 Heyan Wu
Heyan Wu Min Zhou
Min Zhou Xiaoting Ye1
Xiaoting Ye1 Hongxin Lin
Hongxin Lin Li Wang
Li Wang Xing Nie
Xing Nie Lidan Zhang
Lidan Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 08 April 2025
Sec. Pediatric Neurology
Volume 13 - 2025 | https://doi.org/10.3389/fped.2025.1571426
This article is part of the Research TopicNew Insights into Pediatric Neurology: Neurological Disorders and Epileptic EncephalopathiesView all 4 articles
Background: Biallelic variants in NARS2 that encodes the mitochondrial asparaginyl-tRNA synthetase are associated with a wide spectrum of clinical phenotypes. Herein, we report on two siblings carrying the same compound heterozygous missense variants in NARS2, to improve the understanding of the phenotypic heterogeneity of NARS2 variants.
Case presentation: The two probands, a 3-year-old female (Patient 1) and a 16-month-old male (Patient 2), were clinically suspected of Combined oxidative phosphorylation deficiency 24 (COXPD24). Both presented with neurological manifestations, including refractory epilepsy, developmental delay and motor developmental regression, within the first year of life, accompanied by symmetrical brain lesions identified on magnetic resonance imaging (MRI). To elucidate the underlying genetic etiology, whole-exome sequencing (WES) was performed, followed by Sanger sequencing validation in the patients and their non-consanguineous parents. Genetic analysis revealed that both probands harbored identical compound heterozygous variants in the NARS2 gene: c.1253G>A (p.Arg418His) and c.1163C>T (p.Thr388Met). Notably, the c.1163C>T (p.Thr388Met) variant in NARS2 represents a novel finding, further expanding the genetic spectrum associated with this disorder.
Conclusions: Our findings expand the mutational spectrum of NARS2 and highlight the associated phenotypic heterogeneity, underscoring the critical role of NARS2 in epilepsy and neurodevelopmental processes. For pediatric patients with refractory epilepsy, early genetic testing is essential to improve diagnostic accuracy, refine prognostic stratification, and guide personalized treatment strategies. Additionally, mitochondrial drug cocktail therapy may be beneficial for epilepsy caused by NARS2 mutations.
Mitochondrial diseases encompass a broad spectrum of disorders, arising from genetic defects that impair mitochondrial function, leading to ATP synthesis impairment and subsequent energy depletion (1). Although various possible clinical phenotypes can result, neurological and neuromuscular affection is most frequently encountered. Among these complex conditions, refractory epilepsy stands as a prominent and challenging aspect, intricately intertwined with mitochondrial dysfunction (2). On 11q14.1, NARS2 encodes mitochondrial aminoacyl-tRNA synthetases (mt-ARSs), which catalyzes the ligation of asparagine to tRNA molecules and is essential for protein synthesis (3). While NARS2 is widely expressed in human tissues, variants in the gene seem to preferentially affect tissues with high energy demand, such as the brain, cochlea and muscle, similar to other mitochondrial disorders (4). Biallelic variants in NARS2 have been linked to a spectrum of clinical phenotypes, ranging from isolated hearing loss to severe neurodevelopmental disorders, with well-known examples being Combined oxidative phosphorylation deficiency 24 (COXPD24), Autosomal recessive deafness-94 (DFNB94), Alpers syndrome and Leigh syndrome. COXPD24 is an autosomal recessive disorder characterized by early in life with seizures, hypotonia, myopathy, hearing impairment, and overall delay and/or regression of cognitive and motor development (5–9). DFNB94 causes patients to exhibit bilateral nonsyndromic sensorineural hearing loss (10, 11). Despite its fundamental function, the full spectrum of NARS2-related disorders remains elusive due to the limited number of reported cases, the genotype-phenotype relationships remain unpredictable due to a limited number of known cases. Furthermore, the underlying mechanisms of NARS2-related disease have not been comprehensively studied. The purpose of this report is to provide detailed clinical, laboratory, and imaging data for two siblings carrying the same heterozygous missense mutations in the NARS2 gene, so as to enhance the understanding of the phenotypes of NARS2 gene variants.
The two probands were born to non-consanguineous parents, as shown in Figure 1. Notably, their eldest sister showed no clinical manifestations of the condition.
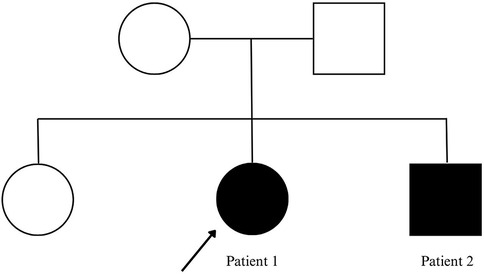
Figure 1. Family pedigree. Probands with status epilepticus are marked by filled symbols and arrows.
Patient 1: a 3-year-old girl is the second child of healthy nonconsanguineous Chinese parents. She was delivered at term without asphyxia. The infant exhibited typical developmental progress until reaching 6 months of age, at which point generalized epilepsy with myoclonic seizures activity manifested. A subsequent episode of convulsions occurred 1 month later. Following a third instance of seizures at 8 months, the child experienced a decline in developmental milestones, accompanied by diminished muscular strength and reduced muscle tone. The MRI performed at 8 months of age indicated delayed myelination, symmetrical abnormal signals in the lentiform nucleus, and diffuse cerebral atrophy (Figure 2). The electroencephalogram (EEG) examination revealed an abnormal pattern, characterized by the presence of a significant number of spike-and-slow-wave complexes in the bilateral occipital regions. Unfortunately, as the test was conducted at an external hospital, we were unable to obtain the EEG images. Topiramate tablets were administered as a means to curb the progression of epilepsy in this child. Despite titrating topiramate tablets to the maximum tolerated dose, the child continued to exhibit progression of seizures and developed abnormal liver function, leading to the decision to discontinue the antiepileptic medication. To minimize the risk of withdrawal symptoms, a structured tapering regimen was implemented, reducing the dosage by 25% every two weeks, with close monitoring for seizure recurrence and potential adverse effects. This approach was designed to ensure a safe and controlled discontinuation process while prioritizing the patient's clinical stability. Subsequently, there was a gradual decrease in both the frequency and intensity of epileptic seizures, though partial onset seizures persisted. As the condition advanced, the child eventually progressed into flaccid quadriplegia by the age of two. Despite the recommendation for a muscle biopsy as a diagnostic measure, the parents opted against the invasive procedure. In an effort to unravel the underlying pathology, Whole Exome Sequencing (WES) was undertaken for both the child and her family members. The whole exon detected two novel compound heterozygotes variants: NM_024678.6:c.1253G>A(p.Arg418His) mutation (from father) and NM_024678.6:c.1163C>T(p.Thr388Met) mutation (from mother) in NARS2 gene, diagnosed with COXPD24.
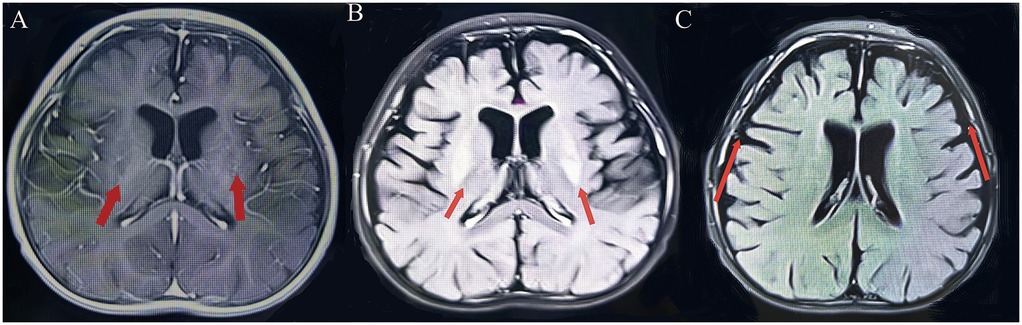
Figure 2. (Patient 1) Brain MRI. (A) T1-weighted image: symmetrical lenticular nucleus hyperintensity. (B) T2-weighted image: symmetrical lenticular nucleus hyperintensity. (C) Diffuse cerebral atrophy.
Patient 2, a 14-month-old boy and the younger brother of Patient 1, had an uneventful birth history and showed normal growth and development until the onset of his condition. At 6 months old, he achieved a developmental milestone by sitting unsupported. The patient was admitted to the hospital at 6 months of age due to seizures following a high fever. Upon physical examination upon admission, it was found that the patient was unable to concentrate or track objects, did not make eye contact, and was unresponsive to auditory stimuli. During the seizure episodes, the patient exhibited a tilt of the head and face to the right side and twitching of the right corner of the mouth, with the episodes lasting approximately 2 h before resolving spontaneously. During the hospital stay, the patient experienced 2 seizure episodes, characterized by a tilt of the head and face to the right side and twitching of the right corner of the mouth. Despite initial efforts to manage the seizures with oral chloral hydrate, the treatment proved ineffective, prompting the initiation of Levetiracetam therapy for seizure control. However, even with this intervention, occasional seizures persisted. Brain MRI imaging revealed a crucial clue: symmetrical abnormal signals in the bilateral posterior putamen (Figure 3). Additionally, EEG demonstrated a marked abnormality, featuring a slowed background rhythm and a proliferation of diffuse slow waves throughout the brain's conduction pathways (Figure 4). During his hospital stay, it became evident that Patient 2's motor development had regressed, making it difficult for him to maintain steady head control. Fortunately, other laboratory parameters, such as liver and cardiac enzyme levels, remained within normal limits. Genetic testing ultimately identified two mutations in the NARS2 gene (c.1253G>A/p.Arg418His and c.1163C>T/p.Thr388Met), mirroring the findings in Patient 1, suggesting a familial link. Considering that this disease is a genetic disorder, we discontinued all antiepileptic drugs when the child was 8 months old and adopted a mitochondrial drug cocktail therapy (12), which included oral administration of coenzyme Q10 (5 mg/kg), L-carnitine(30 mg/kg), and vitamin B1 (10 mg/kg). Surprisingly, during the six—month follow—up, the child did not experience any further seizures.
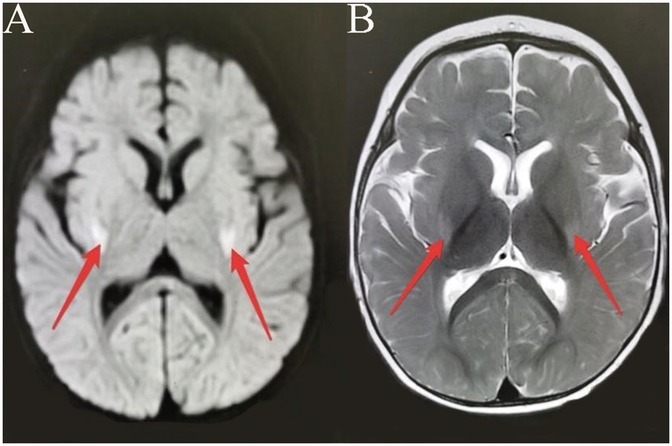
Figure 3. (Patient 2) Brain MRI. (A) T1-weighted image: bilateral posterior putamen hyperintensity. (B) T2-weighted image: bilateral posterior putamen hyperintensity.
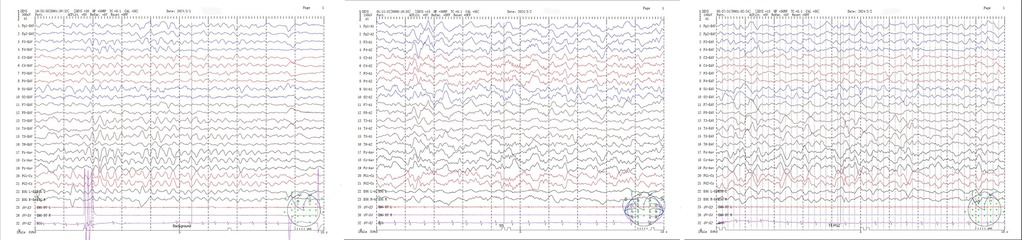
Figure 4. (Patient 2) EEG: slow background activity.
To further explore the inheritance pattern and characteristics of these mutations, genetic verification was conducted on the healthy elder sister. The results revealed a unique genetic profile, as she carried only one of the two mutations identified in her siblings (c.1163C>T/p.Thr388Met), suggesting that a compound heterozygous mutation was necessary to cause the disease (Figure 5).
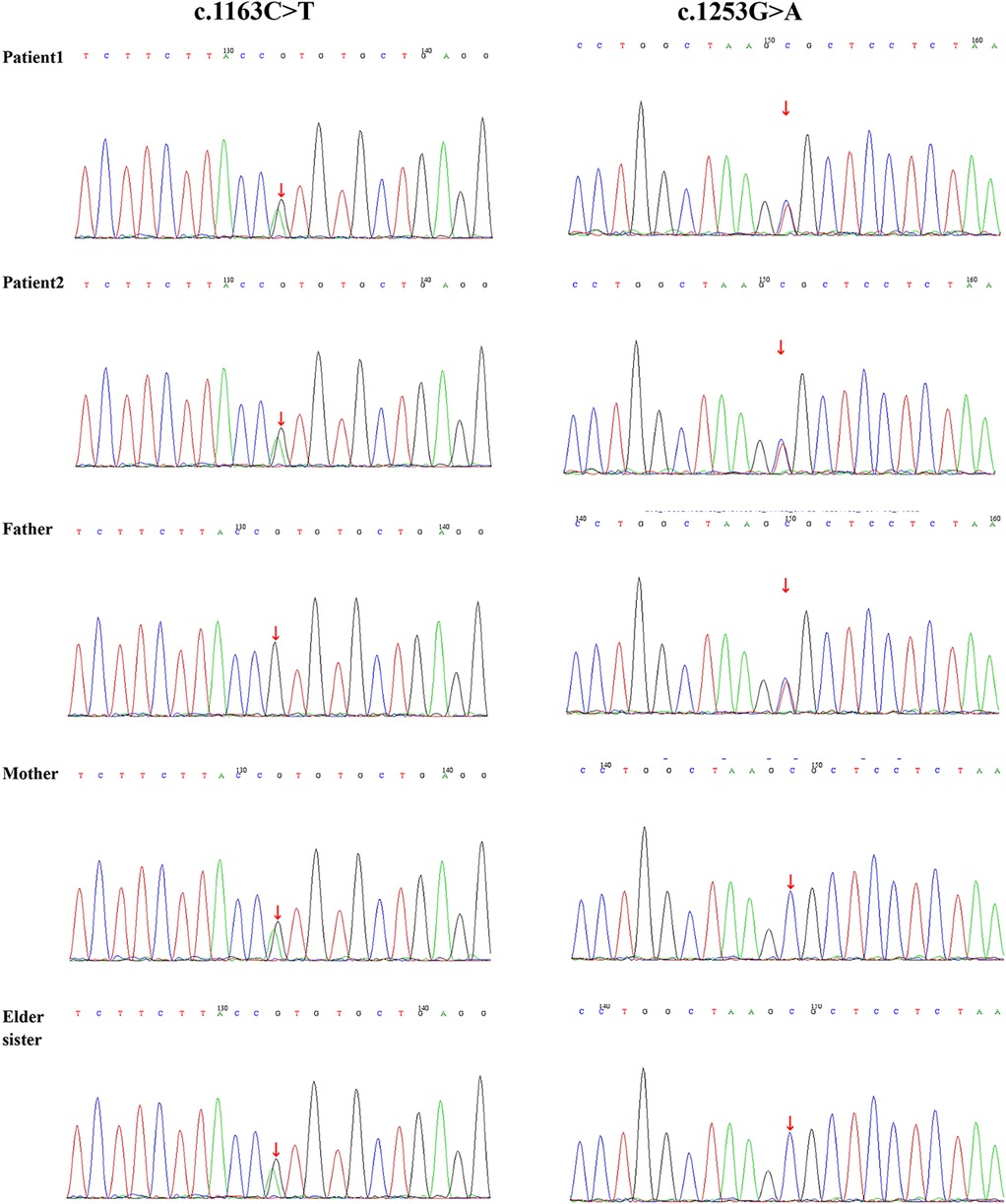
Figure 5. Sanger sequencing electropherograms validating the WES-identified variant in the proband.
A comprehensive and thorough review of NARS2 mutations has been undertaken. As of the conclusion of December 2024, a total of 38 patients with a variant of NARS2 have been documented and reported. (Table 1) (5–11, 13–25), which included 32 missense variants, 3 truncation Missense, 2 splicing and 1 Synonymous. All the reported variants adhere to an autosomal recessive mode of inheritance, with their diagnosis, phenotypic manifestations, variant types, and survival outcomes comprehensively outlined in Table 1. The age of onset spans from the neonatal period to 50 years, with a notable trend towards early-onset disease. The phenotypic spectrum is notably heterogeneous, encompassing a range of conditions. Among the most salient phenotypic features previously reported in NARS2-related diseases are COXPD24, DFNB94, infantile-onset neurodegenerative disorder (NDD), and refractory seizures. Less frequently encountered manifestations include neonatal diabetes, microcephaly, and fatigability-associated ptosis. The disease outcomes vary greatly, with some patients passing away within a few weeks of birth, whereas others, particularly those with milder symptoms, reach adulthood. Nevertheless, based on the current genotype-phenotype correlation analysis, it remains challenging to accurately predict whether the phenotype will be mild or severe. Moreover, the research (13) has revealed that the phenotype does not appear to exhibit a substantial correlation with the location or type of mutation, underscoring the imperative need for the establishment of comprehensive genotype and phenotype databases. This underscores the importance of continued efforts to bridge the gap in our understanding of the intricate relationships between genetic variations and their phenotypic manifestations.
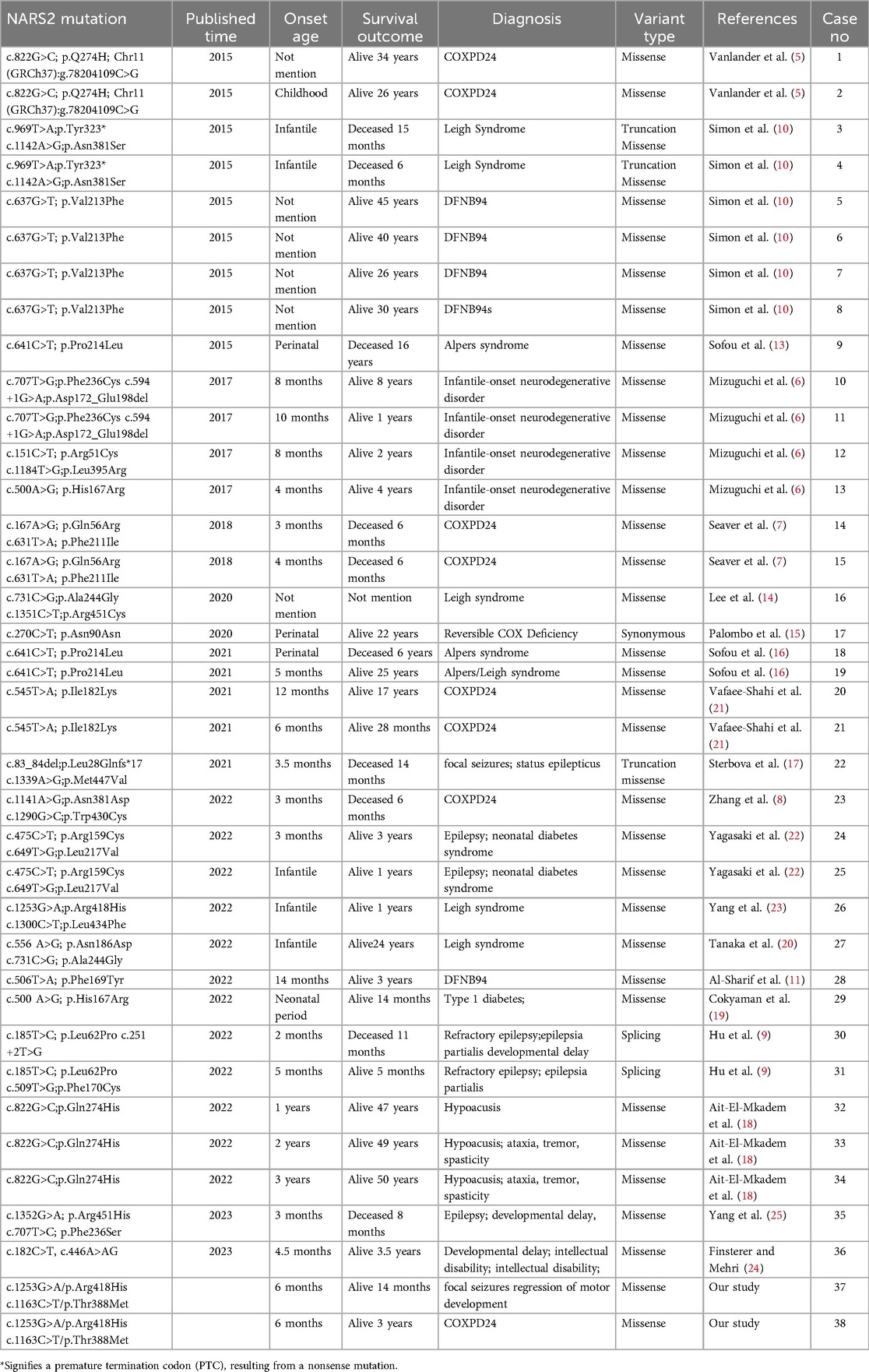
Table 1. Patients with mitochondrial disorders due to NARS2 variants reported as of the end of December 2024.
We report a case of two siblings who carry identical compound heterozygous pathogenic mutations in the NARS2 gene, yet their prognoses are different because the treatment regimens applied to them vary. Both of them presented with neurological manifestations, such as refractory epilepsy, developmental delay, and motor developmental regression within the first year of life, while MRI showed symmetrical brain lesions and neither of them had impaired hearing. The slight difference was that the elder sister had generalized epilepsy with myoclonic seizures, while the younger brother had focal seizures. In our study, Patient 2 received prompt mitochondrial drug cocktail therapy shortly after the disease etiology was identified, which effectively controlled his epilepsy without the need for excessive antiepileptic drugs. Notably, compared to Patient 1, Patient 2 exhibited less pronounced neurodevelopmental regression. Mitochondrial cocktail therapy refers to a combination of supplements, such as coenzyme Q10, L-carnitine, and B vitamins, designed to enhance mitochondrial function, improve energy production, and reduce oxidative stress, primarily used in the treatment of mitochondrial and metabolic disorders (26–28). The positive response to mitochondrial drug cocktail therapy underscores the role of mitochondrial dysfunction in epileptogenesis, implicating NARS2 mutations as a key pathogenic mechanism. NARS2 encodes a mitochondrial aminoacyl-tRNA synthetase crucial for protein synthesis, and its mutations disrupt energy metabolism, leading to epilepsy. This therapeutic success further supports NARS2-related mitochondrial dysfunction as central to the disorder.
The c.1163C>T(p.Thr388Met) is a newly discovered variant in the NARS2 gene, which enrich our comprehension of the mutational spectrum of NARS2, enabling a more profound exploration into its intricacies. As invaluable additions to the gene mutation database, they lay a solid foundation for theoretical constructs that will underpin the development and implementation of precise gene therapeutic strategies in the future.
Previously, there had been report (23) of patient with the same mutation site, c.1253G>A (p.Arg418His), presenting with hyperlactatemia and myocardial dysfunction. However, our patient did not exhibit the same clinical phenotype. This striking demonstration of genetically identical variants resulting in different clinical manifestations underscores the intricate relationship between genotype and phenotype in NARS2-associated disorders, echoing previous observations in the literature (5). We posit that this phenotypic variability stems from a complex interplay of multiple factors, encompassing the specific nature and location of the mutations, as well as their differential effects on the functionality of tRNA synthetase. This finding is congruent with the intricate and multifaceted nature of other mitochondrial disorders, where genetic variations often lead to a diverse spectrum of clinical manifestations, underscoring the importance of nuanced and comprehensive genotype-phenotype analyses in these complex disease processes (29).
The NARS2 encodes a protein of 477 amino acids that contains an anticodon binding domain (amino acids 44–118) and a catalytic domain (amino acids 135–472) of tRNAAsn (http://www.uniprot.org/uniprot/). Our study identified two novel NARS2 mutations, including the two missense mutations Arg418His and Thr388Met located in the catalytic domain (Figure 6). Although the comprehensive exploration of the pathophysiological implications of the detected compound heterozygous variants remains to be undertaken, parallels drawn from similar NARS2 deficiencies hint at the likelihood that homozygous or compound heterozygous variants of NARS2 lead to diminished enzyme synthesis, impaired translocation into mitochondria, compromised asparaginase ligation to tRNA molecules, and a cumulative oxidative phosphorylation insufficiency, collectively referred to as Combined Oxidative Phosphorylation Deficiency 24 (3). We posit that the mutation c.1253G>A, resulting in the amino acid substitution p.Arg418His within exon 12, involves a crucial shift from the phylogenetically conserved non-aromatic arginine (Arg) to the aromatic histidine (His). This transition is hypothesized to diminish the stability of the NARS2 protein dimer through reduced binding free energy, with potential ramifications for the normal function of the NARS2 enzyme in biological pathways. The variant c.1163C>T, leading to the amino acid substitution p.Thr388Met within exon 11, involves a transformation from the polar amino acid threonine to the non-polar methionine. This substitution may elicit a structural reconfiguration within the AsnRS homodimer, potentially altering its conformation and impacting the intricate arrangements necessary for optimal enzyme function. The PolyPhen program predicted the c.1253G>A and c.1163C>T mutation of NARS2 to be “probably damaging” and the SIFT program anticipated these mutations to be “deleterious” (Table 2). Notably, these mutations were absent from any publicly accessible databases of single nucleotide polymorphisms (SNPs), further emphasizing their rarity and potential significance.
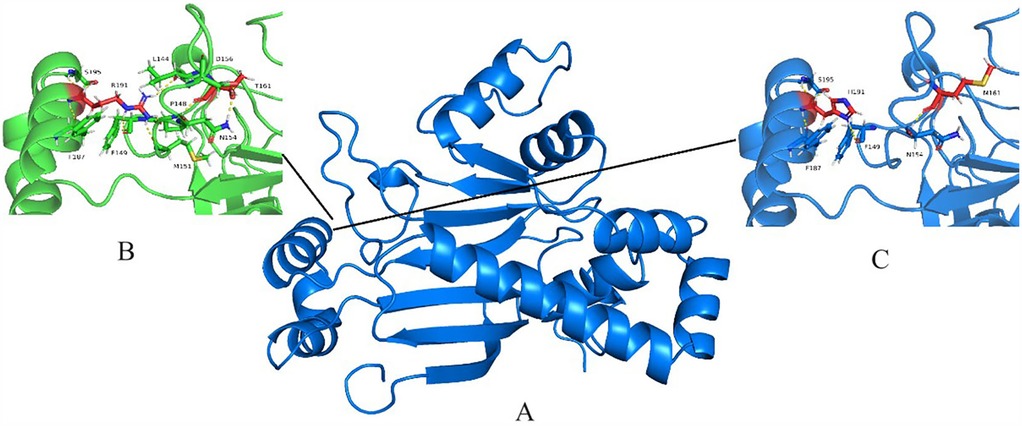
Figure 6. NARS2 protein structural modeling. (A) Alphafold2-predicted Asn-RS homodimer. (B) Close-up of wild-type region. (C) Mutated residues (yellow/green) in monomeric structure.

Table 2. NARS2 mutation information in this study.
In this rigorous medical study, we have identified novel compound mutations in the NARS2 gene that display a striking correlation with a diverse spectrum of clinical phenotypes. Although predictive computational tools, such as PolyPhen and SIFT, offer valuable preliminary assessments of the potential effects of these variants, definitive functional validation remains the gold standard for determining their mutational status. However, it is imperative to acknowledge certain limitations in our investigation. Due to the patient family's reluctance to undergo invasive diagnostic procedures, we were unable to directly assess mitochondrial respiratory chain function in patient-derived tissue specimens, thereby limiting the extent of our correlation analysis between the identified mutations and their underlying cellular functional consequences. Moreover, the lack of subcellular localization experiments and the inability to empirically verify the impact of these novel mutations on protein dimerization represent further constraints. Despite these limitations, the findings of this research represent a substantial contribution to our understanding of the mutational landscape of the NARS2 gene and its intricate associations with a broad range of clinical presentations.
In conclusion, our study notably expands the phenotypic and genotypic spectrum of NARS2 disorders through the identification of a novel disease-causing variant in two siblings. For pediatric refractory epilepsy, early genetic screening may expedite diagnosis and inform prognosis, facilitating personalized treatment. Additionally, to progress targeted therapies for NARS2-related diseases, intensified research is imperative to elucidate the intricate mechanisms underlying the complex effects of NARS2 variants on mitochondrial protein synthesis.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, Bioproject accession number: PRJNA1170432.
The studies involving humans were approved by the Medical Ethics Committee of the Seventh Affiliated Hospital of Sun Yat-sen University (No. KY-2024-320-01). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
HW: Data curation, Formal analysis, Methodology, Project administration, Writing – original draft, Writing – review & editing. MZ: Writing – original draft. XY: Writing – review & editing. HC: Writing – review & editing. HL: Writing – review & editing. LW: Writing – review & editing. XN: Supervision, Writing – review & editing. LZ: Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by funds from Shenzhen Science and Technology Program (No. JCYJ20220530145001002).
The authors thank the patients and their family for their kind cooperation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
mt-ARSs, mitochondrial aminoacyl-tRNA synthetases; NARS2, mitochondrial asparaginyl-tRNA synthetase; MRI, magnetic resonance imaging; COXPD24, Combined Oxidative Phosphorylation Deficiency 24; DFNB94, Autosomal recessive deafness-94; EEG, Electroencephalogram; NDD, neurodegenerative disorder; Arg, arginine; His, histidine.
1. Zhu Z, Wang X. Significance of mitochondria DNA mutations in diseases. Adv Exp Med Biol. (2017) 1038:219–30. doi: 10.1007/978-981-10-6674-0_15
2. Lim A, Thomas RH. The mitochondrial epilepsies. Eur J Paediatr Neurol. (2020) 24:47–52. doi: 10.1016/j.ejpn.2019.12.021
3. Rajendran V, Kalita P, Shukla H, Kumar A, Tripathi T. Aminoacyl-tRNA synthetases: structure, function, and drug discovery. Int J Biol Macromol. (2018) 111:400–14. doi: 10.1016/j.ijbiomac.2017.12.157
4. Fine AS, Nemeth CL, Kaufman ML, Fatemi A. Mitochondrial aminoacyl-tRNA synthetase disorders: an emerging group of developmental disorders of myelination. J Neurodev Disord. (2019) 11(1):29. doi: 10.1186/s11689-019-9292-y
5. Vanlander AV, Menten B, Smet J, De Meirleir L, Sante T, De Paepe B, et al. Two siblings with homozygous pathogenic splice-site variant in mitochondrial asparaginyl-tRNA synthetase (NARS2). Hum Mutat. (2015) 36(2):222–31. doi: 10.1002/humu.22728
6. Mizuguchi T, Nakashima M, Kato M, Yamada K, Okanishi T, Ekhilevitch N, et al. PARS2 and NARS2 mutations in infantile-onset neurodegenerative disorder. J Hum Genet. (2017) 62(5):525–9. doi: 10.1038/jhg.2016.163
7. Seaver LH, Deroos S, Andersen NJ, Betz B, Prokop J, Lannen N, et al. Lethal NARS2-related disorder associated with rapidly progressive intractable epilepsy and global brain atrophy. Pediatr Neurol. (2018) 89:26–30. doi: 10.1016/j.pediatrneurol.2018.07.014
8. Zhang Y, Zhao X, Xu Y, Chen L, Li N, Yao R, et al. Study of novel NARS2 variants in patient of combined oxidative phosphorylation deficiency 24. Transl Pediatr. (2022) 11(4):448–57. doi: 10.21037/tp-21-570
9. Hu W, Fang H, Peng Y, Li L, Guo D, Tang J, et al. Clinical and genetic analyses of premature mitochondrial encephalopathy with epilepsia partialis continua caused by novel biallelic NARS2 mutations. Front Neurosci. (2022) 16:1076183. doi: 10.3389/fnins.2022.1076183
10. Simon M, Richard EM, Wang X, Shahzad M, Huang VH, Qaiser TA, et al. Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and leigh syndrome. PLoS Genet. (2015) 11(3):e1005097. doi: 10.1371/journal.pgen.1005097
11. Al-Sharif F, Alsadeq H, Rozan A, Halabi MB, Badwilan H, Mohammed AA, et al. Bilateral nonsyndromic sensorineural hearing loss caused by a NARS2 mutation. Cureus. (2022) 14(11):e31467. doi: 10.7759/cureus.31467
12. Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, et al. Mitochondrial disease: a practical approach for primary care physicians. Pediatrics. (2007) 120(6):1326–33. doi: 10.1542/peds.2007-0391
13. Sofou K, Kollberg G, Holmstrom M, Davila M, Darin N, Gustafsson CM, et al. Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with alpers syndrome. Mol Genet Genomic Med. (2015) 3(1):59–68. doi: 10.1002/mgg3.115
14. Lee JS, Yoo T, Lee M, Lee Y, Jeon E, Kim SY, et al. Genetic heterogeneity in leigh syndrome: highlighting treatable and novel genetic causes. Clin Genet. (2020) 97(4):586–94. doi: 10.1111/cge.13713
15. Palombo F, Graziano C, Al WN, Nouri N, Marconi C, Magini P, et al. Autozygosity-driven genetic diagnosis in consanguineous families from Italy and the greater Middle East. Hum Genet. (2020) 139(11):1429–41. doi: 10.1007/s00439-020-02187-7
16. Sofou K, Kollberg G, Hedberg-Oldfors C, Oldfors A. The phenotypic variability and natural history of NARS2 associated disease. Eur J Paediatr Neurol. (2021) 31:31–7. doi: 10.1016/j.ejpn.2021.01.012
17. Sterbova K, Vlckova M, Hansikova H, Sebronova V, Sedlackova L, Pavlicek P, et al. Novel variants in the NARS2 gene as a cause of infantile-onset severe epilepsy leading to fatal refractory status epilepticus: case study and literature review. Neurogenetics. (2021) 22(4):359–64. doi: 10.1007/s10048-021-00659-0
18. Ait-El-Mkadem SS, Kaphan E, Morales JA, Fragaki K, Chaussenot A, Bannwarth S, et al. Splicing variants in NARS2 are associated with milder phenotypes and intra-familial variability. Eur J Med Genet. (2022) 65(12):104643. doi: 10.1016/j.ejmg.2022.104643
19. Cokyaman T, Cetin H, Dogan D, Silan F. A new entity in the NARS2 variant: the first reported case of type 1 diabetes mellitus associated with the phenotype. J Trop Pediatr. (2022) 69(1):fmac108. doi: 10.1093/tropej/fmac108
20. Tanaka R, Takeguchi R, Kuroda M, Suzuki N, Makita Y, Yanagi K, et al. Novel NARS2 variant causing leigh syndrome with normal lactate levels. Hum Genome Var. (2022) 9(1):12. doi: 10.1038/s41439-022-00191-z
21. Vafaee-Shahi M, Farhadi M, Razmara E, Morovvati S, Ghasemi S, Abedini SS, et al. Novel phenotype and genotype spectrum of NARS2 and literature review of previous mutations. Ir J Med Sci. (2022) 191(4):1877–90. doi: 10.1007/s11845-021-02736-7
22. Yagasaki H, Sano F, Narusawa H, Watanabe D, Kaga Y, Kobayashi K, et al. Compound heterozygous variants of the NARS2 gene in siblings with developmental delay, epilepsy, and neonatal diabetes syndrome. Am J Med Genet A. (2022) 188(8):2466–71. doi: 10.1002/ajmg.a.62873
23. Yang Z, Cao J, Song Y, Li S, Jiao Z, Ren S, et al. Whole-exome sequencing identified novel variants in three Chinese leigh syndrome pedigrees. Am J Med Genet A. (2022) 188(4):1214–25. doi: 10.1002/ajmg.a.62641
24. Finsterer J, Mehri S. Progressive mitochondrial encephalopathy due to the novel compound heterozygous variants c.182C>T and c.446A>ag in NARS2: a case report. Cureus. (2023) 15(8):e43969. doi: 10.7759/cureus.43969
25. Yang N, Chen L, Zhang Y, Wu X, Hao Y, Yang F, et al. Novel NARS2 variants in a patient with early-onset status epilepticus: case study and literature review. BMC Pediatr. (2024) 24(1):96. doi: 10.1186/s12887-024-04553-0
26. Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. (2012) 2012(4):CD004426. doi: 10.1002/14651858.CD004426.pub3
27. Fukui KO, Kubota M, Terashima H, Ishiguro A, Kashii H. Early administration of vitamins B1 and B6 and l-carnitine prevents a second attack of acute encephalopathy with biphasic seizures and late reduced diffusion: a case control study. Brain Dev. (2019) 41(7):618–24. doi: 10.1016/j.braindev.2019.02.015
28. Omata T, Aoyama H, Murayama K, Takayanagi M, Kawaguchi R, Fujimoto R, et al. Efficacy of a mitochondrial drug cocktail in preventing acute encephalopathy with biphasic seizures and late reduced diffusion. J Neurol Sci. (2024) 466:123245. doi: 10.1016/j.jns.2024.123245
Keywords: combined oxidative phosphorylation deficiency 24 (COXPD24), NARS2, refractory epilepsy, biallelic variants, pediatric, mitochondrial drug cocktail therapy
Citation: Wu H, Zhou M, Ye X, Chen H, Lin H, Wang L, Nie X and Zhang L (2025) Compound heterozygous variants of the NARS2 gene in siblings with refractory seizures: two case report and literature review. Front. Pediatr. 13:1571426. doi: 10.3389/fped.2025.1571426
Received: 5 February 2025; Accepted: 24 March 2025;
Published: 8 April 2025.
Edited by:
Antonio Gennaro Nicotera, University of Messina, ItalyReviewed by:
Lorenzo Pavone, University of Catania, ItalyCopyright: © 2025 Wu, Zhou, Ye, Chen, Lin, Wang, Nie and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xing Nie, bmlleDIzQG1haWwuc3lzdS5lZHUuY24=; Lidan Zhang, emhsZGFuQG1haWwuc3lzdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.