
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 25 March 2025
Sec. Pediatric Surgery
Volume 13 - 2025 | https://doi.org/10.3389/fped.2025.1497203
This article is part of the Research TopicAdvances and Challenges in Neonatal Surgery: Congenital and Acquired ConditionsView all 4 articles
 Marta Tedesco1,2*†
Marta Tedesco1,2*† Simonetta Costa1,2,†
Simonetta Costa1,2,† Pierpaolo Agresti1,2Francesca Priolo1,2
Pierpaolo Agresti1,2Francesca Priolo1,2 Alessandro Perri1,2Annamaria Sbordone1,2
Alessandro Perri1,2Annamaria Sbordone1,2 Stefano Nobile1,2Filomena Valentina Paradiso2,3Maria Vittoria Stern2,3
Stefano Nobile1,2Filomena Valentina Paradiso2,3Maria Vittoria Stern2,3 Riccardo Rizzo2,3
Riccardo Rizzo2,3 Maria Cristina Giustiniani2,4Lorenzo Nanni2,3,‡
Maria Cristina Giustiniani2,4Lorenzo Nanni2,3,‡ Giovanni Vento1,2,‡
Giovanni Vento1,2,‡
Objective: Hepatopulmonary fusion (HPF) is a rare congenital malformation, frequently associated to right-sided congenital diaphragmatic hernia (CDHR). The presence of HPF often leads to a fatal outcome. The most effective approach to managing this condition remains uncertain due to the limited number of documented cases in the literature.
Study design: This case presents a 11-day old full-term female neonate with HPF associated to CDHR. The definitive diagnosis of HPF was made during surgery for CDHR. Our team opted for a simple repair of the diaphragmatic defect and no attempts were made to separate the liver from the right lung.
Results: Our approach was successful, as our patient not only survived the procedure but also showed favorable cardiorespiratory adaptation, consistent growth, and regular neurodevelopment, according to follow-up data, available at six months of life.
Conclusion: The adopted surgical management strongly suggests that when the diagnosis is made intraoperatively and detailed knowledge of the vascularization is lacking, partial separation of the viscera, preserving the medial hepatopulmonary fusion and suturing the diaphragm, is the successful approach.
Congenital diaphragmatic hernia (CDH) is a rare congenital malformation, with an estimated incidence of 2.4–4.2 per 10,000 births in the world, with right-sided CDH (CDHR) being the rarest form, accounting for approximately 15% of cases of diaphragmatic hernia (1). In comparison to left-sided lesions, the prognosis for CDHR is generally worse (2).
Hepatopulmonary fusion (HPF) is a rare congenital malformation associated with CDHR. Its prevalence is approximately 3 in 1,000 newborns affected by CDHR, and it affects both sexes equally. The anomaly can encompass a spectrum of fusion levels, spanning from fibrovascular connections to the complete merging of the pulmonary and hepatic tissues (3, 4). The presence of HPF often leads to a fatal outcome when combined with CDHR (5). The overall mortality associated with HPF or its complications is around 49% (6).
Due to the rarity of this condition, there is a lack of well-established guidelines for the optimal management strategy for HPF. In this study, we present a case of successful management of CDHR accompanied by HPF and provide an extensive review of the existing literature.
A female infant was born at 37 weeks and 3 days of gestational age (GA) from an uneventful pregnancy, with a birth weight of 2,370 g (small for GA, 1.39 z-score, according to Intergrowth-21). No clinical problems occurred at and after birth, and the baby was discharged after 3 days of Rooming-in. At 5 days of life, the infant experienced mild respiratory distress, which required an outpatient visit to the attending pediatrician. Upon examination, the pediatrician observed tachypnea and performed blood tests for acid-base balance and C-reactive protein, both of which yielded negative results. At 11 days of life, the baby was admitted to the emergency room due to a sudden episode of apnea during feeding. Soon after the arrival, the newborn appeared pale and hypotonic, with no respiratory activity and an oxygen saturation of 73%. Blood gas analysis revealed severe respiratory acidosis (pH 6.9, pCO2 114 mmHg, lactate 10 mmol/L, base excess −10 mmol/L). Therefore, the baby was intubated, and ventilation was continued with 100% FiO2. Subsequently, she was transferred to our Neonatal Intensive Care Unit (NICU) with a suspected diagnosis of aspiration pneumonia.
Upon admission to the ward, a chest x-ray was performed, which revealed areas of consolidation in both lung fields (Figure 1a). After a few hours, a follow-up thoracic and abdominal ultrasound (US) was performed, showing a large hypoechoic mass with liver-like parenchyma and vascularization, starting from the fifth intercostal space. The suspicion of a CDHR was raised, and a chest computed tomography (CT) scan was performed, confirming the presence of a posterior CDHR. The hernia defect measured approximately 25 mm, with the cranial ascent of the right liver into the thoracic cavity on the same side, reaching up to the middle-upper third of the right lung field (Figure 1b). There was agenesis of the inferior vena cava in the subhepatic suprarenal segment, with venous return from the lower venous system ensured by slightly ectasic azygos/hemiazygos veins, which drained cranially into the intrathoracic superior vena cava.
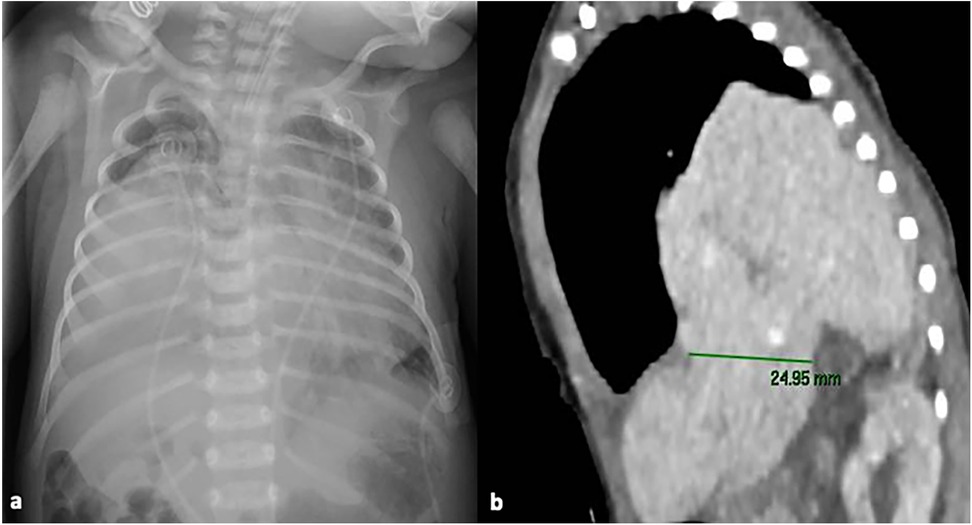
Figure 1. Chest x-ray at NICU admission showing areas of consolidation in both lung fields (a); chest computed tomography scan, showing the presence of a posterior CDHR, through a diaphragmatic defect measuring approximately 25 mm: the liver was herniated into the intrathoracic space, reaching up to the middle-upper third of the right lung field (b) written informed consent was obtained from the patient.
Then, an echocardiogram was performed, which showed regular levocardia, normal atrioventricular and ventriculoarterial connections, and regular systemic and pulmonary venous connections.
Two days after admission to the NICU, at 15 days of life, surgical correction of the diaphragmatic hernia was performed, with the child in stable conditions both from a respiratory point of view (HFOV parameters: MAP 10 cm H2O, FiO2 0.25, RR 10 Hz, Volume guarantee 1.8 ml/Kg requiring ΔP of 20–23 cm H2O and corresponding optimal values of pH and blood gases) and hemodynamic (no need for inotropic drugs or pulmonary vasodilators). A right subcostal laparotomy was performed. At exploration no clear plane of cleavage between the diaphragm and the liver could be identified; additionally, the liver appeared firm, and it was impossible to move it towards the abdomen. On the lateral aspect of the liver a flap of tissue was identified with a consistency and color resembling both liver and lung tissue. Bubbles were noted on the thoracic side of this tissue flap. The histological examination of a fragment of this tissue showed hepatic parenchyma connected to lung tissue displaying significant congestion and blood extravasation (Figure 2). The diaphragmatic defect was repaired without separating the lung and the liver: the medial margin of the diaphragm was fixed to the liver surface with three stitches to close the defect. Soon after surgery, the baby developed pulmonary hypertension, leading to endotracheal nitric oxide administration, which was discontinued after 48 h due to resolution of the condition. The remaining postoperative course progressed uneventfully, except for a Staphylococcus epidermidis pneumonia diagnosed 72 h after surgery, which required prolongation of invasive respiratory support for a further 7 days, followed by successful extubation and non-invasive respiratory support for 14 days. Postoperative imaging and laboratory results indicated normal liver function, with no evidence of impairment due to its prior herniation into the thoracic cavity.
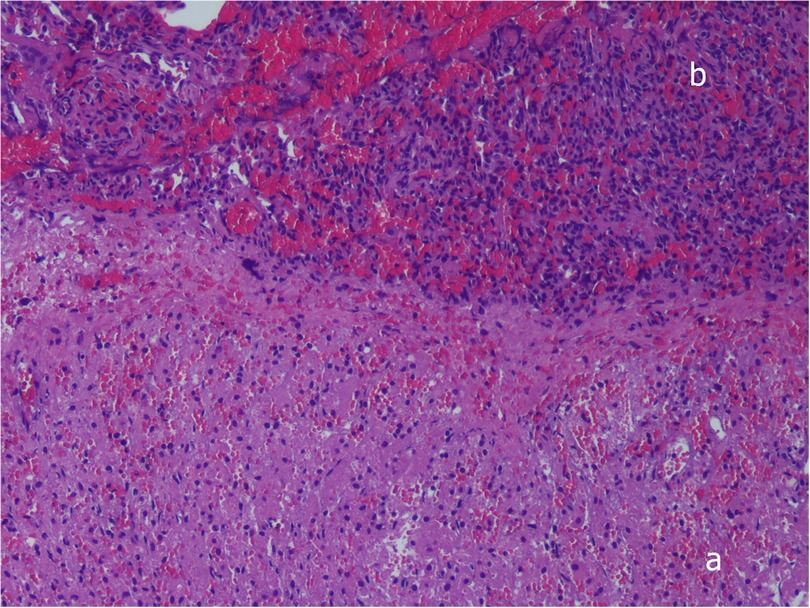
Figure 2. Histopathologic examination, showing complete fusion between liver (a) and pulmonary tissue (b), without a plane of separation. Hematoxylin-and-eosin stain, ×10. Written informed consent was obtained from the patient.
At 48 days of life, approximately one month after surgery, an angio-CT scan was performed to obtain a comprehensive assessment of the involved visceral anatomy and associated vascular anomalies. The scan confirmed the presence of a CDHR with intrathoracic herniation of part of the liver, without any vascular communication between the liver and the lung. It also confirmed agenesis of the retrohepatic inferior vena cava, with continuation of the inferior vena cava through the azygos vein.
Considering the achieved clinical stability, no further surgery was performed, and at 58 days of life, the baby was discharged without any respiratory support, with good feeding autonomy, and with the plan to follow a close clinical follow-up.
At 64 days of life, and on the 49th postoperative day, respiratory function tests—Tidal Breathing Flow Volume test and Multiple Breath Nitrogen Washout test with the Exhalyzer D (Ecomedics, Switzerland)—were performed according to published guidelines (7) which showed tachypnea and tidal volume lower than predicted (Table 1).
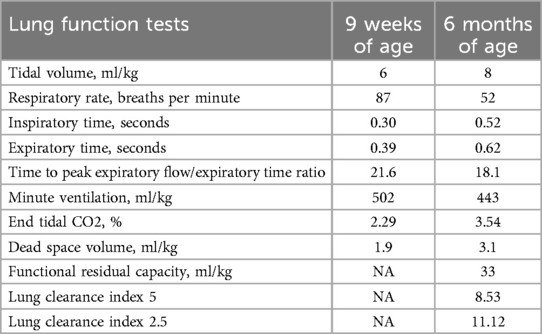
Table 1. Results of the lung function tests.
Follow-up evaluations are ongoing, and the infant is doing well apart from a mild episode of bronchiolitis which did not require hospitalization. She is getting complementary feeding and palivizumab prophylaxis, and her neurological examination is normal. Growth parameters are satisfactory (last weight was at 2nd percentile, length at 46th percentile, head circumference at 24th percentile). Lung function tests were repeated at 6 months of age: tidal volume was normal indicating catch-up growth, time to peak expiratory flow/expiratory time ratio (tPTEF/tE) was mildly reduced, and lung clearance index (LCI) was high, indicating ventilation inhomogeneity (Table 1).
In our child, the definitive diagnosis of HPF was made during surgery and, because of the abnormal vascular anatomy (Table 2), simple repair of the diaphragmatic defect was chosen, and no attempts were made to separate the liver from the right lung.
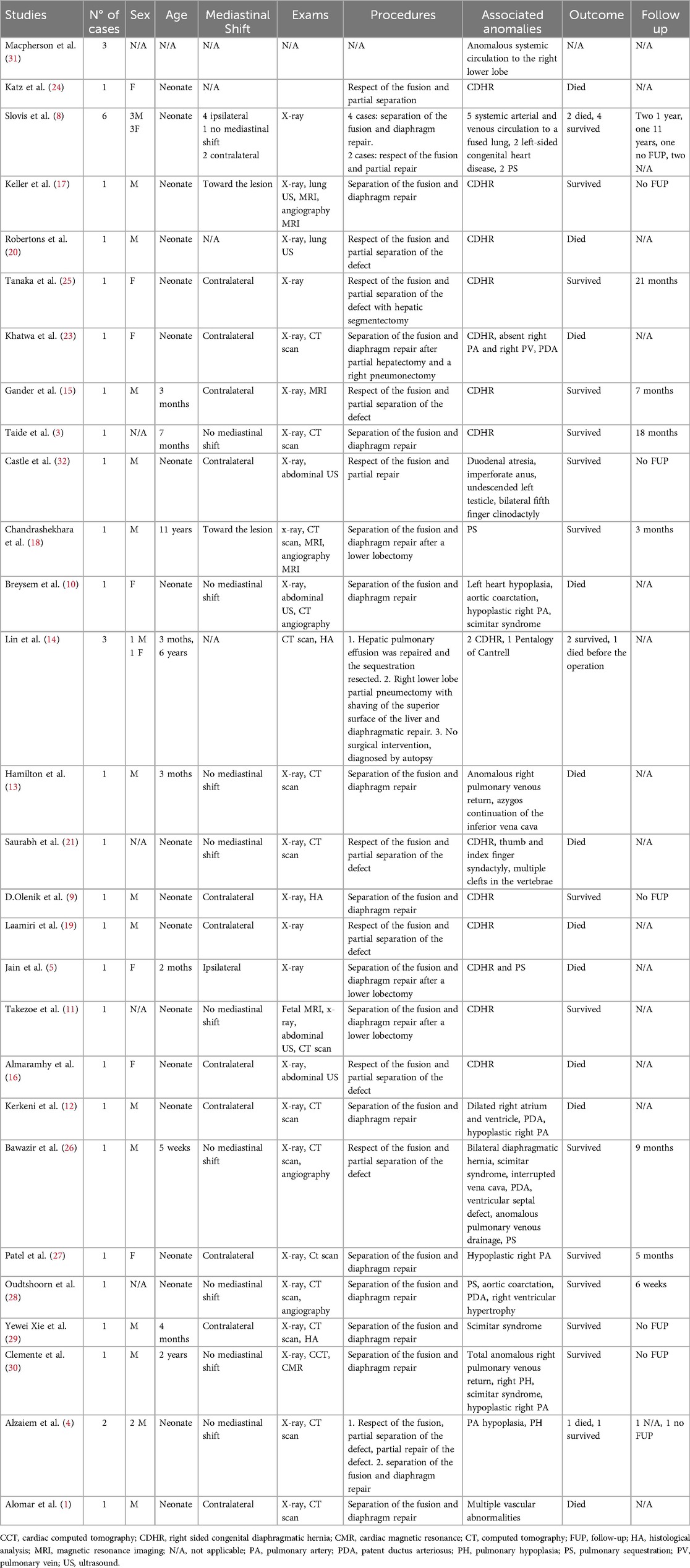
Table 2. Synopsis of the 38 cases reported in the literature.
Our approach was successful, as our patient not only survived the procedure but also showed favorable cardiorespiratory adaptation, consistent growth, and regular neurodevelopment, according to follow-up data, available at six months of life.
The optimal management of this condition is still not clear, given the limited number of documented cases in literature (Table 2). Attempts to separate the liver from the lung parenchyma can be challenging and may require partial pneumonectomy or atypical hepatic resection in some cases. By restoring negative intrathoracic pressure, this procedure should enhance lung development in the affected side and restrict the entry of hepatic tissue into the thoracic cavity (8).
Several surgical approaches have been described for the surgical treatment of HPF. Some surgeons have attempted complete organ separation through resection of the involved tissues (partial hepatectomy or pulmonary lobectomy) (5, 9–14), while others have opted for partial separation and suturing of the diaphragm to the remaining fusion margins (4, 15–21).
A 2019 review by Ferguson described nine cases of HPF identified in the Congenital Diaphragmatic Hernia Registry. Among the reported cases, partial separation of pulmonary and hepatic parenchyma was performed in 6 patients, and among them, one patient did not survive. Complete separation was performed in 2 cases, but both patients did not survive, one due to pulmonary hypertension and the other due to postoperative hemorrhage and renal failure. Finally, separation was not attempted in 1 case, and the surgeon chose to plicate the pleura and peritoneum that were present around the area of fusion. The patient survived to hospital discharge but remained ventilator dependent, and she ultimately expired (6).
In a recent study by Terp et al. (22), complete separation of the lung and liver was possible due to prenatal identification of the anomaly and preoperative characterization of vascular abnormalities. The importance of preoperative diagnosis has been also highlighted by Keller et al. (17), who utilized preoperative chest x-ray, thoracic US, and magnetic resonance imaging (MRI) to establish the diagnosis of CDHR with HPF. In this case, diagnostic and therapeutic cardiac catheterization and preoperative CT were also found to be very useful for preparation and surgical planning.
However, HPF can be missed prenatally and most of the time, it is not diagnosed until surgical exploration (9). HPF, as in our infant, can be asymptomatic at birth or present with cyanosis and respiratory distress. Subsequently, the diagnosis may be incidental, or the most frequent manifestations include recurrent respiratory infections, pleural effusions, and mediastinal compression.
The diagnosis of HPF should be considered when thoracic US or chest x-ray reveals the presence of an opacity in the right hemidiaphragm without mass effect, such as contralateral mediastinal shift or lung compression, due to pulmonary hypoplasia (15). If these findings are associated with cardiac or vascular malformations, they further support the diagnostic suspicion of HPF (5). Exceptions to the above description of HPF can occur, as contralateral or ipsilateral mediastinal shift primarily depends on the size of the diaphragmatic defect and the amount of herniated viscera, regardless of the presence of HPF (16).
The preoperative diagnostic definition is important, above all, because HPF can be associated with cardiac or vascular defects in 10%–30% of patients (4). The association of HPF with cardiac and vascular anomalies can be partially explained by the fact that the hepatic diverticulum, septum transversum, and aortopulmonary septum form near each other during the 4th and 6th gestational week (8).
Among the cases of HPF reported by Ferguson et al., 60% exhibited aberrant right pulmonary vascularization. The most common anomalies include: hypoplastic right pulmonary artery, abnormal pulmonary venous return, inferior vena cava abnormalities (hypoplasia, partial absence of the vena cava, anomalous drainage of the suprahepatic veins), and pulmonary veins that drain into the suprahepatic veins (6).
Due to the uncommon occurrence and anatomical complexity of the condition, some authors suggest that a comprehensive preoperative evaluation with MRI or CT with multiplanar and 3D reconstruction would be beneficial in cases where there is suspicion of HPF. These imaging modalities would enable a comprehensive assessment of the visceral anatomy and provide the most accurate mapping of vascular and bronchial structures (17, 23).
Mortality among cases of HPF is high, and often patients die during the perioperative period. Mortality is mainly related to postoperative complications such as pulmonary hypoplasia, respiratory failure, persistent pulmonary hypertension, right heart failure, congenital heart diseases, and inferior vena cava thrombosis (9, 15).
Our clinical report and the literature review raise several important observations: (a) prenatal diagnosis is crucial, and suspicion should be raised in all cases of CDHR prenatally diagnosed; (b) preoperative diagnosis is not always feasible but the absence of contralateral mediastinal shift or the presence of rightward mediastinum can act as red flags, prompting additional investigations such as MRI, CT scan, or cardiac catheterization. These examinations allow for a thorough evaluation of the malformation's anatomical aspects and associated vascular abnormalities, to plan the optimal surgical approach.
In our infant, in the absence of prenatal diagnosis, complete separation did not appear to be ideal, given the presence of vascular anomalies that could interfere with perfusion/venous drainage of the liver ant the lung after separation. The adopted surgical management strongly suggests that when the diagnosis is made intraoperatively and detailed knowledge of the vascularization is lacking, partial separation of the viscera, preserving the medial hepatopulmonary fusion and suturing the diaphragm, is the successful approach. One of the strengths of our work is the provision of serial functional respiratory evaluations during the follow-up of the presented patient; these data support the feasibility of our approach and provide further details about lung adaptations in hepatopulmonary fusion. We will continue to follow-up the child in the next years by evaluating respiratory and neurodevelopmental function.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
MT: Writing – original draft, Writing – review & editing. SC: Writing – original draft, Writing – review & editing. PA: Writing – original draft, Writing – review & editing. FPr: Funding acquisition, Investigation, Methodology, Writing – review & editing. AP: Formal Analysis, Funding acquisition, Investigation, Methodology, Writing – review & editing. AS: Investigation, Methodology, Writing – review & editing. SN: Writing – original draft, Writing – review & editing. FPa: Conceptualization, Investigation, Writing – review & editing. MS: Conceptualization, Investigation, Writing – review & editing. RR: Investigation, Methodology, Writing – review & editing. MG: Investigation, Writing – review & editing. LN: Supervision, Writing – review & editing. GV: Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Alomar K, Alghazal LK, Alloun M, Dawarah M, Alkhayer G, Alkader MA. A rare case of hepatopulmonary fusion associated with a right congenital diaphragmatic hernia: case report and review of the literature. Int J Surg Case Rep. (2023) 111:108845. doi: 10.1016/j.ijscr.2023.108845
2. Burgos CM, Frenckner B, Luco M, Harting MT, Lally PA, Lally KP, et al. Right versus left congenital diaphragmatic hernia—what’s the difference? J Pediatr Surg. (2017) S0022–3468(17):30649–8. doi: 10.1016/j.jpedsurg.2017.10.027
3. Taide DV, Bendre PS, Kirtane JM, Mukunda R. Hepatic pulmonary fusion: a rare case. Afr J Paediatr Surg. (2010) 7(1):28–9. doi: 10.4103/0189-6725.59357
4. Al-Zaiem M, Alzahrani A, Raml E I, Alsulaimani N, Alzahrani L, Turki A. Right congenital diaphragmatic hernia associated with hepatic pulmonary fusion. J Pediatr Surg Case Rep. (2023) 89:102540. doi: 10.1016/j.epsc.2022.102540
5. Jain V, Yadav DK, Kandasamy D, Gupta DK. Hepatopulmonary fusion: a rare and potentially lethal association with right congenital diaphragmatic hernia. BMJ Case Rep. (2017) 2017:bcr2016218227. doi: 10.1136/bcr-2016-218227
6. Ferguson DM, Congenital Diaphragmatic Hernia Study Group. Hepatopulmonary fusion: a rare variant of congenital diaphragmatic hernia. J Pediatr Surg. (2020) 55(9):1903–7. doi: 10.1016/j.jpedsurg.2019.09.037
7. Nguyen TT, Hoo AF, Lum S, Wade A, Thia LP, Stocks J. New reference equations to improve interpretation of infant lung function. Pediatr Pulmonol. (2013) 48(4):370–80. doi: 10.1002/ppul.22656
8. Slovis TL, Farmer DL, Berdon WE, Rabah R, Campbell JB, Philippart AI. Hepatic pulmonary fusion in neonates. AJR Am J Roentgenol. (2000) 174(1):229–33. doi: 10.2214/ajr.174.1.1740229
9. Olenik D, Codrich D, Gobbo F, Travan L, Zennaro F, Dell'Oste C, et al. Hepatopulmonary fusion in a newborn. An uncommon intraoperatory finding during right congenital diaphragmatic hernia surgery: case description and review of literature. Hernia. (2014) 18(3):417–21. doi: 10.1007/s10029-012-1042-y
10. Breysem L, Vanhaesebrouck S, Gewillig M, Dymarkowski S, Smet MH. Multidetector CT of right-sided con- genital diaphragmatic hernia associated with hepatopulmonary fusion in a new- born. Pediatr Radiol. (2012) 42:1138–41. doi: 10.1007/s00247-012-2379-1
11. Takezoe T, Nomura M, Ogawa K, Tomonaga K, Ohno M, Tahara K, et al. Prenatally diagnosed, right-sided congenital diaphragmatic hernia complicated by hepatic pulmonary fusion and intrathoracic kidney. Birth Defects. (2017) 1. doi: 10.15761/BDJ.1000104
12. Kerkeni Y, Farhani R, Sassi N, Hamzaoui M. How to treat hepatic pulmonary fusion: case re- port with review of literature. Acta Chir Belg. (2018):1–3.28669280
13. Hamilton J, Jaroszewski D, Notrica D. Fatal complication after repair of a congenital diaphragmatic hernia associated with hepatopulmonary fusion, anomalous right pulmonary venous return, and azygos continuation of the inferior vena cava. Eur J Pediatr Surg. (2012) 24:350–2. doi: 10.1055/s-0032-1324695
14. Lin J, Durham MM, Ricketts R, Abramowsky CR, Steelman CK, Shehata BM. Hepatic pulmonary fusion: two cases with diaphragmatic hernia and one case with pentalogy of cantrell. Fetal Pediatr Pathol. (2012) 31(6):401–9. doi: 10.3109/15513815.2012.659406
15. Gander JW, Kadenhe-Chiweshe A, Fisher JC, Lampl BS, Berdon WE, Stolar CJ, et al. Hepatic pulmo- nary fusion in an infant with a right-sided congenital diaphrag- matic hernia and contralateral mediastinal shift. J Pediatr Surg. (2010) 45(1):265–8. doi: 10.1016/j.jpedsurg.2009.10.090
16. Almaramhy HH. Hepatopulmonary fusion associated with right-sided congenital diaphragmatic hernia: management of this rare anomaly and a review of the literature. J Int Med Res. (2018) 46(12):5278–84. doi: 10.1177/0300060518759892
17. Keller RL, Aaroz PA, Hawgood S, Higgins CB. MR Imaging of hepatic pulmonary fusion in neonates. AJR Am J Roentgenol. (2003) 180(2):438–40. doi: 10.2214/ajr.180.2.1800438
18. Chandrashekhara SH, Bhalla As, Gupta AK, Sharma PK, Agarwala S, Srinivas M, et al. Hepatic pulmonary fusion: case report with review of literature. J Pediatr Surg. (2011) 46:e23–7. doi: 10.1016/j.jpedsurg.2010.11.032
19. Laamiri R, Belhassen S, Ksia A, Ben Salem A, Kechiche N, Mosbahi S, et al. Right congenital diaphragmatic hernia associated with hepatic pulmonary fusion: a case report. J Neonatal Surg. (2016) 5:35. doi: 10.21699/jns.v5i3.370
20. Robertson DJ, Harmon CM, Goldberg S. Right congenital diaphragmatic hernia asso- ciated with fusion of the liver and the lung. J Pediatr Surg. (2006) 41:e9–10. doi: 10.1016/j.jpedsurg.2006.02.031
21. Saurabh K, Kumar S, Chellani H, Aarya S. Hepatic pulmonary fusion: a rare association of right-sided congenital diaphragmatic hernia. Ann Gastroenterol. (2013) 26:95–6.24714321
22. Terp KL, Roberts BK, Alonso D, Pevsner Crum RM, Crombleholme T, Karakas SP, et al. Prenatal recognition of hepatopulmonary fusion in right-sided congenital diaphragmatic hernia for successful operative planning. Fetal Diagn Ther. (2022) 49(11-12):451–8. doi: 10.1159/000527802
23. Khatwa U, Lee EY. Multidetector computed tomography evaluation of secondary hepatopulmonary fusion in a neonate. Clin Imaging. (2010) 34(3):234–8. doi: 10.1016/j.clinimag.2009.07.005
24. Katz S, Kidron D, Litmanovitz I, Erez I, Dolfin Z. Fibrous fusion between the liver and the lung: an unusual complication of right congenital diaphragmatic hernia. J Pediatr Surg. (1998) 33(5):766–7. doi: 10.1016/S0022-3468(98)90214-7
25. Tanaka S, Kubota M, Yagi M, Okuyama N, Ohtaki M, Yamazaki S, et al. Treatment of a case with right-sided diaphragmatic hernia associated with an abnormal vessel communication between a herniated liver and the right lung. J Pediatr Surg. (2006) 41(3):e25–8. doi: 10.1016/j.jpedsurg.2005.12.032
26. Bawazir OA. Surgical repair of bilateral congenital diaphragmatic hernia associated with hepatopulmonary fusion. Saudi Med J. (2019) 40(9):949–53. doi: 10.15537/smj.2019.9.24378
27. Patel S, Rael J. Right-Sided congenital diaphragmatic hernia caused by hepatopulmonary fusion. Case Rep Pediatr. (2020) 2020:8851341.33178472
28. van Oudtshoorn S, Gikenye N, Kikiros C, Gera P. Muddle in the middle: a rare case of a hepatopulmonary fusion and lung sequestration in a neonate with a right-sided congenital diaphragmatic hernia. J Paediatr Child Health. (2021) 57(10):1692–4. doi: 10.1111/jpc.15342
29. Xie Y. Hepatic pulmonary fusion: a rare case report. Transl Pediatr. (2021) 10(4):1034–8. doi: 10.21037/tp-20-356
30. Clemente A, Viganò G, Festa L, Remoli E, Marrone C, Federici D, et al. Multimodality approach to a complex scimitar syndrome: how advanced diagnostics can guide therapeutic strategies. JACC Case Rep. (2022) 4(10):596–603. doi: 10.1016/j.jaccas.2022.03.027
Keywords: hepatopulmonary fusion, congenital diaphragmatic hernia, right-sided congenital diaphragmatic hernia, management, neonatal intensive care unit
Citation: Tedesco M, Costa S, Agresti P, Priolo F, Perri A, Sbordone A, Nobile S, Paradiso FV, Stern MV, Rizzo R, Giustiniani MC, Nanni L and Vento G (2025) Case Report: Hepatopulmonary fusion: to separate or not to separate? From a clinical case to A literature review. Front. Pediatr. 13:1497203. doi: 10.3389/fped.2025.1497203
Received: 20 January 2025; Accepted: 7 March 2025;
Published: 25 March 2025.
Edited by:
Juan A. Tovar, Department de Cirugía Pediátrica Hospital Universitario La Paz, SpainReviewed by:
Sanja Miodrag Sindjic Antunovic, University of Belgrade, SerbiaCopyright: © 2025 Tedesco, Costa, Agresti, Priolo, Perri, Sbordone, Nobile, Paradiso, Stern, Rizzo, Giustiniani, Nanni and Vento. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Tedesco, bWFydGEudGVkZXNjbzEwQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.