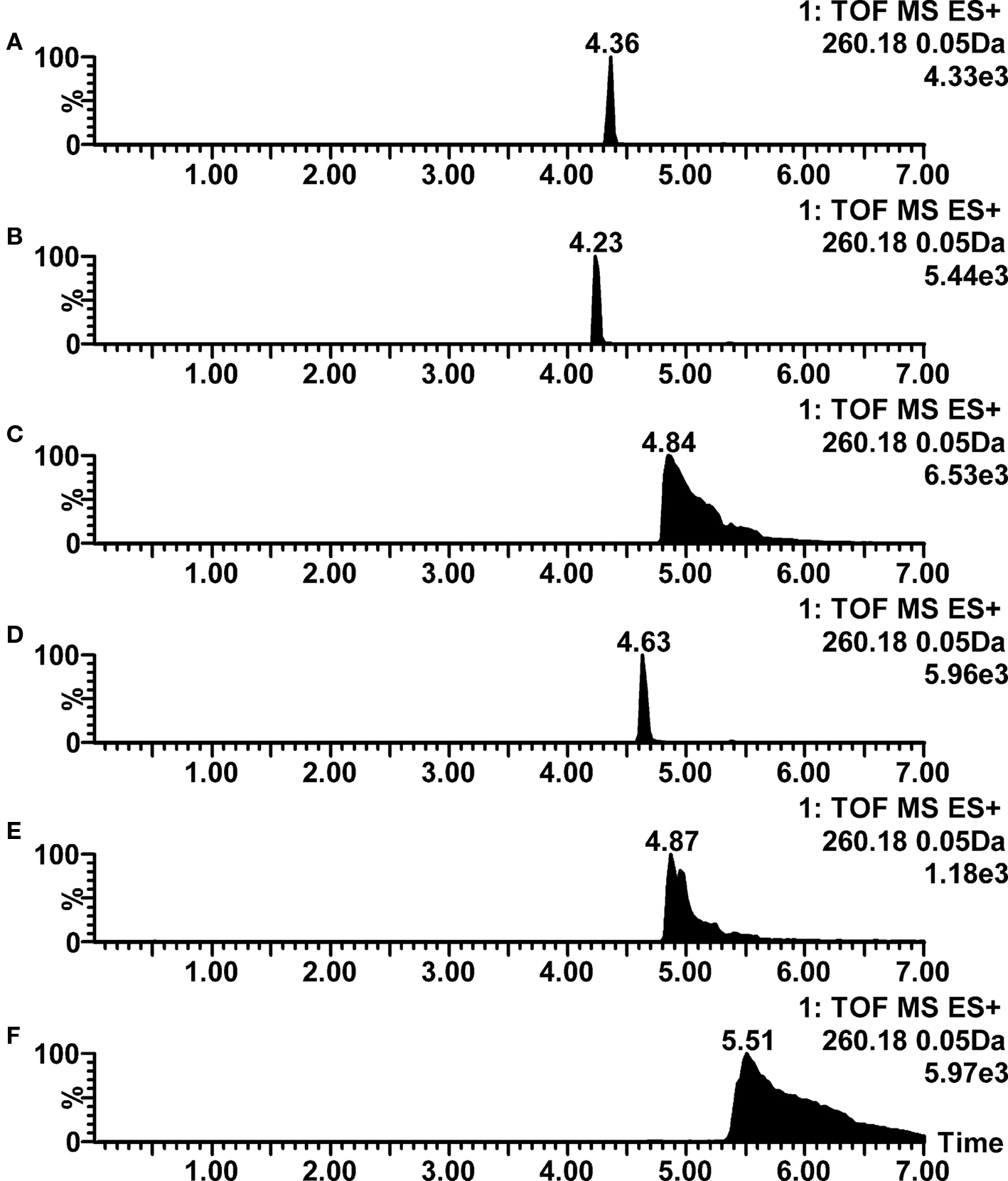
Figure 1. Positive ion mode LC–ESI–TOF–MS ion chromatograms for propranolol at various mobile phase pH conditions. (A) 0.1% formic acid (pH 2.8); (B) 0.1% acetic acid (pH 3.2); (C) 2 mM ammonium formate (pH 6.0); (D) 0.05% acetic acid with 5 mM ammonium acetate (pH 4.4); (E) 2 mM ammonium acetate (pH 6.7); (F) 10 mM ammonia (pH 9.8).
The injection conditions may have their effect to the poor peak shapes in some cases. The acetonitrile content (50%) in injection solvent has clearly increased elution strength with respect to the initial gradient conditions (2%), causing some fronting in the peak shape if too high injection volumes are used. This phenomenon is clearly more pronounced with analytes that have low retention, which again in turn increases the importance of good chromatographic retention behavior. We however deliberately did not change the injection solvent, as in in vitro metabolism studies the samples usually contain high content of organic solvent used for quenching the enzymatic reactions in the incubations, and in high throughput analyses it is often a waste of time to evaporate the organic solvent or dilute the sample with aqueous solvent. Moreover, also in the high throughput in vivo plasma or serum analyses the samples are often prepared with protein precipitation with acetonitrile, using ratios 2:1–4:1 between solvent and sample, leading to even stronger injection solvents than used here.
The column chemistries (particle surface modification) have slight effect to the observed results. As stated above, our column selection was based on need for wide range pH tolerance for the columns, and good experiences with the used column chemistries in various demanding analyses. It may however be the case that some columns with narrower pH range (traditionally pH 2–7) might have produced better chromatographic behavior for certain compounds, but then again those could not have been used in all tested pH conditions. Also, the use of even higher pH (>12) than the highest pH used here could have provided better chromatographic performance for strong bases, but this again would have led to poor applicability with most columns available.
Considering that the mass spectrometer used is from the beginning of the decade, and taking into account the dramatic increase in detection sensitivity with the recent TOF instruments, it is also clear that if such high study compound concentrations are used in metabolism studies as described in this paper, the new instruments are capable for detection sensitivity adequate for metabolite profiling even in slightly misoptimized analytical conditions. However, to obtain more meaningful results, especially regarding the parent compound disappearance (substrate depletion for the estimation of metabolic stability), the incubation concentrations should reflect more truthfully the concentrations observed in circulation in vivo, i.e. around 1 μM. With these kinds of parent compound levels even the modern instruments need again optimal conditions in order to provide high-quality data for metabolism studies.
Acids
The test group contained 10 compounds classified as acids. Obviously, the negative ion mode ESI was clearly more sensitive for almost all of them in all tested pH conditions, only exceptions being observed with acetaminophen, cycloheximide, and warfarin. Observed sensitivity was adequate for metabolite profiling in negative ion mode for all other acidic compounds except acetaminophen when aqueous phase used was 0.1% acetic acid (pH 3.2), 2 mM ammonium formate (pH 6.0), or mixture of 0.05% acetic acid and 5 mM ammonium acetate (pH 4.4). Naturally, the acidic pH provided also good chromatographic peak shape and retention behavior for these compounds. When eluent pH was <3 or >7, the ionization efficiency started to decrease for some of the acids. Also as expected, the neutral or basic conditions (pH ≥ 7) led to inadequate chromatographic retention for some of the acidic compounds.
Acetaminophen was only poorly ionized in negative ion mode, as the sensitivity was adequate only for monitoring metabolic stability, probably because the acidity of acetaminophen is due to a phenol group that is only a weak acid. The positive ion mode ionization was not very good for acetaminophen in any pH conditions either. For cycloheximide and warfarin, good ionization efficiency in positive ion mode electrospray was observed when 2 mM ammonium formate or 10 mM ammonia was used as aqueous mobile phase even though the imide functionality in the compounds would suggest acidic behavior. Warfarin also showed different behavior in comparison with other acids, as it was very well ionized in both positive and negative ion mode, being therefore easy to analyze by LC–ESI–MS in practically all conditions. The ion chromatograms of warfarin are shown in Figure 2. An opposite example of chromatographic performance of acidic compounds is shown in Figure 3, where large differences in negative ion mode ionization efficiency for ibuprofen are observed as a function of pH.
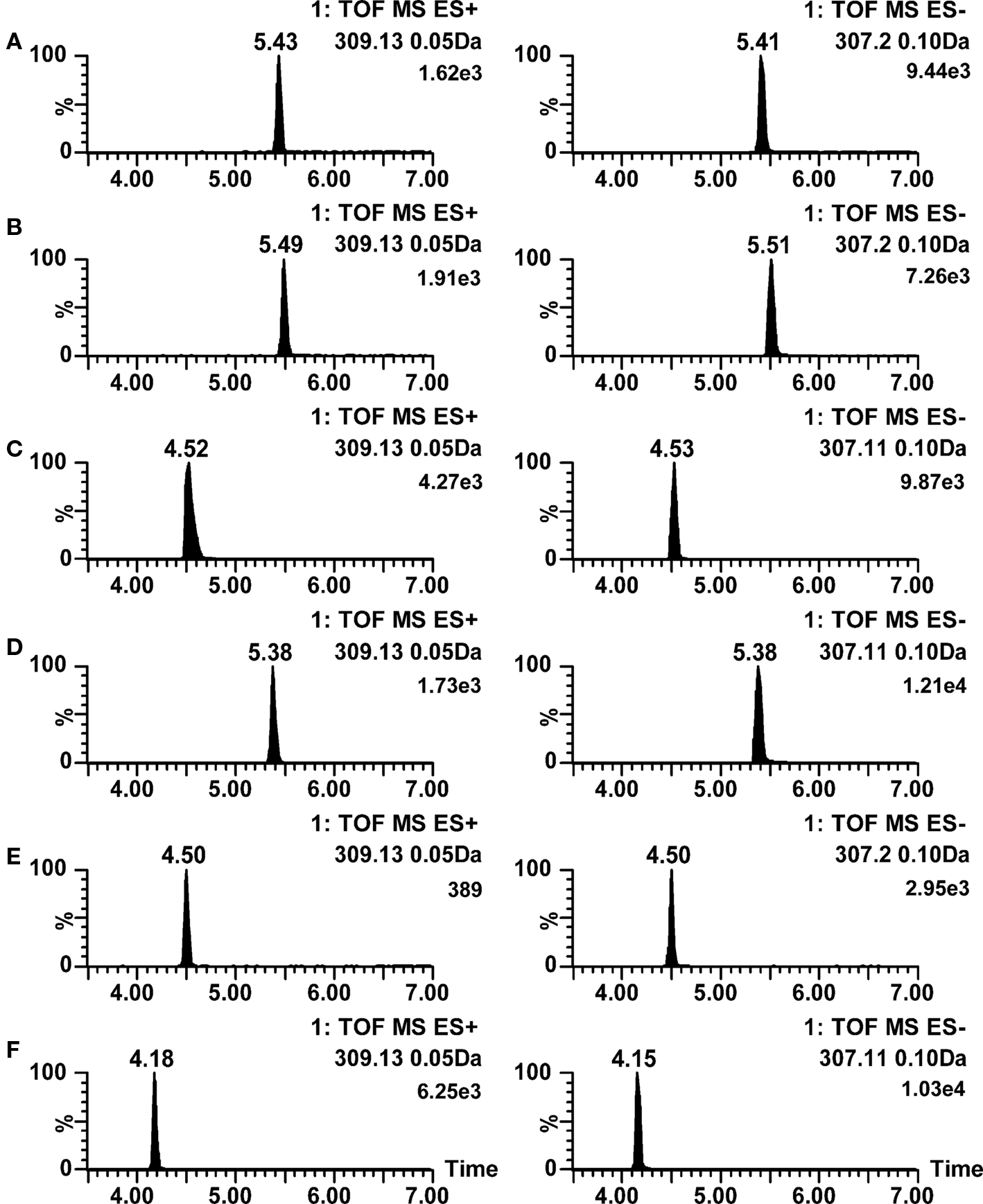
Figure 2. Positive and negative ion mode LC–ESI–TOF–MS ion chromatograms for warfarin at various mobile phase pH conditions. (A) 0.1% formic acid (pH 2.8); (B) 0.1% acetic acid (pH 3.2); (C) 2 mM ammonium formate (pH 6.0); (D) 0.05% acetic acid with 5 mM ammonium acetate (pH 4.4); (E) 2 mM ammonium acetate (pH 6.7); (F) 10 mM ammonia (pH 9.8). Ionization polarities are shown in each trace.
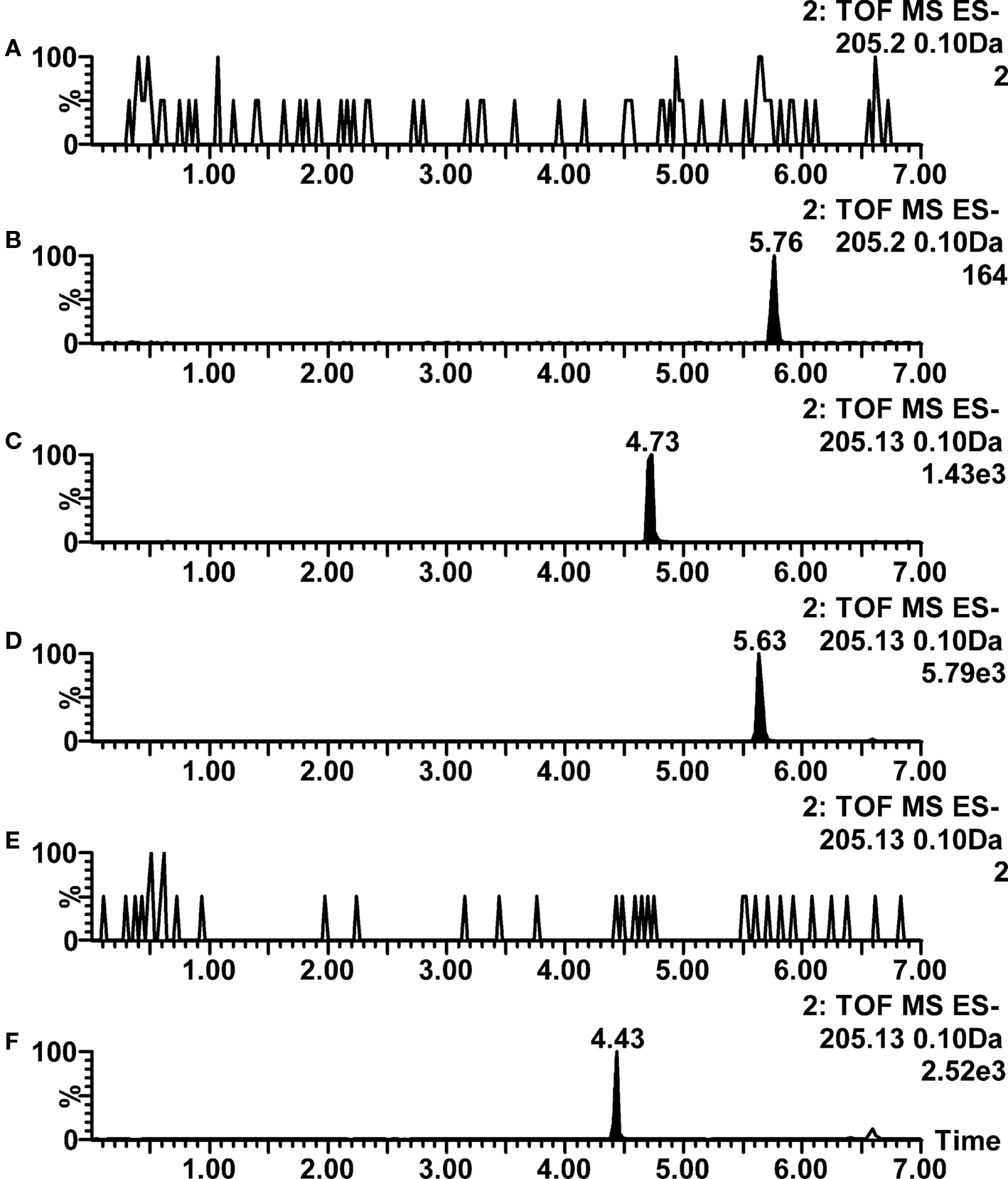
Figure 3. Negative ion mode LC–ESI–TOF–MS ion chromatograms for ibuprofen at various mobile phase pH conditions. (A) 0.1% formic acid (pH 2.8); (B) 0.1% acetic acid (pH 3.2); (C) 2 mM ammonium formate (pH 6.0); (D) 0.05% acetic acid with 5 mM ammonium acetate (pH 4.4); (E) 2 mM ammonium acetate (pH 6.7); (F) 10 mM ammonia (pH 9.8).
Aminoglutethimide, aminopterin, and nalidixic acid were classified as amphoterics rather than acids, in spite of the presence of acidic imide functionality in aminoglutethimide and carboxylic acid functionality in aminopterin and nalidixic acid. In addition to acidic functionalities, aminoglutethimide contains also basic aniline moiety, whereas aminopterin contains primary amines and aromatic imines, and nalidixic acid contains tertiary amine and aromatic imine functionalities. These basic functionalities also show their effect in observed results, as all of these three compounds were ionized clearly better in positive than in negative ion mode electrospray. With aminopterin also good negative ion mode ionization efficiency was observed in most of the acidic conditions tested. Due to the ionization of the mentioned functionalities, also some chromatographic peak shape and retention problems were observed in acidic and as well as in basic conditions, while the mixture of 0.05% acetic acid and 5 mM ammonium acetate (pH 4.4) showed clearly the best performance with these three compounds. Positive ion mode chromatograms of nalidixic acid are as an example in Figure 4, showing very good peak shapes and retention when formic acid, acetic acid or mixture of acetic acid and ammonium acetate are used in mobile phase (traces A, B, and D), but poor peak shape when ammonium formate and ammonium acetate (without acetic acid) are used (traces C and E), and poor peak shape and decreased retention when ammonia is used in mobile phase (trace F).
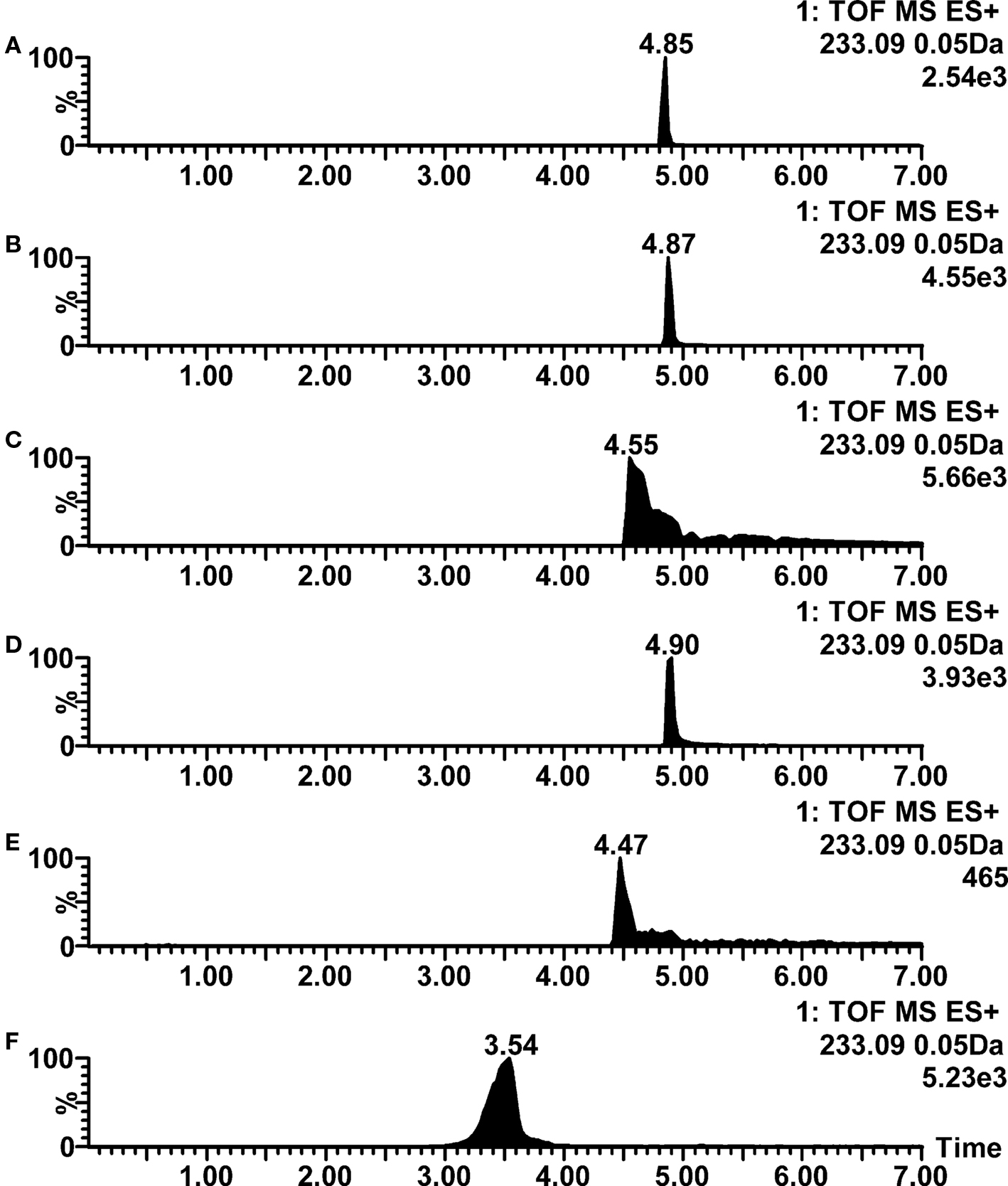
Figure 4. Positive ion mode LC–ESI–TOF–MS ion chromatograms for nalidixic acid at various mobile phase pH conditions. (A) 0.1% formic acid (pH 2.8); (B) 0.1% acetic acid (pH 3.2); (C) 2 mM ammonium formate (pH 6.0); (D) 0.05% acetic acid with 5 mM ammonium acetate (pH 4.4); (E) 2 mM ammonium acetate (pH 6.7); (F) 10 mM ammonia (pH 9.8).
Bases
Based on the amine functionalities in the compounds, 25 of them were classified as bases. As expected, a clear majority of them showed ionization efficiency adequate for metabolite profiling in positive ion mode practically in all tested pH conditions. However, when using basic pH conditions or ammonium acetate or ammonium formate in eluents, surprisingly high number of chromatographic peak shape problems were observed. In high pH these are mostly explained by interactions between basic functionalities and free silanol groups remaining in the column, even though the columns used were manufactured with ethylene-bridged silica, resulting in low number of non-end capped silanols. As with acidic compounds, the generally best chromatographic performance was thus obtained when using low pH eluents, especially with the mixture of 0.05% acetic acid and 5 mM ammonium acetate (pH 4.4). The peak shape problem (good ionization but poor peak shape) is described in Figure 1 with propranolol, whereas example concerning retention behavior problems is shown in Figure 5 with nicotine. For nicotine, a satisfactory peak shape but clearly inadequate retention is observed when formic acid, acetic acid, or mixture of acetic acid and ammonium acetate are used in mobile phase (traces A, B, and D). Increased retention but extremely poor peak shape is obtained when using ammonium formate in mobile phase, whereas with ammonium acetate the ionization is suppressed below detection limit. However, when using 10 mM ammonia in mobile phase, an excellent peak shape and good ionization efficiency in positive ion mode are obtained.
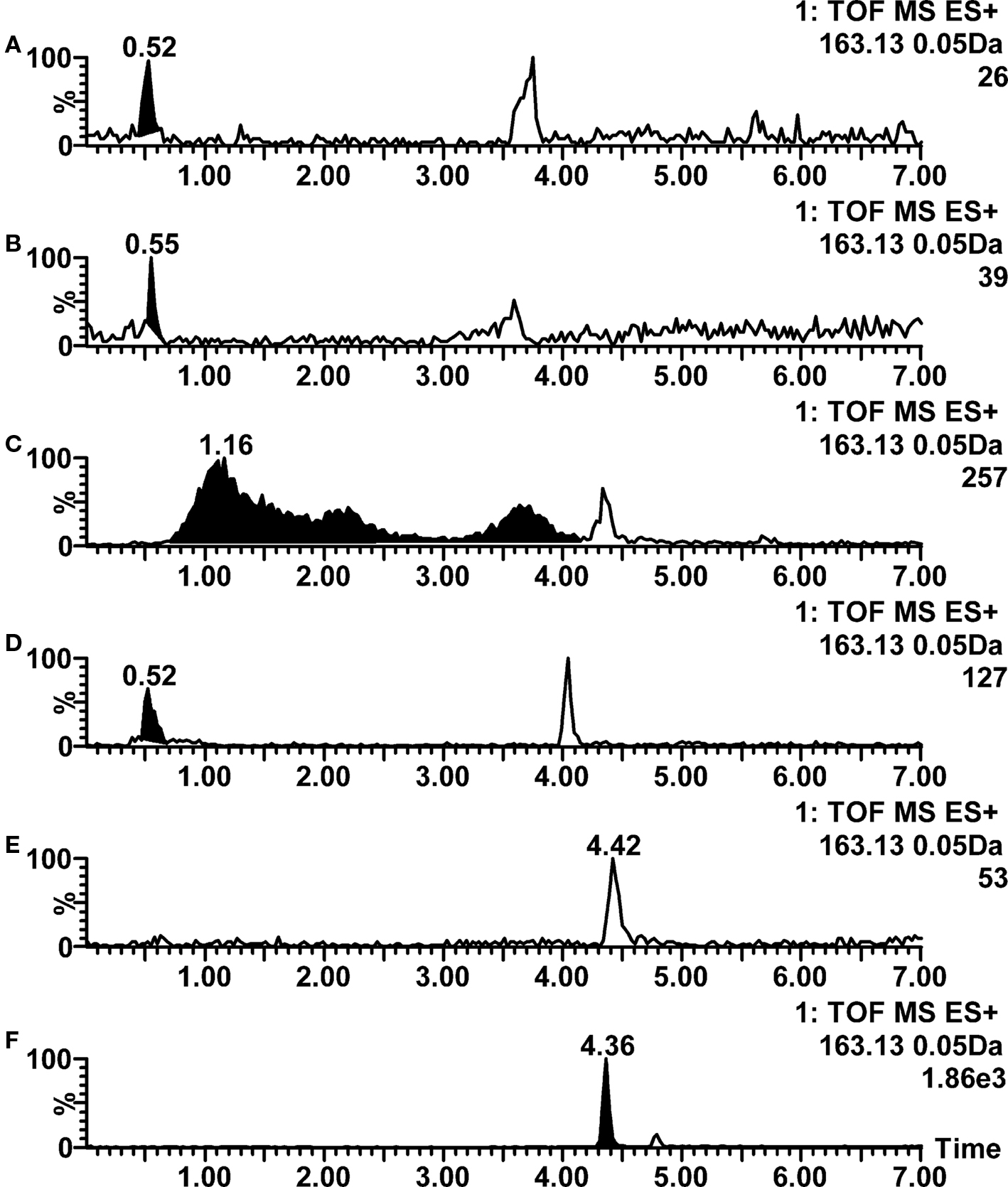
Figure 5. Positive ion mode LC–ESI–TOF–MS ion chromatograms for nicotine at various mobile phase pH conditions. (A) 0.1% formic acid (pH 2.8); (B) 0.1% acetic acid (pH 3.2); (C) 2 mM ammonium formate (pH 6.0); (D) 0.05% acetic acid with 5 mM ammonium acetate (pH 4.4); (E) 2 mM ammonium acetate (pH 6.7); (F) 10 mM ammonia (pH 9.8). Peaks not integrated are not nicotine-related.
In the negative ion mode ionization, a detection sensitivity adequate even for monitoring the parent compound disappearance was rarely obtained for bases, the exception being haloperidol, for which good ionization efficiency was obtained especially with the mixture of 0.05% acetic acid and 5 mM ammonium acetate.
Neutrals
The remaining 17 compounds lacking clearly basic or acidic functional groups, or having no change in calculated logD value over pH range 2–11, were classified as neutrals. Their behavior with respect to ionization efficiency in positive or negative ion mode was generally not so clearly divided, as some showed better ionization in positive and others in negative ion mode. The compounds with better positive ion mode ionization were however in majority over those with more efficient negative ion mode ionization. The slightly acidic 2 mM ammonium formate was observed to be the most suitable of the tested mobile phase conditions for this group of compounds, as the performance with all of the 17 compounds fulfilled the set criteria for monitoring substrate depletion, and with 14 of them the set criteria for metabolite profiling was fulfilled.
Within this class of compounds, problems in chromatography were not observed due to the compounds’ poor liquid state ionization properties. Instead, good retention behaviors and excellent peak shapes in all mobile phase pHs were obtained. An example of this is shown in Figure 6 as ion chromatograms of diuron, where the pH hardly has any effect to chromatography, and only decreasing effect to ionization in negative ion mode with 0.1% formic acid (trace A ESI−) and in positive ion mode with ammonium acetate in mobile phase (trace E ESI+).
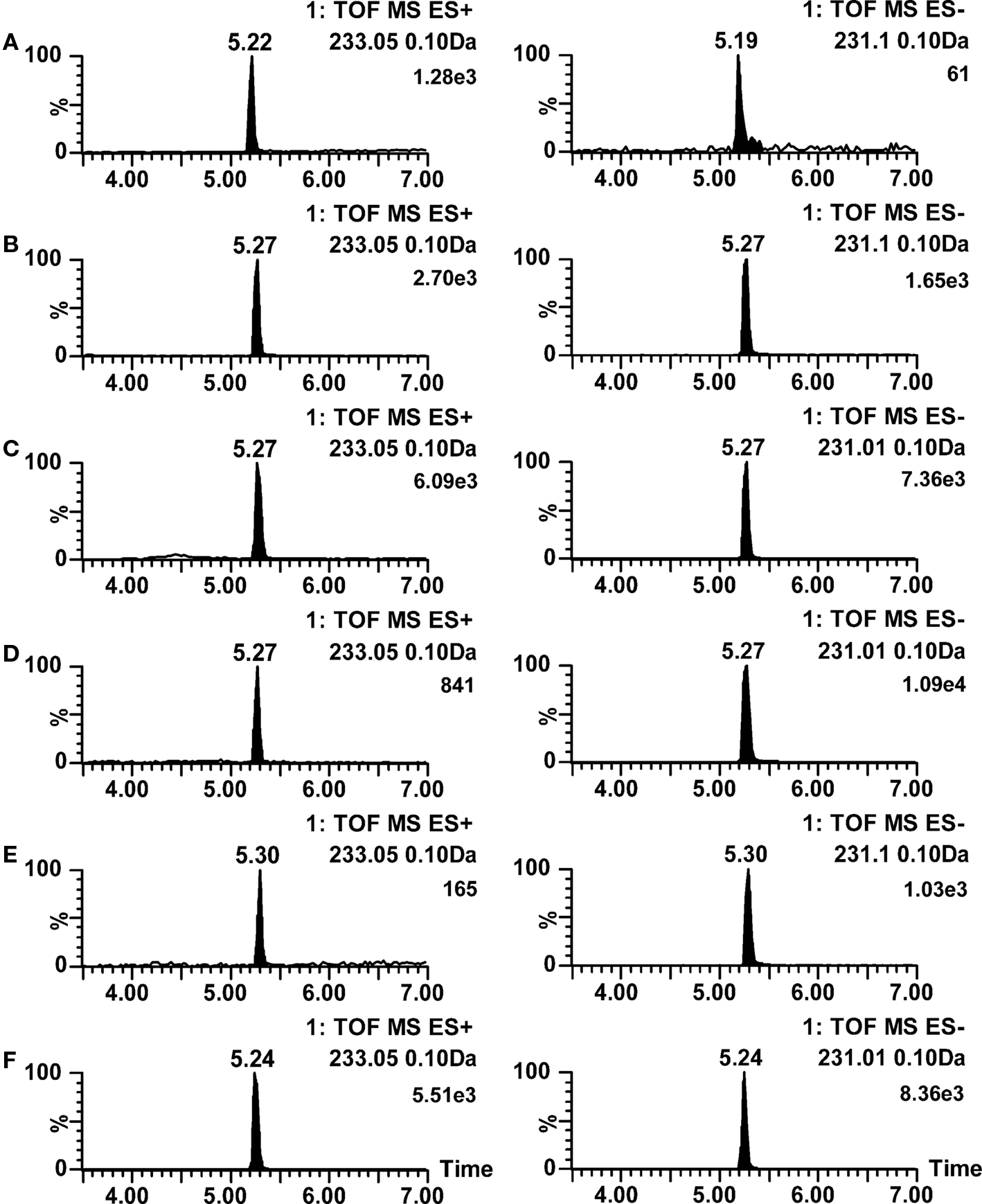
Figure 6. Positive and negative ion mode LC–ESI–TOF–MS ion chromatograms for diuron at various mobile phase pH conditions. (A) 0.1% formic acid (pH 2.8); (B) 0.1% acetic acid (pH 3.2); (C) 2 mM ammonium formate (pH 6.0); (D) 0.05% acetic acid with 5 mM ammonium acetate (pH 4.4); (E) 2 mM ammonium acetate (pH 6.7); (F) 10 mM ammonia (pH 9.8). Ionization polarities are shown in each trace.
Conclusions
Various mobile phase conditions were tested in developing methods for LC–ESI–TOF–MS analysis for metabolite profiling of 55 structurally different compounds. No single or even two different conditions were suitable for covering the whole set of compounds, stressing out that the testing of optimum conditions for each individual compound leads always to clearly more high-quality data. Of the tested conditions, mobile phase with 0.05% acetic acid and 5 mM ammonium acetate provided generally the best performance with easily ionizable compounds (acids and bases), whereas for neutral compounds the best overall performance was obtained with 2 mM ammonium formate as an aqueous mobile phase. The results also suggest that with the used injection solvent conditions, column chemistries, and positive ion mode LC–ESI–MS, poor chromatographic performance, rather than the poor ionization efficiency, may often be the limiting factor in analysis of basic compounds. Noteworthy is also that two very commonly used aqueous mobile phases, i.e. 0.1% formic acid and 2 mM ammonium acetate, showed practically the poorest suitability for LC–ESI–MS analysis for all chemical classes (acids, neutrals, bases) among the different aqueous mobile phases tested.
This in turn suggests that slightly acidic, but not too acidic, pH conditions would be the most optimum for generic LC–ESI–MS analysis of a large set of compounds. However, in this respect recent advances in liquid chromatographic techniques, utilizing sub-2 μm particles and usually referred with names such as ultrahigh performance liquid chromatography (UPLC) (Churchwell et al., 2005; Yu et al., 2006; Pedraglio et al., 2007) or rapid resolution liquid chromatography (Yoshida and Majors, 2006), are to some extent improving the problem with poor peak shapes and may lead to a situation where the generic chromatography works increasingly better and more emphasis can be put to ionization conditions. Still, this technical remedy does not however improve the situation with retention behavior but only with peak shapes.
The conditions found here as most suitable for each individual test compound were used in metabolite profiling studies with rat and human liver microsomes and homogenates, providing high-quality data and enabling identification of high number of metabolites for most of the compounds (data will be published elsewhere).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The expert technical assistance of Mrs Birgitta Paldanius and Mrs Katri Sunnari is gratefully acknowledged. The study was partially funded by European Center for Validation of Alternative Methods, project “Metabolic stability and metabolite identification of 55 ECVAM/ICCVAM validation compounds in human and rat liver homogenates and microsomes” (Contract No CCR.IHCP.C432889.X; principal scientific officers Dr Sandra Coecke and Dr Michel Bouvier d’Yvoire).
References
Ayrton, J., Dear, G. J., Leavens, W. J., Mallett, D. N., and Plumb, R. S. (1998). Use of generic fast gradient liquid chromatography-tandem mass spectroscopy in quantitative bioanalysis. J. Chromatogr. B 709, 243–253.
Cappiello, A., Famiglini, G., Palma, P., Pierini, E., Termopoli, V., and Trufelli, H. (2008). Overcoming matrix effects in liquid chromatography-mass spectrometry. Anal. Chem. 80, 9343–9348.
Castro-Perez, J., Plumb, R., Granger, J. H., Beattie, I., Joncour, K., and Wright, A. (2005). Increasing throughput and information content for in vitro drug metabolism experiments using ultra-performance liquid chromatography coupled to a quadrupole time-of-flight mass spectrometer. Rapid Commun. Mass Spectrom. 19, 843–848.
Cheng, Y. F., Lu, Z., and Neue, U. (2001). Ultrafast liquid chromatography/ultraviolet and liquid chromatography/tandem mass spectrometric analysis. Rapid Commun. Mass Spectrom. 15, 141–151.
Chu, I., Liu, F., Soares, A., Kumari, P., and Nomeir, A. A. (2002). Generic fast gradient liquid chromatography/tandem mass spectrometry techniques for the assessment of the in vitro permeability across the blood-brain barrier in drug discovery. Rapid Commun. Mass Spectrom. 16, 1501–1505.
Churchwell, M. I., Twaddle, N. C., Meeker, L. R., and Doerge, D. R. (2005). Improving LC-MS sensitivity through increases in chromatographic performance: comparisons of UPLC-ES/MS/MS to HPLC-ES/MS/MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. B 825, 134–143.
Delatour, C., and Leclercq L. (2005). Positive electrospray liquid chromatography/mass spectrometry using high-pH gradients: a way to combine selectivity and sensitivity for a large variety of drugs. Rapid Commun. Mass Spectrom. 19, 1359–1362.
Hop, C. E., Tiller, P. R., and Romanyshyn, L. (2002). In vitro metabolite identification using fast gradient high performance liquid chromatography combined with tandem mass spectrometry. Rapid Commun. Mass Spectrom. 16, 212–219.
Kamel, A., and Prakash, C. (2006). High performance liquid chromatography/atmospheric pressure ionization/tandem mass spectrometry (HPLC/API/MS/MS) in drug metabolism and toxicology. Curr. Drug Metab. 7, 837–852.
Kostiainen, R., Kotiaho, T., Kuuranne, T., and Auriola S. (2003). Liquid chromatography/atmospheric pressure ionization mass spectrometry in drug metabolism studies. J. Mass Spectrom. 38, 357–372.
Kostiainen, R., and Kauppila, T. J. (2009). Effect of eluent on the ionization process in liquid chromatography-mass spectrometry. J. Chrom. A. 1216, 685–699.
Mallet, C. R., Lu, Z., and Mazzeo, J. R. (2004). A study of ion suppression effects in electrospray ionization from mobile phase additives and solid-phase extracts. Rapid Commun. Mass Spectrom. 18, 49–58.
Mensch, J., Noppe, M., Adriaensen, J., Melis, A., Mackie, C., Augustijns, P., and Brewster, M. E. (2007). Novel generic UPLC/MS/MS method for high throughput analysis applied to permeability assessment in early drug discovery. J. Chromatogr. B, 847, 182–187.
Law, B. (2004). The effect of eluent pH and compound acid-base character on the design of generic-gradient reversed-phase high-performance liquid chromatography (RP-HPLC) methods for use in drug discovery. J. Pharm. Biomed. Anal. 34, 215–219.
Lee, M. S., and Kerns, E. H. (1999). LC/MS applications in drug discovery. Mass Spectrom. Rev. 18, 187–279.
Pedraglio, S., Rozio, M. G., Misiano, P., Reali, V., Dondio, G., and Bigogno, C. (2007). New perspectives in bio-analytical techniques for preclinical characterization of a drug candidate: UPLC-MS/MS in in vitro metabolism and pharmacokinetic studies. J. Pharm. Biomed. Anal. 44, 665–673.
Seto, C., Bateman, K. P., and Cunter, B. (2002). Development of generic liquid chromatography-mass spectrometry methods using experimental design. J. Am. Soc. Mass Spectrom. 13, 2–9.
Yoshida, T., and Majors, R. E. (2006). High-speed analyses using rapid resolution liquid chromatography on 1.8-microm porous particles. J. Sep. Sci. 29, 2421–2432.
Yu, K., Little, D., Plumb, R., and Smith, B. (2006). High-throughput quantification for a drug mixture in rat plasma-a comparison of ultra performance liquid chromatography/tandem mass spectrometry with high-performance liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 20, 544–552.
Keywords: LC–MS, drug metabolism, human liver, in vitro, generic assays, drugs, pesticides
Citation: Rousu T, Hokkanen J, Pelkonen OR and Tolonen A (2010) Applicability of generic assays based on liquid chromatography-electrospray mass spectrometry to study in vitro metabolism of 55 structurally diverse compounds. Front. Pharmacol. 1:10. doi: 10.3389/fphar.2010.00010
Received: 27 May 2010;
Paper pending published: 15 June 2010;
Accepted: 06 July 2010;
Published online: 06 August 2010
Edited by:
Petr Pavek, Charles University, Czech RepublicReviewed by:
Petr Pavek, Charles University, Czech RepublicIvan Vokral, Charles University in Prague, Czech Republic
Copyright: © 2010 Rousu, Hokkanen, Pelkonen and Tolonen. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Olavi R. Pelkonen, Department of Pharmacology and Toxicology, University of Oulu, POB 5000 (Aapistie 5 B), FIN-90014 Oulu, Finland. e-mail:b2xhdmkucGVsa29uZW5Ab3VsdS5maQ==