
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 04 April 2025
Sec. Pediatric Gastroenterology, Hepatology and Nutrition
Volume 13 - 2025 | https://doi.org/10.3389/fped.2025.1562573
 Chiara Gagliano1Olga Burattini1Luigi Paradisi1Sarah Recchione1Lucia Santoro1Laura Caponi1Annamaria Ciaschini2Maria Elena Lionetti1
Chiara Gagliano1Olga Burattini1Luigi Paradisi1Sarah Recchione1Lucia Santoro1Laura Caponi1Annamaria Ciaschini2Maria Elena Lionetti1 Simona Gatti1*
Simona Gatti1*
Neonatal cholestasis can be caused by several conditions, with biliary atresia being the major cause. Genetic and endocrinological etiologies represent other possibilities, with most of them requiring a rapid diagnosis and a specific treatment. We describe a neonatal case of severe cholestasis with low gamma glutamyl transferase in a child presenting with multiple abnormalities, including pituitary stalk interruption syndrome and consequent hypopituitarism. The cholestasis was rapidly resolved with hormone therapy. Genetic analysis showed a de novo 17q chromosome deletion, including the HNF1β gene implicated in liver damage, and this was considered causative of the complex clinical phenotype. Our case highlights the relationship between congenital hypopituitarism and HNF1β gene deletion in 17q12 deletion syndrome as a severe neonatal cholestasis etiology, emphasizing the need to be especially vigilant in cases with associated hypoglycemia. Prompt endocrine evaluation and genetic testing are crucial in neonatal cholestasis to start targeted therapy and long-term monitoring, which could mitigate serious complications.
Neonatal cholestasis (NC), characterized by conjugated hyperbilirubinemia, is not a benign condition, and the severity of its underlying causes should be promptly investigated (1). Biliary atresia (BA), occurring in 35%–41% of cases, is the major cause, but many other surgical and medical conditions (some potentially treatable) should be quickly excluded, with metabolic and endocrinological disorders representing up to 7% of cases (1). Endocrine diseases underlying NC are associated with significant morbidity and mortality risk due to severe hypoglycemia, acute adrenal insufficiency, and secondary hypothyroidism and require rapid hormone replacement treatment. Many of these conditions, manifesting soon after birth, are the result of a congenital pituitary malformation and/or are related to a specific genetic defect. Pituitary stalk interruption syndrome (PSIS), which is overall a very rare malformation with a reported incidence of 0.5/100,000 (2), is one the most frequent radiological presentations of congenital hypopituitarism (CH). Recently, an increasing proportion of neonatal/infantile cholestasis, previously defined as “idiopathic neonatal hepatitis,” has been recognized as a monogenic liver disorder. Genetic investigations are currently recommended early after the exclusion of BA in the workup of NC, in parallel with a metabolic workup (3). We describe the workup and follow-up of an infantile case of severe cholestasis in a patient with syndromic features (including hypopituitarism secondary to PSIS) and a final diagnosis of 17q12 deletion syndrome (including the HNF1β gene). While adult liver disease has been well described as a consequence of a HNF1β gene mutation/deletion, NC has rarely been reported and never in association with PSIS. This novel case is compared with reported cases of NC and HNF1β gene mutations.
A 43-day-old boy born at term from non-consanguineous parents (birth weight of 3,080 g, 11th percentile; birth length of 49 cm, 13th percentile) was referred to our attention for persistent cholestatic jaundice (for the timeline, see Figure 1). His family history included autoimmune thyroid disorder in the mother. At 10 h of life, he developed tachypnea, severe hypoglycemia, and jaundice, requiring a dextrose infusion and antibiotics due to a suspicion of sepsis, with full recovery. At 1 month of age, blood tests revealed cholestatic liver disease [total bilirubin: 19.9 mg/dl, direct bilirubin: 9.6 mg/dl, aspartate aminotransferase (AST): 4,330 U/L, alanine aminotransferase (ALT) 705 U/L, gamma glutamyl transferase (GGT): 50 U/L, serum bile acids: 480 μmol/L]. There was no history of vomiting, diarrhea, or acholic stools. The baby was transferred to our Pediatric Gastroenterology Department for further management. At admission, he had icteric skin and sclera, an enlarged liver (2 cm below the right costal margin) without splenomegaly, and normal cardiac and neurological examinations. Minor dysmorphisms, including a prominent forehead, extruded tongue, and an appendix on his left fifth finger, were observed. His weight and length were below the third percentile. His stools appeared normochromic but were occasionally hypocolic. Cholestatic jaundice was confirmed with mild derangement of liver synthetic function [hypoalbuminemia: 2.6 g/dl, mild coagulopathy with international normalized ratio (INR) 1.56] and ammonia values at the upper limit for his age. Serum GGT levels were almost within normal limits (55 U/L, normal 0–73 U/L). Plasma glucose level was frequently at the lower limit of normal (nadir: 36 mg/dl).
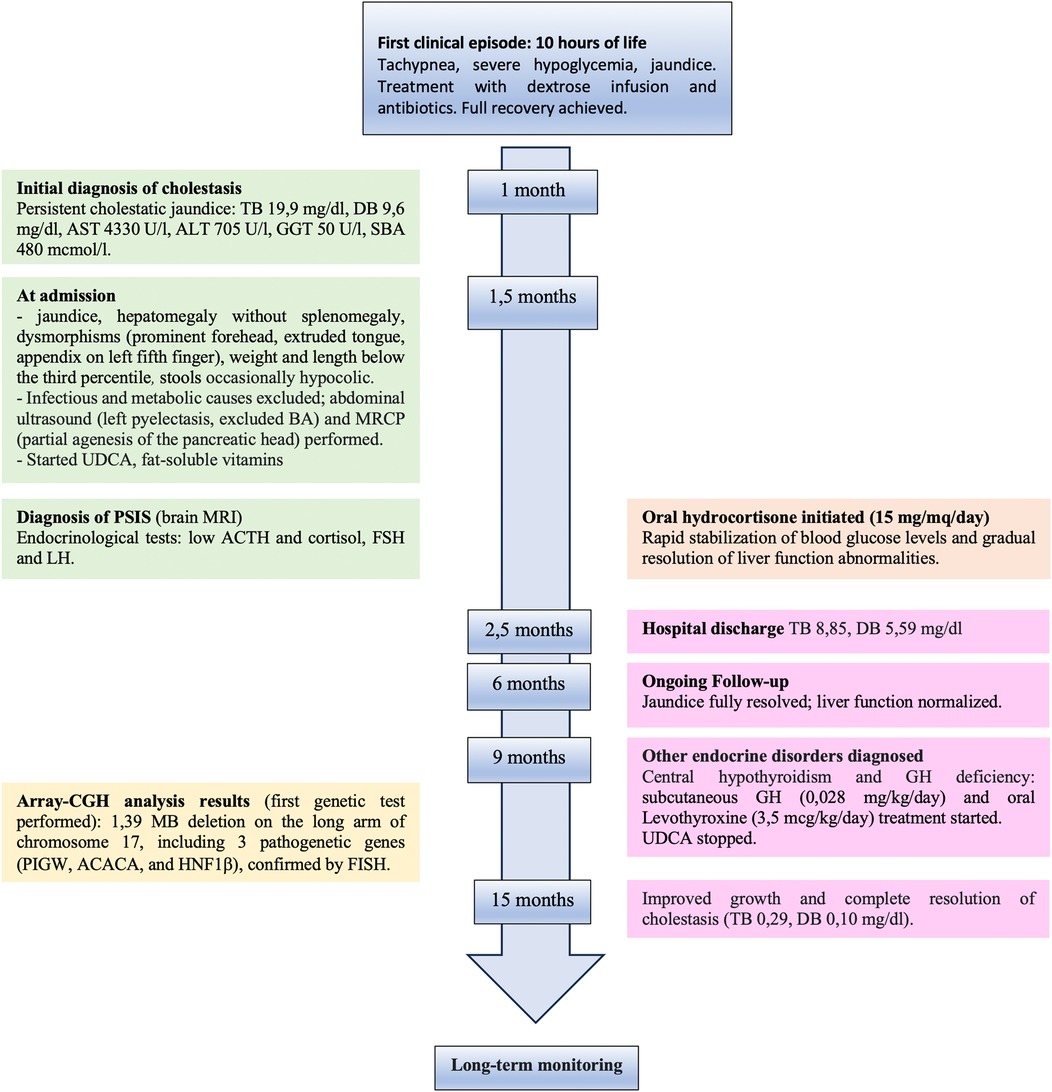
Figure 1. Timeline with chronological data and events from first clinical episode of severe hypoglycemia and hyperbilirubinemia to complete resolution after hormone replacement treatment. TB, total bilirubin; DT, direct bilirubin; AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, gamma glutamyl transferase; SBA, serum bile acid; BA, biliary atresia; MRCP, magnetic resonance cholangiopancreatography; UDCA, ursodeoxycholic acid; PSIS, pituitary stalk interruption syndrome; MRI, magnetic resonance imaging.
The principal infective causes (viral hepatitis, toxoplasmosis, parasites, and bacterial infections) and metabolic liver diseases (normal urine organic acids, alpha1-antitrypsin, galactosemia, plasmatic amino acids profile) were quickly excluded. An abdominal ultrasound demonstrated left pyelectasis, a distended edematous gallbladder, and hepatomegaly with hyperechoic parenchyma, with normal-appearing bile ducts. The absence of acholic stools and the low levels of GGT did not suggest the possibility of a biliary atresia. Magnetic resonance cholangiopancreatography (MRCP) showed partial agenesis of the pancreatic head; these findings were not considered explanatory of the laboratory abnormalities. The endocrinological workup revealed low cortisol and adrenocorticotropic hormone (ACTH) levels (0.7 μg/dl and 2 pg/ml, respectively) with undetectable luteinizing hormone (LH) and follicle-stimulating hormone (FSH) (LH <0.2 mU/ml, FSH <0.2 mU/ml). Thyroid function tests and growth hormone (GH) levels were within the norms. Prolactin values were elevated (45.3 ng/ml). A head magnetic resonance imaging (MRI) scan demonstrated an absent pituitary stalk and an ectopic posterior pituitary, compatible with PSIS. During hospitalization, IV fluids, oral ursodeoxycholic acid, and fat-soluble vitamins were started; albumin and packed red blood cells were transfused. Oral hydrocortisone (15 mg/mq/day) was initiated, with a rapid improvement in the patient's general clinical conditions, a quick stabilization of blood glucose levels, and gradual improvement of liver function tests. At hospital discharge (2.5 months of age), total and direct bilirubin were 8.85 and 5.59 mg/dl, respectively. At 6 months, the jaundice had fully disappeared, and clinical and biological parameters associated with cholestasis were resolved. At 9 months, his weight and length remained below the 3rd percentile. Repeated endocrinological tests showed central hypothyroidism [thyroid-stimulating hormone (TSH): 0.460 μU/ml; T4: 0.87 ng/dl] and GH deficiency [GH: 0.92 ng/ml, insulin-like growth factor 1 (IGF-1) <15 μg/L]; therefore, subcutaneous GH (0.028 mg/kg/day) and oral levothyroxine (3.5 μg/kg/day) were started. Considering the presence of multiple malformations, array-comparative genomic hybridization (aCGH) analysis was performed as the initial genetic test, and a 1.39 MB deletion was detected on the long arm of chromosome 17 (17q12), containing the HNF1β gene (Figure 2). The deletion was confirmed by fluorescence in situ hybridization (FISH) and was not found in the parents; therefore, it was considered causative of the clinical phenotype. At 15 months of age, the child showed improved growth (in the 3rd percentile) and a complete clinical and laboratory resolution of cholestasis was observed (Table 1). Repeated abdominal ultrasounds confirmed the partial agenesis of the pancreatic head and left pyelectasis; pancreatic elastase was within the normal range.
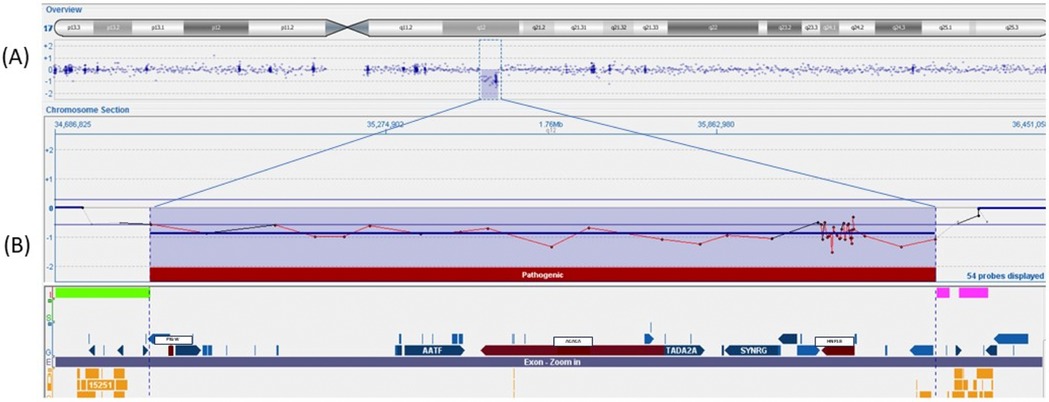
Figure 2. Array-CGH 400K analysis of the patient in which a deletion [arr(hg19) 17q12(34856305_36248926)x1] of 1.39 Mb in the region17q12 was identified. (A) Overview of the genes (refSeq) included in this deleted region. (B) The region contains 13 genes, and the three pathogenetic genes (PIGW, ACACA, and HNF1β) are marked in red.
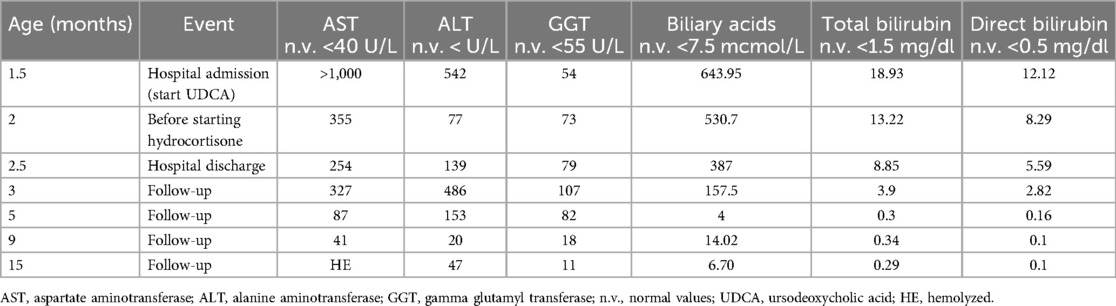
Table 1. Results of liver function tests during hospital stay and at follow-ups.
We have reported a case of severe NC caused by HNF1β deficiency related to CH, with a favorable outcome after hormone replacement therapy. This case report is the first to describe the combination of these two clinical entities (CH and NC) in the context of 17q12 deletion syndrome. Only two previous reports have described the association of CH with chromosome 17q deletion (4, 5), and a total of seven cases, apart from ours, have reported NC with this genetic defect (related to the deletion of the HNF1β gene) (Table 2) (6–12).
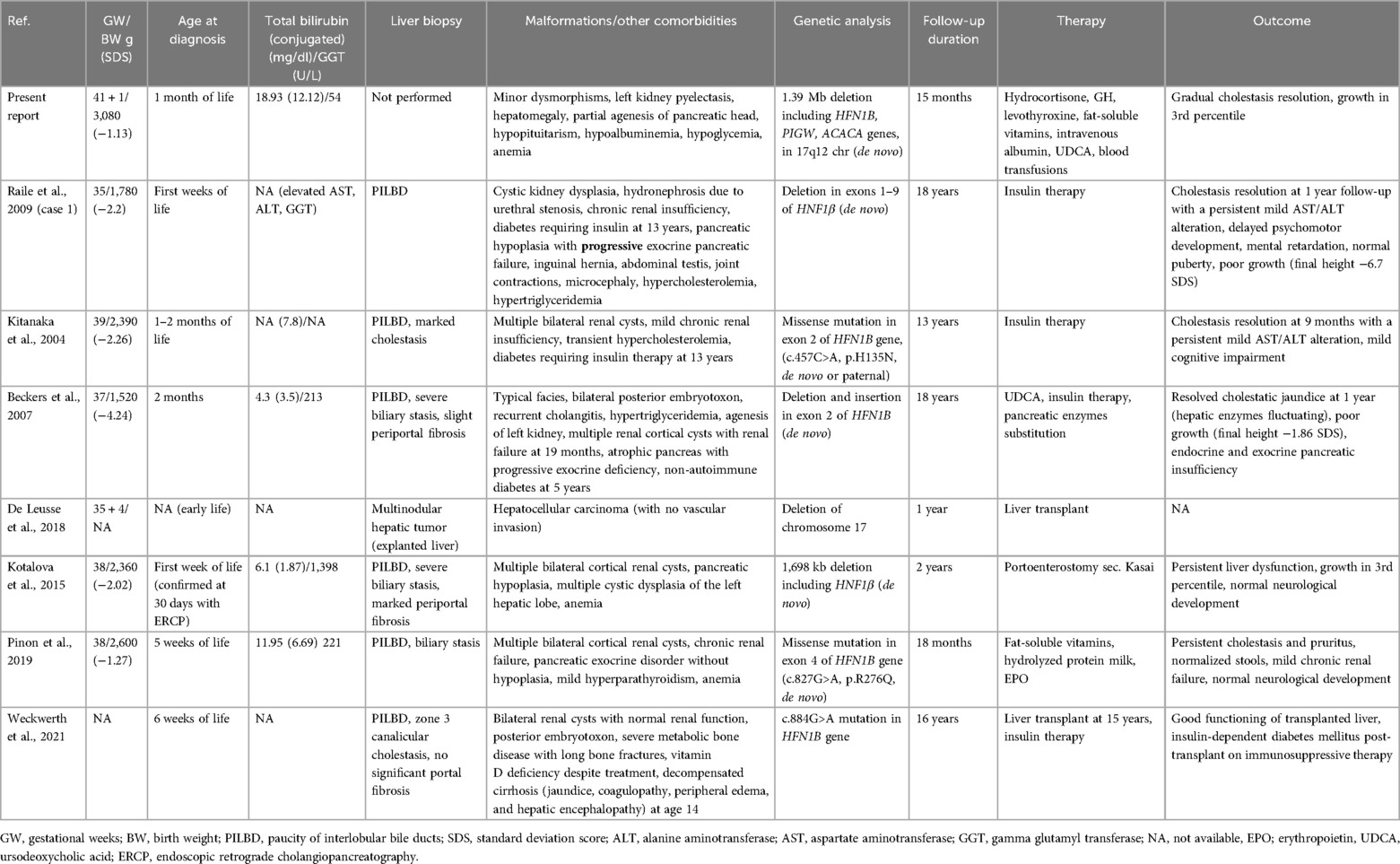
Table 2. Clinical and biochemical features of patients with HNF1β gene mutation-related disease and neonatal cholestasis.
Our clinical report underlines the importance of evaluating endocrine disorders as one of the etiologies of NC. In a systematic review of 1,692 infants with NC, endocrinological disorders represented less than 2% of cases (1). PSIS is a rare developmental pituitary defect, generally presenting at birth or in the first months of life with hypoglycemia, failure to thrive, jaundice, cryptorchidism, and seizures (2). Cholestasis is frequently reported in patients with PSIS, with a prevalence ranging from 6% to 42% in different studies (13). It has been suggested that a decreased plasma cortisol level might be the precipitating factor for cholestasis, even if an effective role of GH and/or the TSH axis cannot be excluded (14). Both the type of liver derangement in our patient, characterized by very high aminotransferases and bilirubin with almost normal GGT, and the timing of resolution were in line with data from the literature, describing normal/low GGT in up to 43%–57% of cholestasis associated with CH, and resolution between 2 and 9 months after replacement therapy initiation (15). We confirm, therefore, that hormone replacement therapy is the basis of treatment for liver cholestatic associated with CH.
CH has been related to known genetic causes in only a small proportion of cases with non-syndromic and syndromic etiologies (16). PSIS, characterized by a triad of a thin/absent pituitary stalk, ectopic posterior pituitary, and aplasia/hypoplasia of the anterior pituitary, is a radiological diagnosis contributing to a high proportion (11.8%–34.2%) of CH cases (17).
The exact etiology of PSIS remains unclear with elusive underlying mechanisms. Many theories were initially proposed, such as adverse perinatal events affecting the hypothalamic-pituitary axis (4). Recent findings suggest genetic origins involving pituitary development, neural development, axonal migration, and other important cellular processes that may act as predisposing factors, combined with environmental effects (2, 18).
CH patients with consistent/syndromic features, family history, or consanguineous parents are candidates for genetic testing, typically through next-generation sequencing (NGS), although some mutations may be detectable on array-CGH. In accordance with a recent study (18), PSIS should be considered part of the phenotypic spectrum of many genetic syndromes; therefore, an exome sequencing approach can more accurately characterize the genetic basis, compared to single gene analysis, retrieving genetic mutations in only 5% of PSIS cases (16).
Our initial genetic approach was based on array-CGH, which showed a de novo 17q12 deletion. The frequency of 17q12 deletion syndromes is estimated to be approximately 1:20.000. The deletion of 17q12 includes several genes, and among these, the HNF1β gene [related to maturity-onset diabetes of the young type 5 (MODY5)] is the best characterized. As in our case report showing left pyelectasis, urinary tract abnormalities have been detected in more than 50% of patients with HNF1β deficiency, with a significant heterogeneity. Neurodevelopmental disorders can occur, especially in large deletions as the one reported in the present case (19, 20). Other typical features include endocrinological disorders, with 25%–50% of patients having MODY5 and hyperparathyroidism; hypomagnesemia and hyperuricemia have also been reported (20). Pancreatic atrophy develops in up to 30% of cases, with the dorsal pancreas generally being more involved (20). Our patient showed early radiological evidence of pancreatic involvement (limited to the pancreatic head) with no signs of pancreatic insufficiency or dysfunction, suggesting the importance of a long-term endocrine follow-up and strict monitoring of pancreatic insufficiency. Mutations or deletions of the HNF1β gene have been described in correlation with different hepatic phenotypes, including NC, adult-onset cholestasis, non-cholestatic liver disease (6–12, 20–22), and, more recently, pediatric hepatocellular carcinoma (9, 23). The cholestasis resolved in all the described neonatal cases, except for one patient who developed hepatocellular carcinoma (9) and one who required a liver transplantation (12). Although our patient fulfilled most of the criteria of 17q12 syndrome, some peculiar features need to be noted, including the association with PSIS, the consequent multiple hormone deficiencies, and the severity of liver impairment.
In the literature, only one report has identified 17q12 deletion (including HNF1β gene) as a pathogenetic copy number variation (CNV), assessed with aCGH, in a non-syndromic patient with PSIS and isolated growth hormone deficiency. There is no reported evidence of CH with or without PSIS in syndromic patients with 17q12 deletion (5).
Another study described an association between 17q21 deletion and PSIS with growth hormone deficiency (GHD) and gonadotropic deficiency but no corticotropic deficiency (4).
The degree of cholestasis was probably the consequence of different etiologies (the pituitary insufficiency and deletion of HNF1β). This is emphasized by the singular biochemical profile of our case with low GGT, which is not reported in HNF1β deficiency (24) but is typical in CH. In our workup of the cholestasis, considering the early and convincing diagnosis of CH, we decided not to perform a liver biopsy; therefore, we cannot completely exclude an underlying histological abnormality of the liver. All the reported neonatal cases showed a paucity of biliary ducts associated with marked cholestasis and variable periportal fibrosis (Table 2). All the cases, except one (9), had concomitant renal malformations (typically multiple renal cysts with a variable degree of chronic renal insufficiency). Similar to our case, pancreas agenesis or hypoplasia was reported in three cases. Four patients subsequently developed diabetes, requiring insulin therapy at 5 years, 13 years, and 15 years, respectively (7, 8, 12). Our patient did not show developmental delay at the last follow-up, while a mild cognitive impairment was found in two patients at their last follow-up (13 and 18 years) (6, 7); however, no brain imaging was reported.
The genetic definition of our case was rapidly achieved by an array-CGH test. Although this was resolutive, we cannot exclude the presence of other genetic mutations that could be revealed by an exome sequencing study.
It is relevant to stress the importance of a periodic biochemical and liver ultrasound follow-up in our patient, even if there was a complete resolution of the cholestasis. In parallel, a high index of suspicion for the development of diabetes and exocrine pancreatic insufficiency should be constantly maintained considering the presence of pancreatic hypoplasia and the concomitant lifelong necessity of steroid replacement in a patient with this genetic predisposition.
In conclusion, our case suggests the possibility of a double etiology for neonatal cholestasis in patients with 17q12 deletion syndrome, including the deletion of the HFN1β gene and concomitant pituitary insufficiency. In the workup of NC, when there is a suspicion for HNF1β deficiency (particularly in patients with syndromic features and severe liver involvement) and NGS is negative, array-CGH or multiplex ligation-dependent probe amplification (MLPA) could be indicated.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The requirement of ethical approval was waived as it was not applicable for this article. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
CG: Data curation, Investigation, Writing – original draft, Writing – review & editing. OB: Data curation, Investigation, Writing – original draft, Writing – review & editing. LP: Data curation, Writing – original draft. SR: Data curation, Writing – original draft. LS: Conceptualization, Supervision, Writing – review & editing. LC: Conceptualization, Supervision, Writing – review & editing. AC: Formal analysis, Investigation, Writing – review & editing. ML: Supervision, Writing – review & editing. SG: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gottesman LE, Del Vecchio MT, Aronoff SC. Etiologies of conjugated hyperbilirubinemia in infancy: a systematic review of 1,692 subjects. BMC Pediatr. (2015) 15:192. doi: 10.1186/s12887-015-0506-5
2. Vergier J, Castinetti F, Saveanu A, Girard N, Brue T, Reynaud R. Diagnosis of endocrine diseases: pituitary stalk interruption syndrome: etiology and clinical manifestations. Eur J Endocrinol. (2019) 181:199–209. doi: 10.1530/EJE-19-0168
3. Ranucci G, Della Corte C, Alberti D, Bondioni M, Boroni G, Calvo P, et al. Diagnostic approach to neonatal and infantile cholestasis: a position paper by the SIGENP liver disease working group. Dig Liv Dis. (2022) 54(1):40–53. doi: 10.1016/j.dld.2021.09.011
4. Chehadeh-Djebbar S, Callier P, Masurel-Paulet A, Bensignor C, Mejean N, Payet M, et al. 17q21.31 microdeletion in a patient with pituitary stalk interruption syndrome. Eur J Med Genet. (2011) 54:369–73. doi: 10.1016/j.ejmg.2011.03.001
5. Correa-Silva SR, Kunii I, Mitne-Neto M, Moreira CM, Dias-da Silva MR, Abucham J. Copy number variation in pituitary stalk interruption syndrome: a large case series of sporadic non-syndromic patients and literature review. J Neuroendocrinol. (2023) 35:13221. doi: 10.1111/jne.13221
6. Raile K, Klopocki E, Holder M, Wessel T, Galler A, Deiss D, et al. Expanded clinical spectrum in hepatocyte nuclear factor 1b-maturity-onset diabetes of the young. J Clin Endocrinol Metab. (2009) 94:2658–64. doi: 10.1210/jc.2008-2189
7. Kitanaka S, Miki Y, Hayashi Y, Igarashi T. Promoter-specific repression of hepatocyte nuclear factor (HNF)-1 beta and HNF-1 alpha transcriptional activity by an HNF-1 beta missense mutant associated with type 5 maturity-onset diabetes of the young with hepatic and biliary manifestations. J Clin Endocrinol Metab. (2004) 89:1369–78. doi: 10.1210/jc.2003-031308
8. Beckers D, Bellanné-Chantelot C, Maes M. Neonatal cholestatic jaundice as the first symptom of a mutation in the hepatocyte nuclear factor-1beta gene (HNF-1beta). J Pediatr. (2007) 150:313–4. doi: 10.1016/j.jpeds.2006.12.006
9. De Leusse C, Maues De Paula AM, Aschero A, Parache C, Hery G, Cailliez M, et al. Hepatocarcinoma and cholestasis associated to germline hemizygous deletion of gene HNF1β. J Pediatr Gastroenterol Nutr. (2019) 68:85. doi: 10.1097/MPG.0000000000002015
10. Kotalova R, Dusatkova P, Cinek O, Dusatkova L, Dedic T, Seeman T, et al. Hepatic phenotypes of HNF1β gene mutations: a case of neonatal cholestasis requiring portoenterostomy and literature review. World J Gastroenterol. (2015) 21:2550–7. doi: 10.3748/wjg.v21.i8.2550
11. Pinon M, Carboni M, Colavito D, Cisaro F, Peruzzi L, Pizzol A, et al. Not only Alagille syndrome. Syndromic paucity of interlobular bile ducts secondary to HNF1β deficiency: a case report and literature review. Ital J Pediatr. (2019) 45:27. doi: 10.1186/s13052-019-0617-y
12. Weckwerth JA, Dahl AR, Pittock ST, Kumar S, Rosen CB, Grothe RM, et al. Liver transplantation and development of diabetes in an adolescent male with HNF1β disease. JPGN Rep. (2021) 2(3):e085. doi: 10.1097/PG9.0000000000000085
13. Mauvais FX, Gonzales E, Davit-Spraul A, Jacquemin E, Brauner R. Cholestasis reveals severe cortisol deficiency in neonatal pituitary stalk interruption syndrome. PLoS One. (2016) 11:0147750. doi: 10.1371/journal.pone.0147750
14. Altay D, Eren E, Ozkan TB, Ozgur T, Tarim O. Liver involvement in congenital hypopituitarism. Indian J Pediatr. (2019) 86(5):412–6. doi: 10.1007/s12098-018-2833-7
15. Braslavsky D, Keselman A, Galoppo M, Lezama C, Chiesa A, Galoppo C, et al. Neonatal cholestasis in congenital pituitary hormone deficiency and isolated hypocortisolism: characterization of liver dysfunction and follow-up. Arq Bras Endocrinol Metabol. (2011) 55(8):622–7. doi: 10.1590/s0004-27302011000800017
16. De Rienzo F, Mellone S, Bellone S, Babu D, Fusco I, Prodam F, et al. Frequency of genetic defects in combined pituitary hormone deficiency: a systematic review and analysis of a multicentre Italian cohort. Clin Endocrinol. (2015) 83:849–60. doi: 10.1111/cen.12849
17. Kalina MA, Kalina-Faska B, Gruszczyńska K, Baron J, Małecka-Tendera E. Usefulness of magnetic resonance findings of the hypothalamic-pituitary region in the management of short children with growth hormone deficiency: evidence from a longitudinal study. Childs Nerv Syst. (2012) 28(1):121–7. doi: 10.1007/s00381-011-1594-7
18. Brauner R, Bignon-Topalovic J, Bashamboo A, McElreavey K. Pituitary stalk interruption syndrome is characterized by genetic heterogeneity. PLoS One. (2020) 15(12):e0242358. doi: 10.1371/journal.pone.0242358
19. Rasmussen M, Vestergaard EM, Graakjaer J, Petkov Y, Bache I, Fagerberg C, et al. 17q12 Deletion and duplication syndrome in Denmark-A clinical cohort of 38 patients and review of the literature. Am J Med Genet A. (2016) 170:2934–42. doi: 10.1002/ajmg.a.37848
20. Gambella A, Kalantari S, Cadamuro M, Quaglia M, Delvecchio M, Fabris L, et al. The landscape of HNF1β deficiency: a syndrome not yet fully explored. Cells. (2023) 12(2):307. doi: 10.3390/cells12020307
21. Dubois-Laforgue D, Cornu E, Saint-Martin C, Coste J, Bellanne-Chantelot C, Timsit J, et al. Diabetes, associated clinical spectrum, long-term prognosis, and genotype/phenotype correlations in 201 adult patients with hepatocyte nuclear factor 1B (HNF1β) molecular defects. Diabetes Care. (2017) 40(11):1436–43. doi: 10.2337/dc16-2462
22. Roehlen N, Hilger H, Stock F, Glaser B, Guhl J, Schmitt-Graeff A, et al. 17q12 deletion syndrome as a rare cause for diabetes mellitus type MODY5. J Clin Endocrinol Metab. (2018) 103(10):3601–10. doi: 10.1210/jc.2018-00955
23. Pinon M, Gambella A, Giugliano L, Chiado C, Kalantari S, Bracciama V, et al. New case of syncytial giant-cell variant of hepatocellular carcinoma in a pediatric patient with HNF1β deficiency: does it fit with the syndrome? BMJ Open Gastroenterol. (2022) 9(1):e001013. doi: 10.1136/bmjgast-2022-001013
Keywords: neonatal cholestasis, liver disease, congenital hypopituitarism, pituitary stalk interruption syndrome, 17q12 deletion, HNF1β
Citation: Gagliano C, Burattini O, Paradisi L, Recchione S, Santoro L, Caponi L, Ciaschini A, Lionetti ME and Gatti S (2025) Severe neonatal cholestasis in HNF1β deficiency: a case report and literature review. Front. Pediatr. 13:1562573. doi: 10.3389/fped.2025.1562573
Received: 17 January 2025; Accepted: 12 March 2025;
Published: 4 April 2025.
Edited by:
Valeria Dipasquale, University of Messina, ItalyReviewed by:
Michele Pinon, Ospedale Pediatrico Regina Margherita, ItalyCopyright: © 2025 Gagliano, Burattini, Paradisi, Recchione, Santoro, Caponi, Ciaschini, Lionetti and Gatti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simona Gatti, cy5nYXR0aUB1bml2cG0uaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.