 Yanping Huang
Yanping Huang Jin Wang
Jin Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 03 March 2025
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2025 | https://doi.org/10.3389/fped.2025.1532561
Adams-Oliver syndrome is a rare congenital disorder with six subtypes that have been identified. Subtypes 1, 3, 5, and 6 have an autosomal dominant inheritance pattern, whereas subtypes 2 and 4 have an autosomal recessive inheritance pattern. The clinical phenotype of Adams–Oliver syndrome is heterogeneous and can be accompanied by abnormalities in other organs, especially the cardiovascular system, such as cutis marmorata telangiectatica congenita, pulmonary hypertension, vascular abnormalities in other organs, and congenital heart defects. Herein, we report a case of Adams–Oliver syndrome caused by a de novo variant in DLL4. The patient was a neonate with clinical manifestations of skin defects who was diagnosed with Adams–Oliver syndrome on the basis of genetic testing.
Adams‒Oliver syndrome (AOS) is a disease characterized mainly by hypoplasia of the skin and terminal transected limb defects that may be accompanied by abnormalities of the cardiovascular and central nervous systems (1, 2). AOS is a type of congenital skin dysplasia (ACC), the mechanism of which is not completely clear, and genetic defects are one of its important causes. The currently known disease-associated genes include the DOCK6, ARHGAP31, RBPJ, NOTCH1 and DLL4 genes (3–5). Because the clinical manifestations of AOS vary widely, treatment needs to be individualized. Comprehensive management of AOS is particularly important.
This article reports a case of AOS that was diagnosed and treated by our team and summarizes the clinical and genetic characteristics of the disease through a review of the literature, aiming to improve the understanding of the disease.
The patient was a 7 h-old male born by cesarean section at 40 weeks' gestation, with a birth weight of 3,500 g. The Apgar scores at 1 min and 5 min were 8 and 9, respectively. The mother was healthy during pregnancy, with no abnormalities in the umbilical cord or placenta. After admission, an approximately 4.5*2.5 cm irregularly shaped area of skin loss was observed at the top of the head, and the surrounding skin was dark red (Figure 1A). All the fingers of both hands were shorter than other newborns of the same gestational age, and no other remarkable symptoms were observed.

Figure 1. Skin manifestations on the top of the head before (A) and after treatment (B).
Blood analysis revealed the following: white blood cell count, 21.84 × 109/L; hemoglobin concentration, 198 g/L; hematocrit, 60.8%; platelet count, 267 × 109/L; lymphocyte percentage, 20.8%; neutrophil percentage, 66.2%; monocyte percentage, 11.9%; and eosinophil percentage, 0.5%. The routine urine, stool, liver and kidney function, and electrolyte test results were normal. The C-reactive protein (CRP) level was 1.08 mg/L (0–3.0 mg/L), and the procalcitonin (PCT) level was 0.26 ng/ml. No pathogenic bacteria were detected in the blood or local secretion cultures. Tests for antibodies against cytomegalovirus, rubella virus, Toxoplasma gondii, herpes simplex virus, human immunodeficiency virus, hepatitis B virus, hepatitis C virus, and syphilis were negative. Head CT revealed a local defect at the top left of the scalp, slight thickening of the surrounding local soft tissue, no lesions with abnormal density in the parenchyma of the bilateral cerebral hemispheres, no expansion of the ventricular system, no ectopia in the midline structure, and no abnormalities in the skull bone window. Cardiac ultrasound revealed a patent ductus arteriosus (2.4 mm) and a central atrial septal defect (3.7 mm). A local skin biopsy of the scalp defect site revealed excessive keratosis of the squamous epithelium, thin epithelium, little inflammatory cell infiltration in the interstitium, necrosis, and calcified tissue. Ultrasound of the liver, spleen, and kidney revealed no abnormalities.
Trio whole-exome sequencing was performed for genetic testing in this study, and Sanger sequencing was performed for validation. A heterozygous variant in DLL4(NM_019074.4): c.2052 + 1G > A, which is an intronic donor splice site variant, was detected in the infant, and neither parent carried the variant (Figure 2). The variant was considered “likely pathogenic” according to the ACMG classification (6). The novel variant is a null variant (PVS1_Moderate); neither parent carried the mutation, which was considered to be a new mutation (PS2); and the incidence of variation in population databases is very low (PM2_Supporting). MaxEntScan was used for mRNA sequence prediction, this variant was predicted to affect the splice donor site and create a new splice donor. As a result, 86 bp may be inserted into the mRNA.
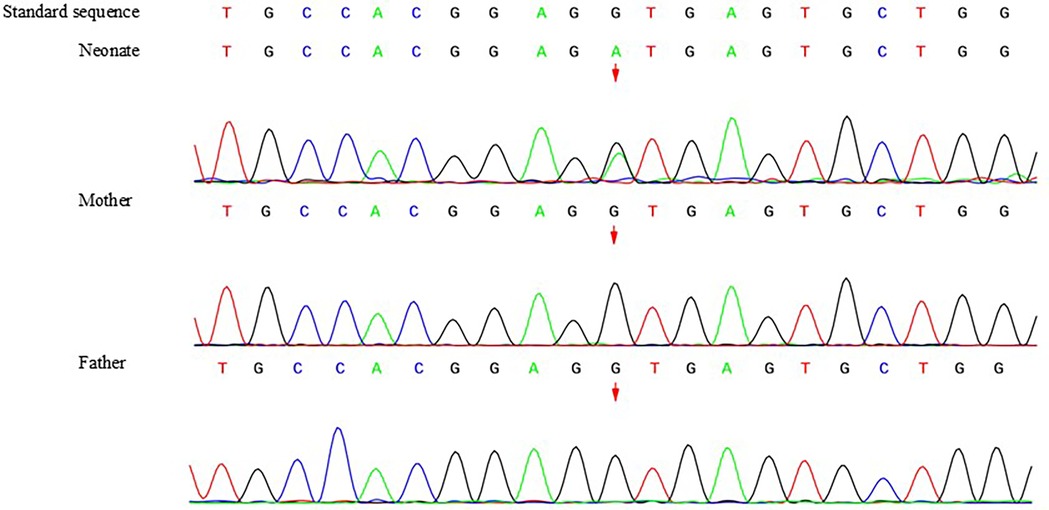
Figure 2. Sanger sequencing result of the whole family members. A heterozygous variant in DLL4(NM_019074.4): c.2052 + 1G > A, which is an intronic donor splice site variant, was detected in the infant, and neither parent carried the variant.
After admission, the child was treated with ampicillin and ceftazidime for anti-infection purposes (stopped after three days of continuous use), and topical drugs, including recombinant human epidermal growth factor topical solution (I) (Shenzhen Huashengyuan Bio, China), collagenase ointment (Liaoning Weibang Bio, China), and hydrocolloid dressing (Coloplast, Denmark), were applied after local debridement. Simultaneously, the scalp wound was contracted and folded for external fixation, and the wound gradually narrowed. After 2 weeks of hospitalization, the wound size was reduced to 2.5 cm*1.5 cm. After discharge, the external drugs were continued, and the wound had completely healed at 3 months after birth (Figure 1B).
ACC, also known as congenital absence of skin (CAS), refers to the absence of skin and/or subcutaneous tissue at birth; the cause of CAS is unclear and may be caused by intrauterine infection, chromosomal abnormalities, single-gene inheritance, or other reasons (1). Currently, nine types of congenital skin defects have been defined, including simple scalp congenital defects, scalp defects combined with limb abnormalities, congenital skin defects combined with papyriform fetuses, congenital skin defects combined with bullous epidermolysis, limb skin defects without bullous epidermolysis, congenital skin defects caused by intrauterine infection, congenital skin defects combined with other malformations, etc (1, 2). AOS is a type of ACC, and gene defects are important factors in AOS. The disease-associated genes identified to date include the DOCK6, ARHGAP31, RBPJ, NOTCH1, EOGT and DLL4 genes (7). AOS can be divided into six types according to genotype: type 1, caused by ARHGAP31 gene variation; type 2, caused by DOCK6 gene variation; type 3, caused by RBPJ gene variation; type 4, caused by EOGT gene variation; type 5, caused by NOTCH gene variation; and AOS, caused by DLL4 gene variation (7, 8).
The incidence of AOS is 1/225 000, and its main clinical feature is scalp dysplasia. Some children may have limb-end defects and cardiovascular development abnormalities. Defects of the autopod include brachydactyly, finger/toe oligodactyly, finger/toe syndactyly, finger/toenail dysplasia, and limb transection. Cardiovascular developmental abnormalities, including pulmonary hypertension, ventricular septal defects, tetralogy of Fallot, and abnormal development of the great arteries, have been detected (9). Terminal transverse limb defects are also more common in AOS caused by DLL4 gene variation, with brachydactyly, finger/toenail dysplasia, and limb transection being the most common defects. The manifestation of limb abnormalities in this study was shortened fingers. In the literature, children with cardiovascular developmental abnormalities have been reported, one child with a ventricular septal defect and one child with a loss of the pulmonary artery branch (10, 11). After admission, the patient underwent a complete cardiac ultrasound examination, which revealed the presence of a patent ductus arteriosus and central atrial septal defect. Reexamination via cardiac ultrasound at 3 months after birth revealed that both the ductus arteriosus and central atrial septal defect were closed, and no other obvious developmental abnormalities of the cardiovascular system were observed. In addition, two children with AOS were found in the literature, both of which presented with marbling skin changes, suggesting that skin manifestations should also be considered in children with AOS caused by a DLL4 gene variant (12, 13).
With increasing research, a preliminary understanding of AOS pathogenesis has been obtained. Among the genes associated with AOS, the NOTCH1, EOGT and RBPJ genes are members of the NOTCH signaling pathway, which is widely involved in the differentiation of neuronal stem cells, hematopoietic stem cells, and other important tissue differentiation processes (14–16). The NOTCH signaling pathway is composed mainly of NOTCH receptors, NOTCH ligands, DNA-binding proteins, and other effector and regulatory molecules. NOTCH receptors include NOTCH1–4, and NOTCH ligands are transmembrane proteins that include multiple regions, such as the N-terminal region, delta/serrate/lag-2 (DSL) region, epidermal growth factor (EGF)-like region, transmembrane region, and intracellular region. Activation of the NOTCH signaling pathway is affected mainly by serrate-like ligands (Jagged 1, Jagged 2) and delta-like ligands, and the DLL4 protein encoded by the DLL4 gene is a member of the delta ligand family. Therefore, the DLL4 gene plays an important role in the formation and differentiation of blood vessels (17–19).
The treatment of AOS focuses on the treatment of skin defects and other symptoms, including infection prevention and the use of topical drugs (3, 7, 11). The drugs used in other studies include recombinant human epidermal growth factor, convalescent ointments, and collagenase ointment (10–13). Owing to the difference in the area of skin lesions, recovery time varies greatly. In this study, recombinant human epidermal growth factor and collagenase ointment were used to treat skin defects, which were completely healed after continuous treatment for 3 months after birth. Other symptoms and signs should be treated according to the various clinical manifestations in children. Limb abnormalities and cardiac dysplasia are the most common abnormalities (10–13). Limb abnormalities can be treated with plastic surgery and functional exercise, whereas cardiac dysplasia often requires surgical treatment to improve cardiac function (1, 11). There are many types of abnormal cardiac development, including atrial septal defects, ventricular septal defects, and the absence of pulmonary arteries. In this study, no significant cardiac abnormalities were observed in the children.
The clinical manifestations of AOS are diverse, and the causative genes are different. Therefore, in clinical practice, genetic testing should be conducted in a timely manner for children with symptoms such as skin hypoplasia and terminal transection limb defects, and the test should focus on multiple genes, including the DLL4 gene, for early identification. The treatment of AOS caused by the DLL4 gene mainly involves infection prevention and topical drug use, and the therapeutic effect in most children is reasonable; for children with congenital heart malformations, early identification and surgical treatment should be performed to alleviate the relevant clinical symptoms (11–13).
In summary, AOS caused by the DLL4 gene mainly manifests as skin hypoplasia, and some children may have terminal transection limb defects and congenital heart malformations. In children with similar clinical manifestations, genetic testing should be performed as soon as possible to identify the cause.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving humans were approved by Ethics Committee of Wuhan Children's Hospital (Wuhan Maternal and Child Healthcare Hospital), Tongji Medical College, Huazhong University of Science & Technology. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
YH: Conceptualization, Writing – original draft. JW: Conceptualization, Writing – original draft. LZ: Investigation, Supervision, Writing – review & editing. SW: Supervision, Writing – review & editing. XZ: Data curation, Validation, Writing – original draft.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We would like to acknowledge the parents for sharing their daughter's diseases.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sathishkumar D, Ogboli M, Moss C. Classification of aplasia cutis congenita: a 25-year review of cases presenting to a tertiary paediatric dermatology department. Clin Exp Dermatol. (2020) 45(8):994–1002. doi: 10.1111/ced.14331
2. Zhu VZ, Hansen-Kiss E, Hecht JT, Payne PE. Adams-Oliver syndrome: vestigial tail and genetics update. Arch Plast Surg. (2022) 49(4):517–22. doi: 10.1055/s-0042-1751107
3. Rashid S, Azeem S, Riaz S. Adams-Oliver syndrome: a rare congenital disorder. Cureus. (2022) 14(3):e23297. doi: 10.7759/cureus.23297
4. Hassed S, Li S, Mulvihill J, Aston C, Palmer S. Adams-Oliver syndrome review of the literature: refining the diagnostic phenotype. Am J Med Genet A. (2017) 173(3):790–800. doi: 10.1002/ajmg.a.37889
5. Nagasaka M, Taniguchi-Ikeda M, Inagaki H, Nagasaka M, Taniguchi-Ikeda M, Inagaki H, et al. Novel missense mutation in DLL4 in a Japanese sporadic case of Adams-Oliver syndrome. J Hum Genet. (2017) 62(9):851–5. doi: 10.1038/jhg.2017.48
6. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
7. Meester JA, Southgate L, Stittrich AB, Venselaar H, Beekmans SJ, Den Hollander N, et al. Heterozygous loss-of-function mutations in DLL4 cause Adams-Oliver syndrome. Am J Hum Genet. (2015) 97(3):475–82. doi: 10.1016/j.ajhg.2015.07.015
8. Chapman G, Moreau JL, Ip E, Szot JO, Iyer KR, Shi H, et al. Functional genomics and gene-environment interaction highlight the complexity of congenital heart disease caused by Notch pathway variants. Hum Mol Genet. (2020) 29(4):566–79. doi: 10.1093/hmg/ddz270
9. Snape KM, Ruddy D, Zenker M, Wuyts W, Whiteford M, Johnson D, et al. The spectra of clinical phenotypes in aplasia cutis congenita and terminal transverse limb defects. Am J Med Genet A. (2009) 149A(8):1860–81. doi: 10.1002/ajmg.a.32708
10. Rojnueangnit K, Phawan T, Khetkham T, Techasatid W, Sirichongkolthong B. A novel DLL4 mutation in Adams-Oliver syndrome with absence of the right pulmonary artery in newborn. Am J Med Genet A. (2022) 188(2):658–64. doi: 10.1002/ajmg.a.62562
11. Yu X, Wang RR, Han SR, Bai X, Habulieti X, Sun Y, et al. A novel DLL4 missense mutation in a Chinese patient with Adams-Oliver syndrome. Chin Med J. (2019) 132(14):1755–7. doi: 10.1097/CM9.0000000000000316
12. Yang XF, Shi SW, Chen K. Case report: recombinant human epidermal growth factor gel plus kangfuxin solution in the treatment of aplasia cutis congenita in a case with Adams-Oliver syndrome. Front Surg. (2023) 9:1072021. doi: 10.3389/fsurg.2022.1072021
13. Meester JAN, Sukalo M, Schröder KC, Schanze D, Baynam G, Borck G, et al. Elucidating the genetic architecture of Adams-Oliver syndrome in a large European cohort. Hum Mutat. (2018) 39(9):1246–61. doi: 10.1002/humu.23567
14. Dours L, Jeanne M, Srour M, Leducq S. A child with a congenital aplasia of the scalp: a quiz. Acta Derm Venereol. (2024) 104:adv39948. doi: 10.2340/actadv.v104.39948
15. Yu X, Wang RR, Han SR, Bai X, Habulieti X, Sun Y, et al. A novel DLL4 missense mutation in a Chinese patient with Adam-Oliver syndrome. Chin Med J. (2019) 132:1755–7. doi: 10.1097/CM9.0000000000000316
16. Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong Z, Li H, et al. Mutations in NOTCH1 cause Adams-Oliver syndrome. Am J Hum Genet. (2014) 95(3):275–84. doi: 10.1016/j.ajhg.2014.07.011
17. Mašek J, Andersson ER. The developmental biology of genetic Notch disorders. Development. (2017) 144(10):1743–63. doi: 10.1242/dev.148007
18. Ogawa M, Okajima T. Structure and function of extracellular O-GlcNAc. Curr Opin Struct Biol. (2019) 56:72–7. doi: 10.1016/j.sbi.2018.12.002
Keywords: Adams-Oliver syndrome, DLL4, case report, new born, genetic disease
Citation: Huang Y, Wang J, Zeng L, Wang S and Zhang X (2025) Case Report: A novel DLL4 variant in a neonate with Adams-Oliver syndrome. Front. Pediatr. 13:1532561. doi: 10.3389/fped.2025.1532561
Received: 22 November 2024; Accepted: 17 February 2025;
Published: 3 March 2025.
Edited by:
Nagwa Elsayed Afify Gaboon, Ain Shams University, EgyptReviewed by:
Anna Sowińska-Seidler, Poznan University of Medical Sciences, PolandCopyright: © 2025 Huang, Wang, Zeng, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuechen Zhang, emhhbmd4dWVjaGVuQHpnd2hmZS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.