
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 11 February 2025
Sec. Children and Health
Volume 13 - 2025 | https://doi.org/10.3389/fped.2025.1487000
 Siri Hauge Opdal1*
Siri Hauge Opdal1* Arne Stray-Pedersen2,3Johanna Marie Lundesgaard Eidahl2Åshild Vege2Linda Ferrante1
Arne Stray-Pedersen2,3Johanna Marie Lundesgaard Eidahl2Åshild Vege2Linda Ferrante1 Torleiv Ole Rognum3
Torleiv Ole Rognum3
Sudden Infant Death Syndrome (SIDS) is the sudden and unexpected death of an otherwise healthy infant less than 1 year of age where the cause of death remains unexplained after a thorough post-mortem investigation and evaluation of the circumstances. Epidemiological, clinical, biochemical, immunological and pathological evidence indicates that three factors must coincide for SIDS to occur: a vulnerable developmental stage of the immune system and central nervous system in the infant, predisposing factors, and external trigger events. This model is referred to as the fatal triangle or triple risk hypothesis. The concept of a vicious spiral in SIDS, starting with the fatal triangle and ending in death, is proposed as a model to understand the death mechanism. The vicious spiral is initiated by a mucosal infection and immune activation in the upper respiratory and digestive tracts, increased production of cytokines, and an overstimulation of the immature and rapidly developing immune system. A second trigger is the prone sleeping position, which may lead to rebreathing and hypercapnia, in addition to intensify the immune stimulation. In susceptible infants, this induces an aberrant cytokine production that affects sleep regulation, induces hyperthermia, and disrupts arousal mechanisms. In turn, this initiates downregulation of respiration and hypoxemia, which is worsened by nicotine. Inefficient autoresuscitation results in severe hypoxia and accumulation of hypoxic markers which, if not prevented by a normally functioning serotonergic network, contribute to a self-amplifying vicious spiral that eventually leads to coma and death. The purpose of this review is to summarize the research that underpins the concept of the vicious spiral.
The first definition of Sudden Infant Death Syndrome (SIDS) was established during “The Second international conference on causes of sudden death in infants” in Seattle in 1969: “The sudden death of any infant or young child which is unexpected by history, and in which a thorough post-mortem examination fails to demonstrate an adequate cause of death” (1). This was followed by a slightly revised definition in 1991, the National Institute of Child Health and Human Development (NICHD) definition, where an investigation of the death scene and also review of the clinical history was included: “The sudden death of an infant under one year of age which remains unexplained after a thorough case investigation, including performance of a complete autopsy, examination of the death scene, and review of the clinical history” (2).
The current commonly used SIDS definition is from 2004, now including an expanded general definition of SIDS and further subdivision into different SIDS categories (IA, IB, or II), depending upon the amount of information available (3). Commonly referred to as the San Diego definition, the definition says “SIDS is defined as the sudden unexpected death of an infant <1 year of age, with onset of the fatal episode apparently occurring during sleep, that remains unexplained after a thorough investigation, including performance of a complete autopsy and review of the circumstances of death and the clinical history”. This definition only includes infants <1 year of age, which is why an additional definition for Sudden Unexplained Death in early Childhood (SUDC) has been proposed (4). According to this, SUDC is defined as “The sudden death of a child older than one year of age which remains unexplained after a thorough case investigation, including review of the clinical history and circumstances of death, and performance of a complete autopsy with appropriate ancillary testing”.
Despite efforts to provide international standards on how to perform autopsy on infants, including ancillary testing and how to interpret findings, there seems to be a lack of consensus among pathologists and death certifiers on how to classify unexplained infant deaths (5–7). In most countries the reduction in SIDS rate follows in parallel the decrease in total post neonatal mortality (8). To a certain extent this reduction may have been exaggerated by the attribution of these deaths to causes, like positional asphyxia (9). A recent study showed that unexplained infant deaths in the US are now more often designated “undetermined” or due to “accidental suffocation” than classified as SIDS (10). Lack of a generally accepted diagnosis for sudden unexpected and unexplained deaths in infants and children hinders effective surveillance, prevention and research (11). Motivated by the increasing rejection of the SIDS diagnosis, an initiative has been taken to develop a unified diagnosis for sudden death in paediatrics which might be accepted by the World Health Organization for inclusion in the upcoming ICD-11. A broader definition of the SIDS diagnosis will most likely include all sudden unexpected deaths not fully explained after a thorough post-mortem investigation (11).
The first version of a triple risk hypothesis for SIDS was introduced by Wedgwood in 1972 (12). He postulated three elements that are involved in these deaths: (1) general risk factors such as low socioeconomic status and sex, (2) age-dependent factors relating to the developmental stage of the infant, including immunological status and the changing physiological responsiveness with regard to cardiopulmonary reflex control, and (3) external stimuli, including sleeping position and viral infections. A few years later Valdés-Dapena proposed a two-hit model for SIDS; a previous hypoxic episode, followed by a second fatal event (13, 14). Her model was based on the studies by Naeye et al., who identified various tissue-markers for hypoxia and hypoxemia, as well as her own observations in SIDS victims (15).
In 1993, Rognum and Saugstad introduced the concept of a “fatal triangle” in SIDS, which was based on morphological, biochemical and immunological findings (Figure 1) (16). They demonstrated elevated levels of the hypoxic marker hypoxanthine in SIDS compared to violent infant death (17, 18), and also increased immune reaction in the upper airways and digestive tract in SIDS compared to infants dying of non-infectious causes (19, 20). Recognizing the typical age peek for SIDS between 1 and 6 months of age and the coinciding rapid development of mucosal immunity during this period prompted the introduction of a vulnerable development stage (21, 22). A possible link between the peripheral immune system and the central nervous system was based on the observation that SIDS victims often exhibited signs of infection prior to death, manifested immune responses in the laryngeal mucosa, and displayed elevated levels of interleukin 6 (IL-6) in the cerebrospinal fluid (23, 24). It was thus postulated a fatal tringle in SIDS. This consists of a vulnerable developmental stage, predisposing factors, including a genetic predisposition, and trigger events such as a stimulation of the mucosal immune system due to a common cold (Figure 1) (16).
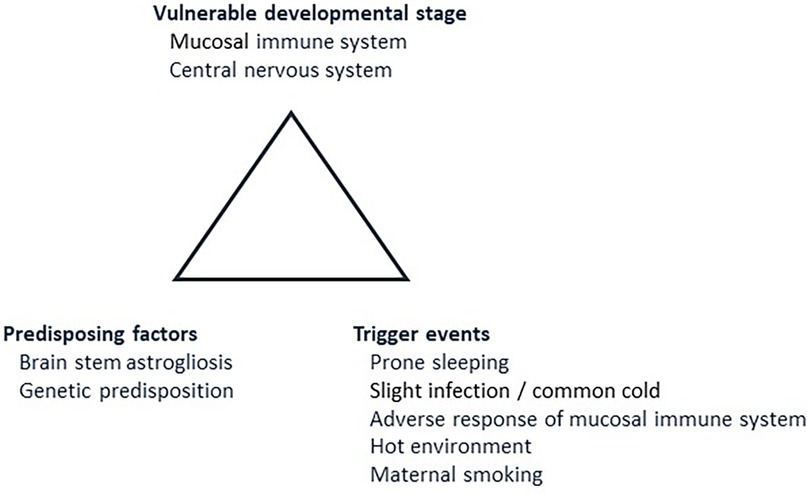
Figure 1. The fatal triangle in SIDS, modified after Rognum and Saugstad (16). This model postulates that SIDS occurs when an infant is at vulnerable developmental stage in maturation of the immune system and central nervous system (2.1). At the same time, predisposing factors, such as brainstem astrogliosis and/or predisposing gene variants must be present (2.2). Lastly, the infant must be exposed to external trigger events such as prone sleeping, slight infection, for example a common cold, and/or maternal smoking (2.3). Numbers in brackets refer to paragraphs in the text.
Filiano and Kinney presented another and slightly different version of the triple risk model (25, 26). According to this model, SIDS occurs when three factors impinge upon the infant simultaneously: an underlying vulnerability in protective responses to hypoxia, a critical developmental phase in homeostatic control, and exogenous stressors that induce life-threatening hypoxia, such as prone or facedown sleep position. The vulnerability remains dormant until the infant reaches a critical stage of development and is exposed to a stressor, such as unsafe sleeping practices like prone sleeping, co-sleeping, excessive bedding, or passive smoking. The underlying vulnerability or abnormality in the brainstem in SIDS victims differentiates them from healthy infants; in the latter, protective brainstem responses are activated when exposed to homeostatic challenges.
The following sections will elaborate the fatal triangle in more detail (Figure 1).
The fatal triangle in SIDS suggests an age-dependent increased risk for sudden death in infancy. During the first months of life, there is a rapid development of both the central nervous system and the immune system, particularly in terms of homeostatic control, as the infant adapts to the microbial environment outside the womb. This period is recognized as a vulnerable developmental stage. Within the central nervous system, significant changes occur in the serotonergic network, which is crucial for chemo-sensitivity, respiration, upper airway reflexes, blood pressure, thermoregulation and arousal (27, 28). Simultaneously, the infant's mucosal immune system undergoes rapid development, making the infant more susceptible to various immune stimuli (21, 22, 29, 30). IgG from the mother disappears quickly. At the same time, the secretory immune system undergoes a rapid development, and the number of IgM and IgA immunocytes, as well as the transport of IgM and IgA by secretory component through the epithelia increase dramatically from the second week after birth (30, 31). It is reasonable that disappearance of maternal IgG and the rapid development of the immune system in the period from the fourth postpartum week makes infants vulnerable for unfortunate and possibly dangerous immune reactions. This suspicion has been strengthened by the observation of enhanced immune responses in the respiratory and gastrointestinal tract in SIDS (19, 32, 33). These observations are supported in a prospective study of immunoglobulins and albumin in saliva sampled repeatedly in 263 infants from day two until eight weeks postpartum (30). One infant in the cohort died from SIDS at the age of eight weeks. Whereas saliva from the cohort showed a relative rapid increase of IgA and IgM from the second week after birth, the SIDS victim experienced a much steeper increase of both IgA and IgM from the fourth week of life. At week eight, the SIDS case had reached a about a nine-fold increase in IgA concentration and a six-fold increase in concentration of IgM, compared to the median in the controls (30).
Increased levels of IL-6 in the cerebrospinal fluid have been found in a large proportion of SIDS victims, and a relationship with immune responses in the respiratory tract has been demonstrated (23, 24, 34). These studies indicate that IL-6 may constitute a link between the mucosal immune system and the central nervous system. During infection, peripherally produced cytokines may cross the blood-brain barrier, either by retrograde axonal transport or by blood born, and bind to endogenous receptors on neuronal populations that mediate stress responses in the hypothalamus and/or brainstem, and thereby determine sickness behavior including fever, blunted arousal and depressed respiration (35, 36). A study from Harvard Medical School on samples from Norwegian SIDS victims disclosed abnormal IL-6R expression in the arcuate nucleus in the SIDS cases, of which 44% had signs of slight infection immediately prior to death (37). This observation seems interesting, since the reduction in SIDS rates in Norway to a large extent is due to less small infants with a history of common cold in the days before they were found dead in prone position (38).
Temporal shift in innate immunity may also contribute to increased vulnerability during the first months after birth. It is known that Surfactant protein A, which plays a role as an opsonic in the lungs by tagging foreign pathogens for elimination by phagocytes, substantially decreases in infants between 1 and 5 months of age (39). This further emphasizes the vulnerability in the immune system during this period.
Multiple predisposing factors contribute to SIDS, including brainstem astrogliosis, neurochemical imbalances in the serotonergic network, and genetic predispositions (Figure 1).
Brainstem astrogliosis may develop as a nonspecific response to injury in the central nervous system as early as in the second trimester of a pregnancy. Studies have reported an increased number of reactive astrocytes in the brainstem in SIDS compared to controls, with a correlation observed between brainstem astrogliosis and maternal smoking during pregnancy (15, 40, 41). Gliosis typically takes 3–4 days to develop, suggesting it is not a consequence of events immediately preceding death but rather linked to an injury occurring at least a few days prior to death.
Compelling evidence implicates the serotonergic network in SIDS, although the question remains whether serotonergic abnormalities originate from prenatal or postnatal development (26, 42). Several findings support a developmental origin, such as immature serotonergic neurons in the medulla, decreased serotonergic receptor binding in the arcuate nucleus in SIDS, and abnormal expression pattern of proteins crucial for neurological development (43, 44).
The genetic predisposition most likely represents a polygenic inheritance pattern; a combination of various gene variants and polymorphisms that collectively increase susceptibility. This genetic predisposition remains dormant and inconsequential until the infant reaches a critical developmental stage and coincides with exposure to external risk factors known to increase the risk for sudden death in infancy.
Over the years, numerous genes and gene regions have emerged as important in the context of SIDS (45–49). Initial investigations focused on genes involved in the function and regulation of the immune system, including complement component C4 and different cytokines and interleukins (50–52). Furthermore, genes associated with brain development and function are likely to contribute to the genetic predisposition for SIDS. The most important genes in this category include genes involved in the serotonergic network and genes encoding different ion channels and aquaporins (53–61). Another crucial area involves genes related to cardiac function and arrhythmia (62–65).
It is crucial, particularly concerning cardiac genes, to distinguish between gene variants that may constitute a predisposition for SIDS in specific contexts and pathogenic gene variants confirmed to induce arrhythmias and possibly death. When genetic heart disease is identified, the cause of death is clarified, and the case is no longer classified as SIDS.
The concept of the fatal triangle and triple risk hypotheses represent classical SIDS theories, where trigger events and risk factors are circumstances that may be demanding for an already vulnerable infant (66). It is important to note that trigger events are not the direct cause of death but rather initiators of a chain of reactions leading to SIDS.
Several of the trigger events are associated with sleeping environment. These include prone sleeping position and hazardous sleeping situations such as soft bedding, head covering, excessive clothing, and unsafe bed sharing (67–69). Public health campaigns promoting supine sleeping began in the early 1990s and significantly contributed to the decline in the SIDS rates (70). The prone sleeping position with face down, soft bedding and bed-sharing may all affect the infant's breathing, leading to rebreathing and thus hypercapnia. In unfortunate cases, the sleeping position may obstruct the airways, leading to positional asphyxia. Such cases should not be misclassified as SIDS. A thorough post-mortem examination, including a detailed investigation of the death scene, preferably with doll reenactment, makes it possible to rule out accidental suffocation as a cause of death in most cases (71). The increased risk for sudden infant death posed by prone sleeping position with face down and environmental risk factors cannot solely be attributed to mechanical breathing obstruction (72, 73).
Multiple studies have demonstrated that both pre- and postnatal nicotine exposure increases the risk of SIDS in a dose-dependent manner (74, 75). While the highest risk is associated with maternal smoking, especially during pregnancy, there is also a small independent risk associated with paternal smoking after the infant's birth (76, 77). A significant association has also been highlighted between smoking exposure and the biochemical lack of detoxification enzymes (78). The risk is further increased in the presence of additional risk factors, such as co-sleeping or prone sleeping combined with an infection (67, 79, 80). Mild upper airway infection during the last week or days prior to death was observed in more than half of the SIDS cases, with around 10%–15% exhibiting fever during the same period (67, 79). Interestingly, the dramatic decrease in SIDS rates in Norway from the 1980s to the 1990s was largely attributed to fewer SIDS in the age group 2–4 months with the combination of prone sleeping position and minor infection prior to death (38).
The overall SIDS mortality rate has decreased during the last three decades, in concordance with overall postnatal mortality (8, 81). The decreased SIDS rate is largely due to back-to-sleep campaigns and reduced rate of maternal smoking, although improvements in perinatal healthcare and infant sleep environment also may have had substantial impact (8, 81).
As early as 1973, Beckwith proposed that SIDS represents a final common pathway in which similar agonal mechanisms of death are shared by the majority of cases (82). In 1999, Vege and Rognum proposed that a potential death mechanism in classical SIDS could be thought of as a vicious circle, starting with the fatal triangle and ending with death (83, 84). A similar sequence of events, focusing on failures in protective mechanisms against life-threatening events during sleep, was proposed by Kinney and Thach in 2009 (85).
The vicious spiral is initiated by a mucosal infection causing immune activation in the upper respiratory and digestive tract, increased production of cytokines, and overstimulation of the rapidly developing immune system. In susceptible infants, this may trigger aberrant cytokine production or a cytokine storm, which, via retrograde axonal transport, affects sleep regulation and arousal. This, in turn, initiates downregulation of respiration and accumulation of hypoxic markers which is not counteracted by rescue mechanisms, resulting in a vicious spiral that ultimately results in death (Figure 2).
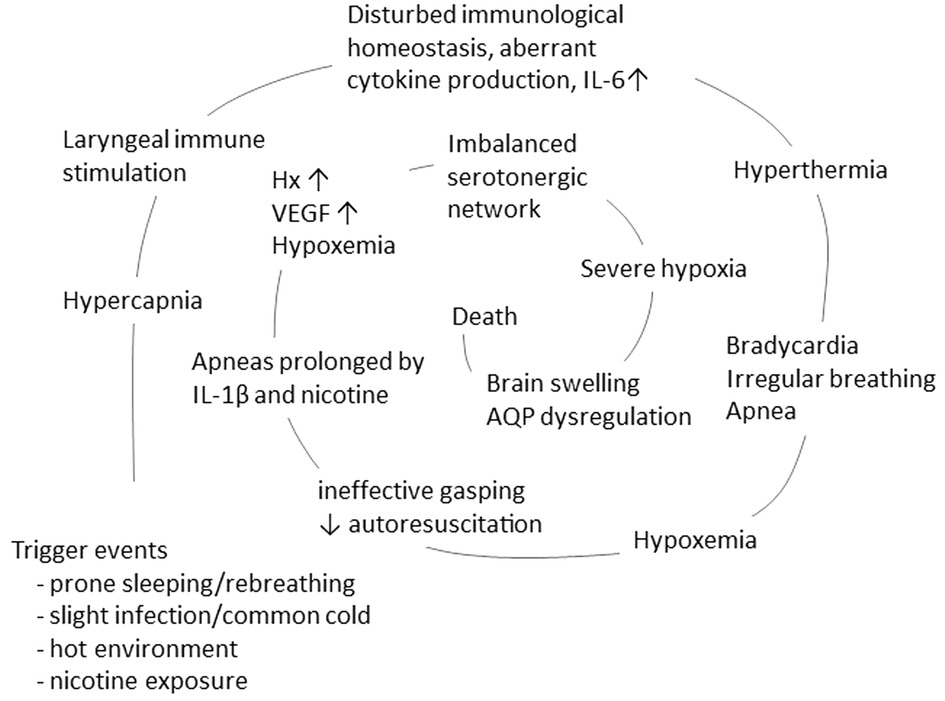
Figure 2. The concept of a vicious spiral of SIDS, suggesting a final common pathway in the majority of SIDS, adapted from Vege et al. 1999 and 2004 (83, 84). The model propose that in the vulnerable infant, a combination of triggers, such as prone position, hot environment and minor infection/common cold, may initiate a spiral leading to death.
Each step of the vicious spiral will be elaborated in the following sections.
The synergistic effect of hazardous sleeping conditions and infection can lead to hypercapnia. Prone sleeping or sleeping in a crib crowded with bedclothes or stuffed animals facilitate rebreathing and increased CO2 level (72). Additionally, even mild upper respiratory infection can increase metabolic rate and consequently CO2 levels in infants older than 3 months (86, 87). Furthermore, hypercapnia have the ability to increase the expression of cytokines and interleukins, including interleukin 1β (IL-1β), tumor necrosis factor α (TNF-α) and IL-6 (88, 89). A possible pathophysiological mechanism initiating the vicious spiral could be the combination of elevated CO2 levels and minor infection, triggering cytokine production and subsequent steps leading to death.
A significant proportion of infants dying in SIDS exhibits signs of infection prior to death, and several risk factors associated with susceptibility to infection have been identified as risk factors for SIDS (84, 90, 91). In a Norwegian SIDS study, it was reported that 46% of the cases had a cold during their last week, and 33% had a cold on the day before death (67). Several studies report findings of bacteria in SIDS, and the common bacterial toxin hypothesis suggests that toxins from common bacteria could be a link between inflammation and SIDS (90–94). Studies have highlighted higher prevalence of Staphylococcus aureus in nasopharyngeal flora from SIDS, as well as staphylococcal endotoxins being more prevalent in samples from intestinal tract from SIDS compared to those from healthy controls (92, 94). Toxic shock syndrome and sepsis may develop rapidly from S. aureus or group A streptococcal infection and cause sudden death. The diagnosis of an infectious cause, rather than SIDS, relies on detection of bacteria in blood or cerebrospinal fluid, as limited pathological signs of inflammation may be detected in the organs with conventional histological methods. Post mortal bacterial growth of uncertain significance is common in samples taken after death, often attributed to post mortal growth. While some bacteria are well established as triggering pathogens and fatal infections, others are not.
Helicobacter pylori infection in childhood is associated with gastrointestinal disease but is mostly asymptomatic (95). However, a relatively large proportion of newborns have H. pylori antigen in stool (96, 97). The same was true for fecal specimen in a high proportion of SIDS victims and infants who died due to severe infection with other microorganisms (97). Interestingly, signs of H. pylori colonization were rarely present in infants who died of violent deaths or due to non-infectious diseases or malformations. The study hypothesized that H. pylori infection in infancy may be involved as a triggering pathogen for sudden death during the first 5 months after birth (97).
Viruses may also affect and compromise the immune response. Respiratory virus RNA has been detected more often in SIDS victims than in non-SIDS cases (98, 99). The seasonal variation involving a winter peak is observed both for virus epidemics causing respiratory infections and SIDS (100). Higher rates of herpesviruses, such as cytomegalovirus and Epstein-Barr virus have been reported in SIDS compared to controls, suggesting a relation and possible contribution in SIDS (101, 102).
Several studies have reported an activation of the mucosal immune system in SIDS. Both a higher number of IgM immunocytes in the tracheal wall, increased IgA immunocytes in the duodenal mucosa, and elevated IgG and IgA immunocyte density in the palatine tonsillar compartments have been reported in SIDS compared to controls (19, 32, 33, 103). In salivary glands an increased number of CD45+ stromal leucocytes, intensified expression of HLA-DR, and increased expression of HLA class I and II have been reported in SIDS compared to controls (20).
Numerous studies have investigated cytokines and interleukins in SIDS (23, 104–107). Initial studies examining IL-6 levels in the cerebrospinal fluid in SIDS disclosed elevated levels compared to controls, with about half of the SIDS victims exhibiting IL-6 levels within the same range as infants dying from severe infection (23, 104). SIDS cases with high IL-6 levels in cerebrospinal fluid and symptoms of mild infection prior to death also showed increased expression of both IgA and HLA-DR in the laryngeal mucosa (24, 34). Another interesting finding is the reported downregulation of myeloid differentiation primary response gene 88 (Myd88) in cerebral tissue from SIDS (108). Myd88 is crucial for both innate and adaptive immune responses, acting as a signal transducer in pathways that regulates the activation of several pro-inflammatory genes. Myd88 plays a pivotal role in initiating and sustaining an effective immune response. A deficiency in this protein may impair the ability to mount an optimal immune reaction, and potentially contributing to vulnerability in SIDS.
In summary, these findings suggest a potential common feature in SIDS; a faulty immunological response to an apparently mild infection (109), initiating an immune reaction in the laryngeal and tracheal mucosa, which either through retrograde axonal transport or by blood borne signals induce an elevated cytokine production within the central nervous system (24, 109, 110). IL-6 induces fever and deeper sleep, and interacts with the serotonergic network in the medulla oblongata (37). This interaction between the immune system and the serotonergic network may interfere with the homeostatic control of cardiorespiratory and arousal responses (26, 37).
Thermal stress and hyperthermia are frequently observed in SIDS. The hyperthermia can result from various external factors such as excessive clothing and high ambient temperature, or intrinsic factors such as mild infection (111, 112). Several studies have documented SIDS victims to be overdressed and/or are discovered in environments with elevated temperatures. Some are described as unusually warm or sweating when found dead (111, 113–115). The prone sleeping position potentiates the risk of overheating by reducing the surface area available for radiant heat loss. One study indicate that the heat loss coefficient can be up to 60% lower in a prone position compared to sleeping supine or on the side (116). In heavily wrapped infants sleeping prone, a combination of infection and a warm environment may be particularly harmful (117).
Thermal stress and overheating can significantly affect respiratory control; even mild thermal stress may potentially lead to an unstable breathing pattern (118). In 12-day-old mouse pups, hyperthermia enhanced the magnitude of bradycardia, suggesting that thermal stress may induce spontaneous bradycardia in an age dependent manner (119). Furthermore, prone sleeping position and subsequent reduced heat loss influence heart rate and blood pressure regulation, especially during the critical age range of 2–3 months when the risk of SIDS is highest (120). High ambient temperature (24°–28°C) could also impede arousal from REM sleep, particularly in the late hours of the night when most SIDS deaths occur (121).
Infection has been demonstrated to disrupt respiratory control mechanisms, alter breathing pattern, and impair arousal (122, 123). Both viruses and bacteria can provoke respiratory disturbances. In infants with respiratory syncytial virus (RSV) infection, it is shown that laryngeal stimulation results in disturbances in the regulation of breathing, most likely through the effect of IL-1β (124). IL-1β has also been found to induce prolonged apnea and modify autoresuscitation in piglets, and even low doses of nicotine and endotoxin have adverse effects on autoresuscitation following apnea by intensifying the effect of IL-1β (125–127).
High IL-1β immunoreactivity have been reported in the brainstem in SIDS (105). This may contribute to molecular interactions causing disturbed homeostatic control of cardiorespiratory and arousal responses. In addition, IL-10 may also play a role. The presence of IL-10 receptors on microglia suggests that IL-10 may affect the control of breathing since microglia modulate respiratory rhythm generation, breathing pattern, and autoresuscitation (128–130). Elevated levels of IL-10 have been reported in thymic tissue from SIDS (107). Another interesting finding, regarding nicotine exposure as a risk factor for SIDS, is that smokers are reported to have lower IL-10 production compared to non-smokers when exposed to infectious agents (131, 132).
An early polysomnographic study of infants who subsequently died of SIDS reported that obstructive and mixed apneas were more frequent and lasted longer in the SIDS victims compared to the control group (133). The study also observed a reduced number of sighs followed by apnea, suggesting a lower peripheral chemoreceptor response in some SIDS cases.
Irregular breathing and apnea may lead to hypoxemia and subsequently hypoxia. In most infants this trigger gasping as a mechanism of autoresuscitation to restore tissue oxygenation. However, in SIDS, several studies have identified incomplete and less frequent arousal from sleep in response to hypoxia, indicating impaired autoresuscitation and a failure of reflexes that are supposed to be protective (134–136). This compromised autoresuscitation fails to restore the respiration, ultimately leading to severe hypoxia, coma and death.
It is notable that several significant risk factors for SIDS, including prone sleeping position, maternal smoking, prematurity and recent infection, all diminish infants’ ability to be aroused from sleep (137). Prone sleeping and maternal smoking have been shown to impair both stimulus-induced and spontaneous arousal, while sleeping in the prone position significantly impairs arousal from both active sleep and quiet sleep in healthy term infants at the age when SIDS incidence is highest (138–141).
A biochemical indication supporting impaired arousal in SIDS is the finding of a reduced orexin immunoreactivity in hypothalamic tissue in SIDS compared to controls (142). Orexin is a neuropeptide involved in respiratory control during sleep and in regulation of the sleep cycle. It has been suggested that hypoxic events occurring before death may be responsible for the decreased orexin immunoreactivity observed in cases of SIDS (142).
Impaired autoresuscitation leads to hypoxemia and if no other rescue mechanisms are activated, it progresses to hypoxia. There are several biochemical markers of acute hypoxia, including hypoxanthine (Hx) (143). In the absence of oxygen, Hx accumulates in body fluids due to hypoxic degradation of adenosine monophosphate (AMP). Studies have shown that vitreous humor Hx levels are elevated in SIDS compared to sudden accidental deaths in infants and children. This suggests that SIDS is preceded by repeated episodes of respiratory failure and hypoxia (17, 18, 144, 145). Animal studies have demonstrated that intermittent periods of hypoxemia result in higher levels of Hx in vitreous humor than chronic hypoxemia (146). In SIDS, a correlation between elevated β-endorphin immunoreactivity in cerebrospinal fluid and increased levels of Hx in the vitreous humor has been reported (147). The neuropeptide β-endorphin is an agonist of opioid receptors in the brain and has the ability to induce respiratory depression. Increased levels may thus aggravate the development of severe hypoxia prior to death in SIDS (147).
Another marker of hypoxia is vascular endothelial growth factor (VEGF). VEGF is highly sensitive to changes in tissue oxygen levels; increased VEGF expression occurs during hypoxia through binding of HIF-1α to a hypoxic responsive element in the VEGF gene (148). A study of VEGF in SIDS found a significantly higher VEGF level in cerebrospinal fluid from cases compared to controls, indicating hypoxia prior to death (149). This finding suggests that the hypoxic episodes occur several hours before death, as this would be the minimum time required for genomic transcription, expression of VEGF and subsequent release into body fluids.
Findings in the brain stem, including gliosis, apoptosis, and reactive astrocytes, also suggest that hypoxemia and episodic hypoxia precedes death in SIDS (150–153). A recent comprehensive study questions the validity of astrogliosis being a feature of SIDS studied glial fibrillary acidic protein (GFAP) expression in astrocytes in tissue from various regions of the brain (154). The findings indicate that GFAP density is high in specific regions of the brain in the first 2 months of life, followed by a decrease during the first year of postnatal development. This was seen in both SIDS and controls. However, the control group were infants who had died of severe infections or cardiovascular disease, conditions that are associated with hypoxia. Thus, what to consider a normal expression of GFAP remains to be clarified.
Neuronal expression of mitochondrial-associated protein-2 (MAP2) is a marker for neuronal damage. It has been shown that when comparing SIDS and controls with and without hypoxic/ischemic injury the level of MAP2-negative reactive neurons was the same in SIDS as in the hypoxic control group (155).
Another key protein in this context is β-amyloid precursor protein (β-APP). When neurons are damaged, the production of β-APP is rapidly upregulated. Similar pattern of β-APP expression have been observed in infants who died from SIDS and infants who died from mechanical asphyxia. Furthermore, it was found higher β-APP staining scores in SIDS sleeping alone compared to SIDS dying when bed-sharing (156, 157). Thus, repeated episodes of hypoxia appear to be a crucial part of the death mechanism in SIDS, leading to neuronal damage and eventually a dysfunction of the serotonergic network.
Serotonin (5-HT), often referred to as the guardian of stable breathing, plays a crucial role in numerous physiological processes. This include recovery from hypoxic reflex-apnea and gasping, restoration of heart rate and blood pressure, termination of apnea, restoration of eupnoea, and arousal. Several studies have presented evidence suggesting a disturbed serotonergic network in cases of SIDS, indicating a failure of normal protective response against life-threatening challenges in these cases (26, 42, 43).
Numerous findings in the brainstem support the hypothesis of a serotonergic dysfunction. These includes a reduced serotonergic 5-HT1A and 5-HT2A receptor binding and immunoreactivity (43, 158, 159), a higher 5-HT neuron count and decreased 5-HT1A and 5-HT2A receptor immunoreactivity (160, 161), as well as a lower ratio of serotonin transporter (5-HTT) binding density to 5-HT neuron count (43, 162). Additionally, there is evidence suggesting abnormal 5-HT2A/C and 5-HT1A signaling across multiple medullary nuclei that are vital for arousal and autoresuscitation in at least a subset of SIDS (163). A study of 5-HT and its primary metabolite 5-hydroxy indoleacetic acid (5-HIAA) in medulla from SIDS found lower 5-HT levels in cases compared to controls, indicating a deficiency in medullary serotonergic activity in SIDS (42). Transgenic mice with over-expression of 5-HT1A in serotonergic neurons exhibit sporadic bradycardia and hypothermia that frequently progress to death (164). This indicates that excessive serotonin autoinhibition could be a risk factor for autonomic dysregulation and thus provide a mechanism for the altered serotonin homeostasis in SIDS.
In addition to 5-HTT, which mediates 5-HT reuptake in the synapsis, brain 5-HT metabolism is also influenced by monoamine oxidase A (MAOA), the latter being responsible for intracellular degradation of 5-HT. The dopaminergic activity is closely modulated by the serotonergic system. A study of dopaminergic neurons in midbrain samples from SIDS and controls reported signs of anatomical and functional degenerations of dopaminergic neurons in a large proportion of the SIDS cases compared to deaths from explained causes (165).
Several studies have investigated genetic variation in genes involved in the serotonergic network, including the genes encoding 5-HTT, MAOA and dopamine transporter (DAT) (53, 54, 166–168). It has been reported significant findings, but as several papers not take into consideration that the MAOA gene is located on the X chromosome and do not divide cases and controls according to sex, the results are inconclusive and difficult to interpret (166, 167).
The γ-aminobutyric acid (GABA) is another essential neurotransmitter in the brainstem. GABA neurons in the medulla oblongata regulate homeostasis through interactions with the medullary serotonergic network, and GABAA receptors are critical markers of GABAergic function. It is interesting that in SIDS it is reported reduced GABAA receptor binding density in nuclei in the medullary serotonergic system (169).
Peripherally produced cytokines can cross the blood-brain barrier and bind to cytokine receptors on neurons in the brainstem, influencing sickness behavior, blunt arousal and also suppress respiration (110, 170). IL-6 affects the central nervous system by activating the hypothalamo-pituitary-adrenocortical axis, thereby increasing brain tryptophan and serotonin metabolism. A study of the expression of IL-6 receptors in the brainstem in SIDS found that the mean IL-6R intensity was significantly higher in SIDS than in controls (37). Hence, aberrant interactions between IL-6 and CO2-sensing regions in the brainstem may contribute to impaired responses to hypercapnia generated by infection combined with prone sleeping position and rebreathing.
Maternal smoking is a recognized major risk factor for SIDS. Nicotine, the primary component in cigarette smoke, has a direct effect on the serotonergic system. Studies on SIDS have reported an effect of both pre- and post-natal maternal smoking in medullary nuclei containing serotonergic neurons (159, 171). Animal studies have demonstrated that prenatal nicotine exposure induces a significant increase in 5-HT turnover. In 5-HT deficient rat pups, it results in impaired arousal (172, 173). Thus, when nicotine exposure interacts with a mild serotonergic deficiency, autoresuscitation failure may be aggravated.
Collectively, these findings have contributed to the formulation of the “Brainstem hypothesis in SIDS” (26). This hypothesis suggests that at least a subset of SIDS is due to abnormal regulation of the serotonergic system and/or impairments in arousal mechanisms. These abnormalities hinder the infant's ability to respond effectively to common sleep-related stressors, such as hypoxia, hypercapnia, asphyxia, and hyperthermia. One potential scenario involves an increased number of serotonergic neurons which could lead to an excess of extracellular 5-HT. This excess will trigger a compensatory down-regulation of serotonergic receptors (43). Cytokines may constitute a link between a dysregulated peripheral immune system and the brainstem serotonergic network (37).
The aquaporin (AQP) family consists of 13 proteins sharing a common structure that facilitate diffusion of water across cell membranes. AQPs are ubiquitously distributed through the organs, including the brain and central nervous system. The most important water channel in the brain is AQP4, but also AQP1 and AQP9 have been reported present (174, 175).
AQP4 plays a crucial role in maintaining brain water homeostasis, neural signal transduction, and development of brain edema (176, 177). AQP4 is co-expressed with the inwardly rectifying potassium channel Kir4.1, forming a multifunctional unit responsible for the clearance of potassium and/or water following neuronal activity (178). Even minor alterations in the expression or function of the AQP4/Kir4.1 complex can disrupt water/ion homeostasis, affecting brain development, increase susceptibility to seizures and contribute to brain swelling and edema formation. This may be particularly unfavorable during the vulnerable developmental stage in the first months of life.
AQP4 also plays a significant role in infection. Studies have demonstrated that IL-1β, IL-6 and TNFα can upregulate the AQP4 expression in astrocytes (179–181). IL-1β has been shown to stimulate the expression of AQP4 in cerebral tissue in a dose-dependent manner. Both AQP4 mRNA and protein levels are increasing with higher doses of IL-1β (182). Furthermore, TNFα and IL-6 secretion is found reduced in astrocyte cultures from AQP4-knockout mice, providing further evidence of a connection between AQP4 water permeability and cytokine release (181). Thus, it is possible that in some SIDS cases, excessive production of cytokines may contribute to a disturbed water/ion homeostasis and development of brain swelling and edema.
Interestingly, AQP4 expression in the hippocampus is lower in infants with the AQP4 genotype rs2075575 CT/TT than the CC genotype, and higher in the youngest infants (≤12 weeks) (183). High-water content of brains of the youngest infants, and finding of association between the CT/TT genotype and the brain/body weight ratio in SIDS victims younger than 12 weeks, may indicate that the rs2075575 CT/TT genotype represents a genetic risk factor for a subgroup of SIDS (56). One possible underlying mechanism is an increased risk of subclinical seizures and a decreased ability to eliminate hypoxia-generated edema fluid.
The aquaporins are important also with regard to the serotonergic network, and it is shown that lack of AQP4 expression is paralleled by alteration in the levels of 5-HT in various areas of the brain (184). It has also been shown that AQP4 participates in regulating K+-stimulated releases of neurotransmitters, including 5-HT (185). This indicates that maintaining a well-functioning water balance is important also for the regulation of neurotransmitters, suggesting a possible connection between the expression and function of aquaporins and the serotonergic imbalances observed in SIDS.
Another common aspect in SIDS is oxidative stress and hypoxia prior to death (17, 18, 144, 145, 149). One key molecule in the oxygen-sensing system is the transcriptional regulator hypoxia-inducible factor (HIF), which controls a range of oxygen responsive target genes such as VEGF and erythropoietin. VEGF has been found upregulated in SIDS, and both VEGF and HIF-1α have the ability to up-regulate the expression of AQP4 (149, 186–188). This up-regulation may serve as a protective response to hypoxia by facilitating elimination of hypoxia-initiated brain edema. However, the upregulation may also induce a molecular pathway leading to upregulation of AQP4, resulting in brain swelling and edema formation through disruption of the blood-brain barrier (187, 188). Furthermore, it is shown that hypercapnia-induced IL-1β overproduction, combined with hypoxia, may increase blood-brain barrier permeability (189).
A few studies have reported higher brain weight in SIDS cases compared to both controls and established standards (190–192). Macroscopic signs of cerebral edema has been described in cases of SIDS, even though these findings are inaccurate. It is important to consider that increased fluid content in postmortal brain tissue might represent congestion associated with the death process, rather than vital edema that developed earlier (193, 194). The brain undergoes significant growth in both size and weight during the first year of life, and one study has indicated an association between specific variants in the AQP4 gene and enlarged brain/body weight ratio in SIDS cases under 3 months of age (56). It is hypothesized that the observed increased brain weight in SIDS, when controlled for age and body weight, may reflect abnormal cerebral development potentially impairing neural control (192).
The vicious spiral is a model suggesting a final common pathway in a large proportion of SIDS (Figure 2). The initiating event is a synergistic effect of trigger events, i.e., prone position with face down in a soft mattress, combined with slight infection/common cold and hot environment, that induce hypercapnia. An adverse response of the mucosal immune system may, via either retrograde axonal transport or blood born, induce cytokine production in the cerebrospinal fluid. The subsequent increase in IL-6 levels induces fever, resulting in thermal stress and hyperthermia. These conditions can cause bradycardia and irregular breathing, leading to ineffective gasping with impaired autoresuscitation. This sequence of events culminates in hypoxemia and downregulation of respiration, further exacerbated by IL-1β and nicotine exposure. Next, hypoxic markers such as hypoxanthine and VEGF are expressed. If the serotonergic network fails to induce an appropriate compensatory mechanism, the infant enters a state of severe hypoxia. This impaired response of the serotonergic network may be worsened by a disturbed water homeostasis in the brain, inducing brain swelling and ultimately coma and death.
The critical step in this model is the downregulation of respiration, followed by impaired autoresuscitation and the accumulation of hypoxic markers. In an infant without vulnerabilities, hypercapnia and hypoxia induce protective responses in the brainstem, ultimately restoring normal oxygen levels. However, in an infant susceptible to SIDS due to a combination of a vulnerable developmental stage and/or having a genetic predisposition, these protective mechanisms are impaired, leading to the progression of hypoxia, subsequent brain swelling, and eventually death.
Understanding the etiology and pathogenesis of SIDS is crucial for preventing these deaths. Prevention of SIDS requires increased knowledge of each step in the vicious spiral, beginning with identifying and avoiding external risk factors. Equally important is a comprehensive understanding of genetic and developmental vulnerabilities, as well as further examination of the mechanisms that, when combined, lead to the death of an infant.
SIDS is a devastating but rare event, and the SIDS rate have steadily decreased since the back-to-sleep campaigns in the early 1990s. This trend is seen reflected in reduction of total post neonatal mortality (8, 81). Because of this very positive reduction, many published papers report relatively few SIDS cases and even fewer suitable controls. Ideally, the most appropriate control group for SIDS research would consist of healthy infants who experience a sudden and rapid death from a violent cause and yet have an uninjured brain. Fortunately, such cases are rare.
The impact of correct controls, as well as the impact of postmortem time and temperature, might be of particular importance with regard to hypoxic markers. Though there are morphological clues for previous episodes of hypoxia in SIDS, it has been difficult to find markers for demonstration of asphyxia in the period preceding death (10). Our study of increased levels of Hx in vitreous humor in SIDS was commented on by Beckwith, who pointed to uncertain postmortem intervals and the “warm room effect” peculiar for SIDS (17, 18, 195). A study from Carpenter et al, which by comparing Hx level in cerebrospinal fluid in SIDS and deaths from a “variety of conditions” concluded that Hx could not be a marker of pre-mortem hypoxia in SIDS (196). The latter study illustrates the challenges posed by lack of appropriate controls in SIDS research. In our opinion, the only suitable controls are infants and small children who have died suddenly from accidents (197). By controlling for postmortem time and ambient temperature, we were able to confirm previous observations of significant higher Hx levels in SIDS than in violent death, whether there were no significant difference between SIDS and infectious death (144, 145). Nevertheless, it is important that these and other findings are confirmed in independent studies.
Although the definition of SIDS always has involved aspects of exclusion, the diagnostic criteria have evolved over the years. This evolution began with the Seattle definition in 1969, continued with the NICHD definition in 1989, and eventually led to the now most widely used San Diego definition in 2004 (1–3). Despite these advancements, a large proportion of SIDS research include infant cohorts without clear specification of definition used or the extent of the investigations performed (6, 198, 199). This lack of standardization complicates the ability to compare data across studies.
As stated for studies of postmortem asphyxia, a limitation of today's research on death mechanisms in SIDS is lack of reproducing studies. Several factors contribute to this, including limited access to tissue and fluid samples from SIDS cases classified according to international standards, suitable controls, and expertise and facilities to perform high quality studies (6, 200). To address this issue, largescale collaborative studies that include well defined SIDS cases and control groups are crucial. These studies should aim to systematically replicate and validate previous findings. Such initiatives should be encouraged.
SO: Conceptualization, Writing – original draft, Writing – review & editing. AS-P: Writing – original draft, Writing – review & editing. JE: Writing – original draft, Writing – review & editing. ÅV: Conceptualization, Writing – original draft, Writing – review & editing. LF: Writing – original draft, Writing – review & editing. TR: Conceptualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Abraham B, Bergman JBB, George Ray C. Sudden infant death syndrome: proceedings of the second international conference on causes of sudden death in infants; Seattle: University of Washington press (1970).
2. Willinger M, James LS, Catz C. Defining the sudden infant death syndrome (SIDS): deliberations of an expert panel convened by the national institute of child health and human development. Pediatr Pathol. (1991) 11(5):677–84. doi: 10.3109/15513819109065465
3. Krous HF, Beckwith JB, Byard RW, Rognum TO, Bajanowski T, Corey T, et al. Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics. (2004) 114(1):234–8. doi: 10.1542/peds.114.1.234
4. Krous HF, Chadwick AE, Crandall L, Nadeau-Manning JM. Sudden unexpected death in childhood: a report of 50 cases. Pediatr Dev Pathol. (2005) 8(3):307–19. doi: 10.1007/s10024-005-1155-8
5. Vege A, Rognum TO. Use of new Nordic criteria for classification of SIDS to re-evaluate diagnoses of sudden unexpected infant death in the Nordic countries. Acta Paediatr. (1997) 86(4):391–6. doi: 10.1111/j.1651-2227.1997.tb09029.x
6. Bajanowski T, Vege A, Byard RW, Krous HF, Arnestad M, Bachs L, et al. Sudden infant death syndrome (SIDS)–standardised investigations and classification: recommendations. Forensic Sci Int. (2007) 165(2–3):129–43. doi: 10.1016/j.forsciint.2006.05.028
7. Byard RW. The autopsy and pathology of sudden infant death syndrome. In: Duncan JR, Byard RW, editors. SIDS Sudden Infant and Early Childhood Death: The Past, the Present and the Future. Adelaide, AU: University of Adelaide Press (2018). p. 497–538.
8. Rognum TO, Vege A, Stray-Pedersen A, Boylestad L. A Scandinavian perspective. In: Duncan JR, Byard RW, editors. SIDS Sudden Infant and Early Childhood Death: The Past, the Present and the Future. Adelaide, AU: University of Adelaide Press (2018). p. 421–9.
9. Matshes EW, Lew EO. An approach to the classification of apparent asphyxial infant deaths. Acad Forensic Pathol. (2017) 7(2):200–11. doi: 10.23907/2017.021
10. Wojcik MH, Poduri AH, Holm IA, MacRae CA, Goldstein RD. The fundamental need for unifying phenotypes in sudden unexpected pediatric deaths. Front Med (Lausanne). (2023) 10:1166188. doi: 10.3389/fmed.2023.1166188
11. Goldstein RD, Blair PS, Sens MA, Shapiro-Mendoza CK, Krous HF, Rognum TO, et al. Inconsistent classification of unexplained sudden deaths in infants and children hinders surveillance, prevention and research: recommendations from the 3rd international congress on sudden infant and child death. Forensic Sci Med Pathol. (2019) 15(4):622–8. doi: 10.1007/s12024-019-00156-9
12. Wedgwood RJ. Sudden and unexpected deaths in infancy (cot deaths). In: Camps FE, Carpenter R, editors. Sudden and Unexpected Deaths in Infancy (Cot Deaths). Bristol, England: John Wright & Sons LTD (1972). p. 22–8.
13. Valdes-Dapena M. Are some crib deaths sudden cardiac deaths? J Am Coll Cardiol. (1985) 5(6 Suppl):113B–7. doi: 10.1016/S0735-1097(85)80539-8
14. Valdes-Dapena M. Sudden infant death syndrome. Morphology update for forensic pathologists–1985. Forensic Sci Int. (1986) 30(2–3):177–86. doi: 10.1016/0379-0738(86)90012-5
15. Naeye RL. Brain-stem and adrenal abnormalities in the sudden-infant-death syndrome. Am J Clin Pathol. (1976) 66(3):526–30. doi: 10.1093/ajcp/66.3.526
16. Rognum TO, Saugstad OD. Biochemical and immunological studies in SIDS victims. Clues to understanding the death mechanism. Acta Paediatr. (1993) 82(Suppl 389):82–5. doi: 10.1111/j.1651-2227.1993.tb12886.x
17. Rognum TO, Saugstad OD, Oyasaeter S, Olaisen B. Elevated levels of hypoxanthine in vitreous humor indicate prolonged cerebral hypoxia in victims of sudden infant death syndrome. Pediatrics. (1988) 82(4):615–8. doi: 10.1542/peds.82.4.615
18. Rognum TO, Saugstad OD. Hypoxanthine levels in vitreous humor: evidence of hypoxia in most infants who died of sudden infant death syndrome. Pediatrics. (1991) 87(3):306–10. doi: 10.1542/peds.87.3.306
19. Thrane PS, Rognum TO, Brandtzaeg P. Increased immune response in upper respiratory and digestive tracts in SIDS. Lancet. (1990) 335(8683):229–30. doi: 10.1016/0140-6736(90)90325-Y
20. Thrane PS, Rognum TO, Brandtzaeg P. Up-regulated epithelial expression of HLA-DR and secretory component in salivary glands: reflection of mucosal immunostimulation in sudden infant death syndrome. Pediatr Res. (1994) 35(5):625–8. doi: 10.1203/00006450-199405000-00017
21. Rognum TO, Thrane S, Stoltenberg L, Vege A, Brandtzaeg P. Development of intestinal mucosal immunity in fetal life and the first postnatal months. Pediatr Res. (1992) 32(2):145–9. doi: 10.1203/00006450-199208000-00003
22. Stoltenberg L, Thrane PS, Rognum TO. Development of immune response markers in the trachea in the fetal period and the first year of life. Pediatr Allergy Immunol. (1993) 4(1):13–9. doi: 10.1111/j.1399-3038.1993.tb00059.x
23. Vege A, Rognum TO, Scott H, Aasen AO, Saugstad OD. SIDS cases have increased levels of interleukin-6 in cerebrospinal fluid. Acta Paediatr. (1995) 84(2):193–6. doi: 10.1111/j.1651-2227.1995.tb13608.x
24. Vege A, Rognum TO, Anestad G. IL-6 cerebrospinal fluid levels are related to laryngeal IgA and epithelial HLA-DR response in sudden infant death syndrome. Pediatr Res. (1999) 45(6):803–9. doi: 10.1203/00006450-199906000-00004
25. Filiano JJ, Kinney HC. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple-risk model. Biol Neonate. (1994) 65(3–4):194–7. doi: 10.1159/000244052
26. Kinney HC, Haynes RL. The serotonin brainstem hypothesis for the sudden infant death syndrome. J Neuropathol Exp Neurol. (2019) 78:765–79. doi: 10.1093/jnen/nlz062
27. Kinney HC, Belliveau RA, Trachtenberg FL, Rava LA, Paterson DS. The development of the medullary serotonergic system in early human life. Auton Neurosci. (2007) 132(1–2):81–102. doi: 10.1016/j.autneu.2006.11.001
28. Perrone S, Lembo C, Moretti S, Prezioso G, Buonocore G, Toscani G, et al. Sudden infant death syndrome: beyond risk factors. Life (Basel). (2021) 11(3):1–14. doi: 10.3390/life11030184
29. Thrane PS, Rognum TO, Brandtzaeg P. Ontogenesis of the secretory immune system and innate defence factors in human parotid glands. Clin Exp Immunol. (2008) 86(2):342–8. doi: 10.1111/j.1365-2249.1991.tb05820.x
30. Gleeson M, Clancy RL, Cripps AW. Mucosal immune response in a case of sudden infant death syndrome. Pediatr Res. (1993) 33(6):554–6. doi: 10.1203/00006450-199306000-00003
31. Brandtzaeg P, Nilssen DE, Rognum TO, Thrane PS. Ontogeny of the mucosal immune system and IgA deficiency. Gastroenterol Clin North Am. (1991) 20(3):397–439. doi: 10.1016/S0889-8553(21)00564-1
32. Forsyth KD, Weeks SC, Koh L, Skinner J, Bradley J. Lung immunoglobulins in the sudden infant death syndrome. Br Med J. (1989) 298(6665):23–6. doi: 10.1136/bmj.298.6665.23
33. Stoltenberg L, Saugstad OD, Rognum TO. Sudden infant death syndrome victims show local immunoglobulin M response in tracheal wall and immunoglobulin A response in duodenal mucosa. Pediatr Res. (1992) 31(4 Pt 1):372–5. doi: 10.1203/00006450-199204000-00013
34. Ferrante L, Opdal SH, Vege A, Rognum TO. Is there any correlation between HLA-DR expression in laryngeal mucosa and interleukin gene variation in sudden infant death syndrome? Acta Paediatr. (2013) 102(3):308–13. doi: 10.1111/apa.12107
35. Maehlen J, Olsson T, Zachau A, Klareskog L, Kristensson K. Local enhancement of major histocompatibility complex (MHC) class I and II expression and cell infiltration in experimental allergic encephalomyelitis around axotomized motor neurons. J Neuroimmunol. (1989) 23(2):125–32. doi: 10.1016/0165-5728(89)90031-3
36. Vollmer-Conna U, Fazou C, Cameron B, Li H, Brennan C, Luck L, et al. Production of pro-inflammatory cytokines correlates with the symptoms of acute sickness behaviour in humans. Psychol Med. (2004) 34(7):1289–97. doi: 10.1017/S0033291704001953
37. Rognum IJ, Haynes RL, Vege A, Yang M, Rognum TO, Kinney HC. Interleukin-6 and the serotonergic system of the medulla oblongata in the sudden infant death syndrome. Acta Neuropathol. (2009) 118(4):519–30. doi: 10.1007/s00401-009-0535-y
38. Vege A, Rognum TO, Opdal SH. SIDS–changes in the epidemiological pattern in eastern Norway 1984–1996. Forensic Sci Int. (1998) 93(2–3):155–66. doi: 10.1016/S0379-0738(98)00048-6
39. Stray-Pedersen A, Vege A, Stray-Pedersen A, Holmskov U, Rognum TO. Post-neonatal drop in alveolar SP-A expression: biological significance for increased vulnerability to SIDS? Pediatr Pulmonol. (2008) 43(2):160–8. doi: 10.1002/ppul.20750
40. Kinney HC, Burger PC, Harrell FE Jr., Hudson RP Jr. “Reactive gliosis” in the medulla oblongata of victims of the sudden infant death syndrome. Pediatrics. (1983) 72(2):181–7. doi: 10.1542/peds.72.2.181
41. Storm H, Nylander G, Saugstad OD. The amount of brainstem gliosis in sudden infant death syndrome (SIDS) victims correlates with maternal cigarette smoking during pregnancy. Acta Paediatr. (1999) 88(1):13–8. doi: 10.1111/j.1651-2227.1999.tb01260.x
42. Duncan JR, Paterson DS, Hoffman JM, Mokler DJ, Borenstein NS, Belliveau RA, et al. Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA. (2010) 303(5):430–7. doi: 10.1001/jama.2010.45
43. Paterson DS, Trachtenberg FL, Thompson EG, Belliveau RA, Beggs AH, Darnall R, et al. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA. (2006) 296(17):2124–32. doi: 10.1001/jama.296.17.2124
44. Hunt NJ, Phillips L, Waters KA, Machaalani R. Proteomic MALDI-TOF/TOF-IMS examination of peptide expression in the formalin fixed brainstem and changes in sudden infant death syndrome infants. J Proteomics. (2016) 138:48–60. doi: 10.1016/j.jprot.2016.02.022
45. Brownstein CA, Poduri A, Goldstein RD, Holm IA. The genetics of sudden infant death syndrome. In: Duncan JR, Byard RW, editors. SIDS Sudden Infant and Early Childhood Death: The Past, the Present and the Future. Adelaide, AU: University of Adelaide Press (2018). p. 711–30.
46. Keywan C, Poduri AH, Goldstein RD, Holm IA. Genetic factors underlying sudden infant death syndrome. Appl Clin Genet. (2021) 14:61–76. doi: 10.2147/TACG.S239478
47. Koh HY, Haghighi A, Keywan C, Alexandrescu S, Plews-Ogan E, Haas EA, et al. Genetic determinants of sudden unexpected death in pediatrics. Genet Med. (2022) 24(4):839–50. doi: 10.1016/j.gim.2021.12.004
48. Bard AM, Clark LV, Cosgun E, Aldinger KA, Timms A, Quina LA, et al. Known pathogenic gene variants and new candidates detected in sudden unexpected infant death using whole genome sequencing. Am J Med Genet A. (2024) 194:e63596. doi: 10.1002/ajmg.a.63596
49. Opdal SH, Rognum TO. Gene variants predisposing to SIDS: current knowledge. Forensic Sci Med Pathol. (2011) 7(1):26–36. doi: 10.1007/s12024-010-9182-9
50. Ferrante L, Opdal SH. Sudden infant death syndrome and the genetics of inflammation. Front Immunol. (2015) 6:63. doi: 10.3389/fimmu.2015.00063
51. Fard D, Laer K, Rothamel T, Schurmann P, Arnold M, Cohen M, et al. Candidate gene variants of the immune system and sudden infant death syndrome. Int J Legal Med. (2016) 130(4):1025–33. doi: 10.1007/s00414-016-1347-y
52. Opdal SH. Cytokines, infection, and immunity. In: Duncan JR, Byard RW, editors. SIDS Sudden Infant and Early Childhood Death: The Past, the Present and the Future. Adelaide, AU: University of Adelaide (2018). p. 689–710.
53. Weese-Mayer DE, Zhou L, Berry-Kravis EM, Maher BS, Silvestri JM, Marazita ML. Association of the serotonin transporter gene with sudden infant death syndrome: a haplotype analysis. Am J Med Genet A. (2003) 122A(3):238–45. doi: 10.1002/ajmg.a.20427
54. Weese-Mayer DE, Berry-Kravis EM, Maher BS, Silvestri JM, Curran ME, Marazita ML. Sudden infant death syndrome: association with a promoter polymorphism of the serotonin transporter gene. Am J Med Genet A. (2003) 117A(3):268–74. doi: 10.1002/ajmg.a.20005
55. Weese-Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Curran ME, Silvestri JM, et al. Sudden infant death syndrome: case-control frequency differences at genes pertinent to early autonomic nervous system embryologic development. Pediatr Res. (2004) 56(3):391–5. doi: 10.1203/01.PDR.0000136285.91048.4A
56. Opdal SH, Vege A, Stray-Pedersen A, Rognum TO. Aquaporin-4 gene variation and sudden infant death syndrome. Pediatr Res. (2010) 68(1):48–51. doi: 10.1203/PDR.0b013e3181df4e7c
57. Opdal SH, Vege A, Stray-Pedersen A, Rognum TO. The gene encoding the inwardly rectifying potassium channel Kir4.1 may be involved in sudden infant death syndrome. Acta Paediatr. (2017) 106(9):1474–80. doi: 10.1111/apa.13928
58. Pfisterer N, Meyer-Bockenkamp F, Qu D, Preuss V, Rothamel T, Geisenberger D, et al. Sudden infant death syndrome revisited: serotonin transporter gene, polymorphisms and promoter methylation. Pediatr Res. (2021) 92:694–9. doi: 10.1038/s41390-021-01773-3
59. Zhou Q, Gong D, Zhang Y, Huang F. Association between monoamine oxidase A promoter polymorphism and the risk of sudden infant death syndrome: a meta-analysis. Int J Legal Med. (2021) 135(4):1179–90. doi: 10.1007/s00414-020-02496-6
60. Qu D, Schurmann P, Rothamel T, Dork T, Klintschar M. Variants in genes encoding the SUR1-TRPM4 non-selective cation channel and sudden infant death syndrome (SIDS): potentially increased risk for cerebral edema. Int J Legal Med. (2022) 136(4):1113–20. doi: 10.1007/s00414-022-02819-9
61. Uzuntas E, Schurmann P, Rothamel T, Dork T, Klintschar M. Polymorphisms of the hypothalamic-pituitary-adrenal axis may lead to an inadequate response to stress and contribute to sudden infant death syndrome. Acta Paediatr. (2023) 112(7):1478–84. doi: 10.1111/apa.16772
62. Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C, et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. (2007) 115(3):361–7. doi: 10.1161/CIRCULATIONAHA.106.658021
63. Tester DJ, Wong LCH, Chanana P, Jaye A, Evans JM, FitzPatrick DR, et al. Cardiac genetic predisposition in sudden infant death syndrome. J Am Coll Cardiol. (2018) 71(11):1217–27. doi: 10.1016/j.jacc.2018.01.030
64. Liebrechts-Akkerman G, Liu F, van Marion R, Dinjens WNM, Kayser M. Explaining sudden infant death with cardiac arrhythmias: complete exon sequencing of nine cardiac arrhythmia genes in Dutch SIDS cases highlights new and known DNA variants. Forensic Sci Int Genet. (2020) 46:102266. doi: 10.1016/j.fsigen.2020.102266
65. Koffer J, Scheiper-Welling S, Verhoff MA, Bajanowski T, Kauferstein S. Post-mortem genetic investigation of cardiac disease-associated genes in sudden infant death syndrome (SIDS) cases. Int J Legal Med. (2021) 135(1):207–12. doi: 10.1007/s00414-020-02394-x
66. Moon RY, Hauck FR. Risk factors and theories. In: Duncan JR, Byard RW, editors. SIDS Sudden Infant and Early Childhood Death: The Past, the Present and the Future. Adelaide, AU: University of Adelaide Press (2018). p. 169–85.
67. Arnestad M, Andersen M, Vege A, Rognum TO. Changes in the epidemiological pattern of sudden infant death syndrome in southeast Norway, 1984–1998: implications for future prevention and research. Arch Dis Child. (2001) 85(2):108–15. doi: 10.1136/adc.85.2.108
68. MacFarlane ME, Thompson JMD, Wilson J, Lawton B, Taylor B, Elder DE, et al. Infant sleep hazards and the risk of sudden unexpected death in infancy. J Pediatr. (2022) 245:56–64. doi: 10.1016/j.jpeds.2022.01.044
69. Pease A, Turner N, Ingram J, Fleming P, Patrick K, Williams T, et al. Changes in background characteristics and risk factors among SIDS infants in England: cohort comparisons from 1993 to 2020. BMJ Open. (2023) 13(10):e076751. doi: 10.1136/bmjopen-2023-076751
70. Sidebotham P, Bates F, Ellis C, Lyus L. Preventive strategies for sudden infant death syndrome. In: Duncan JR, Byard RW, editors. SIDS Sudden Infant and Early Childhood Death: The Past, the Present and the Future. Adelaide, AU: University of Adelaide Press (2018). p. 217–56.
71. Boylestad L, Stray-Pedersen A, Vege A, Osberg S, Rognum T. Death-scene investigations contribute to legal protection in unexpected child deaths in Norway. Acta Paediatr. (2020) 109(12):2627–35. doi: 10.1111/apa.15284
72. Kemp JS, Kowalski RM, Burch PM, Graham MA, Thach BT. Unintentional suffocation by rebreathing: a death scene and physiologic investigation of a possible cause of sudden infant death. J Pediatr. (1993) 122(6):874–80. doi: 10.1016/S0022-3476(09)90010-5
73. Ramirez JM, Ramirez SC, Anderson TM. Sudden infant death syndrome, sleep, and the physiology and pathophysiology of the respiratory network. In: Duncan JR, Byard RW, editors. SIDS Sudden Infant and Early Childhood Death: The Past, the Present and the Future. Adelaide, AU: University of Adelaide Press (2018). p. 616–40.
74. Anderson TM, Lavista Ferres JM, Ren SY, Moon RY, Goldstein RD, Ramirez JM, et al. Maternal smoking before and during pregnancy and the risk of sudden unexpected infant death. Pediatrics. (2019) 143(4):1–8. doi: 10.1542/peds.2018-3325
75. Sun J, Liu X, Zhao M, Magnussen CG, Xi B. Dose-response association between maternal smoking during pregnancy and the risk of infant death: a nationwide, population-based, retrospective cohort study. EClinicalMedicine. (2023) 57:101858. doi: 10.1016/j.eclinm.2023.101858
76. Mitchell EA, Milerad J. Smoking and the sudden infant death syndrome. Rev Environ Health. (2006) 21(2):81–103. doi: 10.1515/REVEH.2006.21.2.81
77. Liebrechts-Akkerman G, Lao O, Liu F, van Sleuwen BE, Engelberts AC, L’Hoir M P, et al. Postnatal parental smoking: an important risk factor for SIDS. Eur J Pediatr. (2011) 170(10):1281–91. doi: 10.1007/s00431-011-1433-6
78. Filonzi L, Magnani C, Lavezzi AM, Vaghi M, Nosetti L, Nonnis Marzano F. Detoxification genes polymorphisms in SIDS exposed to tobacco smoke. Gene. (2018) 648:1–4. doi: 10.1016/j.gene.2018.01.034
79. Helweg-Larsen K, Lundemose JB, Oyen N, Skjaerven R, Alm B, Wennergren G, et al. Interactions of infectious symptoms and modifiable risk factors in sudden infant death syndrome. The Nordic epidemiological SIDS study. Acta Paediatr. (1999) 88(5):521–7. doi: 10.1111/j.1651-2227.1999.tb00168.x
80. Ponsonby AL, Dwyer T, Gibbons LE, Cochrane JA, Wang YG. Factors potentiating the risk of sudden infant death syndrome associated with the prone position. N Engl J Med. (1993) 329(6):377–82. doi: 10.1056/NEJM199308053290601
81. Goldstein RD, Trachtenberg FL, Sens MA, Harty BJ, Kinney HC. Overall postneonatal mortality and rates of SIDS. Pediatrics. (2016) 137(1):1–10. doi: 10.1542/peds.2015-2298
82. Beckwith JB. The sudden infant death syndrome. Curr Probl Pediatr. (1973) 3(8):1–36. doi: 10.1016/S0045-9380(73)80001-5
83. Vege A, Rognum TO. Inflammatory responses in sudden infant death syndrome—past and present views. FEMS Immunol Med Microbiol. (1999) 25(1–2):67–78. doi: 10.1111/j.1574-695X.1999.tb01328.x
84. Vege A, Ole Rognum T. Sudden infant death syndrome, infection and inflammatory responses. FEMS Immunol Med Microbiol. (2004) 42(1):3–10. doi: 10.1016/j.femsim.2004.06.015
85. Kinney HC, Thach BT. The sudden infant death syndrome. N Engl J Med. (2009) 361(8):795–805. doi: 10.1056/NEJMra0803836
86. Fleming PJ, Howell T, Clements M, Lucas J. Thermal balance and metabolic rate during upper respiratory tract infection in infants. Arch Dis Child. (1994) 70(3):187–91. doi: 10.1136/adc.70.3.187
87. Buchanan GF. Impaired CO(2)-induced arousal in SIDS and SUDEP. Trends Neurosci. (2019) 42(4):242–50. doi: 10.1016/j.tins.2019.02.002
88. Ding H, Li Y, Li X, Liu X, Chen S, Liu M, et al. Treatment with 7% and 10% CO(2) enhanced expression of IL-1beta, TNF-alpha, and IL-6 in hypoxic cultures of human whole blood. J Int Med Res. (2020) 48(4):300060520912105. doi: 10.1177/0300060520912105
89. Ding HG, Deng YY, Yang RQ, Wang QS, Jiang WQ, Han YL, et al. Hypercapnia induces IL-1beta overproduction via activation of NLRP3 inflammasome: implication in cognitive impairment in hypoxemic adult rats. J Neuroinflammation. (2018) 15(1):4. doi: 10.1186/s12974-017-1051-y
90. Highet AR. An infectious aetiology of sudden infant death syndrome. J Appl Microbiol. (2008) 105(3):625–35. doi: 10.1111/j.1365-2672.2008.03747.x
91. Alfelali M, Khandaker G. Infectious causes of sudden infant death syndrome. Paediatr Respir Rev. (2014) 15(4):307–11. doi: 10.1016/j.prrv.2014.09.004
92. Blackwell CC, MacKenzie DA, James VS, Elton RA, Zorgani AA, Weir DM, et al. Toxigenic bacteria and sudden infant death syndrome (SIDS): nasopharyngeal flora during the first year of life. FEMS Immunol Med Microbiol. (1999) 25(1–2):51–8. doi: 10.1111/j.1574-695X.1999.tb01326.x
93. Morris JA. The common bacterial toxins hypothesis of sudden infant death syndrome. FEMS Immunol Med Microbiol. (1999) 25(1–2):11–7. doi: 10.1111/j.1574-695X.1999.tb01322.x
94. Highet AR, Berry AM, Bettelheim KA, Goldwater PN. Gut microbiome in sudden infant death syndrome (SIDS) differs from that in healthy comparison babies and offers an explanation for the risk factor of prone position. Int J Med Microbiol. (2014) 304(5–6):735–41. doi: 10.1016/j.ijmm.2014.05.007
95. Matos I A, Oliva SE D, Escobedo AA, Villa Jimenez OM, Velazco Villaurrutia YDC. Helicobacter pylori infection in children. BMJ Paediatr Open. (2020) 4(1):e000679. doi: 10.1136/bmjpo-2020-000679
96. Stray-Pedersen A, Gaustad P, Stray-Pedersen B, Rognum TO. Detection rate of Helicobacter pylori stool antigen in newborn infants and small children. J Perinat Med. (2007) 35(2):155–8. doi: 10.1515/JPM.2007.040
97. Stray-Pedersen A, Vege A, Rognum TO. Helicobacter pylori antigen in stool is associated with SIDS and sudden infant deaths due to infectious disease. Pediatr Res. (2008) 64(4):405–10. doi: 10.1203/PDR.0b013e31818095f7
98. Bajanowski T, Rolf B, Jorch G, Brinkmann B. Detection of RNA viruses in sudden infant death (SID). Int J Legal Med. (2003) 117(4):237–40. doi: 10.1007/s00414-003-0367-6
99. Niklasson B, Almqvist PR, Hornfeldt B, Klitz W. Sudden infant death syndrome and ljungan virus. Forensic Sci Med Pathol. (2009) 5(4):274–9. doi: 10.1007/s12024-009-9086-8
100. Douglas AS, Helms PJ, Jolliffe IT. Seasonality of sudden infant death syndrome in mainland Britain and Ireland 1985–95. Arch Dis Child. (1998) 79(3):269–70. doi: 10.1136/adc.79.3.269
101. Dettmeyer R, Baasner A, Schlamann M, Padosch SA, Haag C, Kandolf R, et al. Role of virus-induced myocardial affections in sudden infant death syndrome: a prospective postmortem study. Pediatr Res. (2004) 55(6):947–52. doi: 10.1203/01.pdr.0000127022.45831.54
102. Alvarez-Lafuente R, Aguilera B, Suarez-Mier MA, Morentin B, Vallejo G, Gomez J, et al. Detection of human herpesvirus-6, Epstein–Barr virus and cytomegalovirus in formalin-fixed tissues from sudden infant death: a study with quantitative real-time PCR. Forensic Sci Int. (2008) 178(2–3):106–11. doi: 10.1016/j.forsciint.2008.02.007
103. Stoltenberg L, Vege A, Saugstad OD, Rognum TO. Changes in the concentration and distribution of immunoglobulin-producing cells in SIDS palatine tonsils. Pediatr Allergy Immunol. (1995) 6(1):48–55. doi: 10.1111/j.1399-3038.1995.tb00258.x
104. Vege A, Rognum TO, Aasen AO, Saugstad OD. Are elevated cerebrospinal fluid levels of IL-6 in sudden unexplained deaths, infectious deaths and deaths due to heart/lung disease in infants and children due to hypoxia? Acta Paediatr. (1998) 87(8):819–24. doi: 10.1111/j.1651-2227.1998.tb01544.x
105. Kadhim H, Kahn A, Sebire G. Distinct cytokine profile in SIDS brain: a common denominator in a multifactorial syndrome? Neurology. (2003) 61(9):1256–9. doi: 10.1212/01.WNL.0000092014.14997.47
106. Vennemann MM, Loddenkotter B, Fracasso T, Mitchell EA, Debertin AS, Larsch KP, et al. Cytokines and sudden infant death. Int J Legal Med. (2012) 126(2):279–84. doi: 10.1007/s00414-011-0638-6
107. Qu D, Preuss V, Hagemeier L, Radomsky L, Beushausen K, Keil J, et al. Age-related cytokine imbalance in the thymus in sudden infant death syndrome (SIDS). Pediatr Res. (2024) 95:949–58. doi: 10.1038/s41390-023-02809-6
108. Ferrante L, Rognum TO, Vege A, Nygard S, Opdal SH. Altered gene expression and possible immunodeficiency in cases of sudden infant death syndrome. Pediatr Res. (2016) 80(1):77–84. doi: 10.1038/pr.2016.45
109. Blackwell C, Moscovis S, Hall S, Burns C, Scott RJ. Exploring the risk factors for sudden infant deaths and their role in inflammatory responses to infection. Front Immunol. (2015) 6:44. doi: 10.3389/fimmu.2015.00044
110. Thrane PS, Maehlen J, Stoltenberg L, Brandtzaeg P. Retrograde axonal cytokine transport: a pathway for immunostimulation in the brain inducing hypoxia and sudden infant death? Med Hypotheses. (1995) 44(2):81–4. doi: 10.1016/0306-9877(95)90074-8
111. Kleemann WJ, Schlaud M, Poets CF, Rothamel T, Troger HD. Hyperthermia in sudden infant death. Int J Legal Med. (1996) 109(3):139–42. doi: 10.1007/BF01369674
112. Bach V, Libert JP. Hyperthermia and heat stress as risk factors for sudden infant death syndrome: a narrative review. Front Pediatr. (2022) 10:816136. doi: 10.3389/fped.2022.816136
113. Stanton AN. Sudden infant death. Overheating and Cot death. Lancet. (1984) 324(8413):1199–201. doi: 10.1016/S0140-6736(84)92753-3
114. Ponsonby AL, Dwyer T, Gibbons LE, Cochrane JA, Jones ME, McCall MJ. Thermal environment and sudden infant death syndrome: case-control study. Br Med J. (1992) 304(6822):277–82. doi: 10.1136/bmj.304.6822.277
115. Fleming PJ, Levine MR, Azaz Y, Wigfield R, Stewart AJ. Interactions between thermoregulation and the control of respiration in infants: possible relationship to sudden infant death. Acta Paediatr. (1993) 82(Suppl 389):57–9. doi: 10.1111/j.1651-2227.1993.tb12878.x
116. Tuffnell CS, Petersen SA, Wailoo MP. Prone sleeping infants have a reduced ability to lose heat. Early Hum Dev. (1995) 43(2):109–16. doi: 10.1016/0378-3782(95)01659-7
117. Gilbert R, Rudd P, Berry PJ, Fleming PJ, Hall E, White DG, et al. Combined effect of infection and heavy wrapping on the risk of sudden unexpected infant death. Arch Dis Child. (1992) 67(2):171–7. doi: 10.1136/adc.67.2.171
118. Riesenfeld T, Hammarlund K, Norsted T, Sedin G. Irregular breathing in young lambs and newborn infants during heat stress. Acta Paediatr. (1996) 85(4):467–70. doi: 10.1111/j.1651-2227.1996.tb14063.x
119. Cummings KJ, Li A, Deneris ES, Nattie EE. Bradycardia in serotonin-deficient pet-1-/- mice: influence of respiratory dysfunction and hyperthermia over the first 2 postnatal weeks. Am J Physiol Regul Integr Comp Physiol. (2010) 298(5):R1333–42. doi: 10.1152/ajpregu.00110.2010
120. Yiallourou SR, Walker AM, Horne RS. Prone sleeping impairs circulatory control during sleep in healthy term infants: implications for SIDS. Sleep. (2008) 31(8):1139–46.18714786
121. Franco P, Scaillet S, Valente F, Chabanski S, Groswasser J, Kahn A. Ambient temperature is associated with changes in infants’ arousability from sleep. Sleep. (2001) 24(3):325–9. doi: 10.1093/sleep/24.3.325
122. Horne RS, Osborne A, Vitkovic J, Lacey B, Andrew S, Chau B, et al. Arousal from sleep in infants is impaired following an infection. Early Hum Dev. (2002) 66(2):89–100. doi: 10.1016/S0378-3782(01)00237-7
123. Pena-Ortega F. Clinical and experimental aspects of breathing modulation by inflammation. Auton Neurosci. (2019) 216:72–86. doi: 10.1016/j.autneu.2018.11.002
124. Lindgren C, Grogaard J. Reflex apnoea response and inflammatory mediators in infants with respiratory tract infection. Acta Paediatr. (1996) 85(7):798–803. doi: 10.1111/j.1651-2227.1996.tb14154.x
125. Stoltenberg L, Sundar T, Almaas R, Storm H, Rognum TO, Saugstad OD. Changes in apnea and autoresuscitation in piglets after intravenous and intrathecal interleukin-1 beta injection. J Perinat Med. (1994) 22(5):421–32. doi: 10.1515/jpme.1994.22.5.421
126. Froen JF, Akre H, Stray-Pedersen B, Saugstad OD. Adverse effects of nicotine and interleukin-1beta on autoresuscitation after apnea in piglets: implications for sudden infant death syndrome. Pediatrics. (2000) 105(4):E52. doi: 10.1542/peds.105.4.e52
127. Froen JF, Akre H, Stray-Pedersen B, Saugstad OD. Prolonged apneas and hypoxia mediated by nicotine and endotoxin in piglets. Biol Neonate. (2002) 81(2):119–25. doi: 10.1159/000047196
128. Ledeboer A, Breve JJ, Wierinckx A, van der Jagt S, Bristow AF, Leysen JE, et al. Expression and regulation of interleukin-10 and interleukin-10 receptor in rat astroglial and microglial cells. Eur J Neurosci. (2002) 16(7):1175–85. doi: 10.1046/j.1460-9568.2002.02200.x
129. Lorea-Hernandez JJ, Morales T, Rivera-Angulo AJ, Alcantara-Gonzalez D, Pena-Ortega F. Microglia modulate respiratory rhythm generation and autoresuscitation. Glia. (2016) 64(4):603–19. doi: 10.1002/glia.22951
130. Giannakopoulou CE, Sotiriou A, Dettoraki M, Yang M, Perlikos F, Toumpanakis D, et al. Regulation of breathing pattern by IL-10. Am J Physiol Regul Integr Comp Physiol. (2019) 317(1):R190–202. doi: 10.1152/ajpregu.00065.2019
131. Gordon AE, El Ahmer OR, Chan R, Al Madani OM, Braun JM, Weir DM, et al. Why is smoking a risk factor for sudden infant death syndrome? Child Care Health Dev. (2002) 28 (Suppl 1):23–5. doi: 10.1046/j.1365-2214.2002.00007.x
132. Moscovis SM, Gordon AE, Al Madani OM, Gleeson M, Scott RJ, Roberts-Thomson J, et al. Interleukin-10 and sudden infant death syndrome. FEMS Immunol Med Microbiol. (2004) 42(1):130–8. doi: 10.1016/j.femsim.2004.06.020
133. Kahn A, Blum D, Rebuffat E, Sottiaux M, Levitt J, Bochner A, et al. Polysomnographic studies of infants who subsequently died of sudden infant death syndrome. Pediatrics. (1988) 82(5):721–7. doi: 10.1542/peds.82.5.721
134. Poets CF, Meny RG, Chobanian MR, Bonofiglo RE. Gasping and other cardiorespiratory patterns during sudden infant deaths. Pediatr Res. (1999) 45(3):350–4. doi: 10.1203/00006450-199903000-00010
135. Kato I, Franco P, Groswasser J, Scaillet S, Kelmanson I, Togari H, et al. Incomplete arousal processes in infants who were victims of sudden death. Am J Respir Crit Care Med. (2003) 168(11):1298–303. doi: 10.1164/rccm.200301-134OC
136. Garcia AJ 3rd, Koschnitzky JE, Ramirez JM. The physiological determinants of sudden infant death syndrome. Respir Physiol Neurobiol. (2013) 189(2):288–300. doi: 10.1016/j.resp.2013.05.032
137. Horne RS, Parslow PM, Ferens D, Bandopadhayay P, Osborne A, Watts AM, et al. Arousal responses and risk factors for sudden infant death syndrome. Sleep Med. (2002) 3(Suppl 2):S61–5. doi: 10.1016/S1389-9457(02)00168-5
138. Franco P, Groswasser J, Hassid S, Lanquart JP, Scaillet S, Kahn A. Prenatal exposure to cigarette smoking is associated with a decrease in arousal in infants. J Pediatr. (1999) 135(1):34–8. doi: 10.1016/S0022-3476(99)70324-0
139. Horne RS, Ferens D, Watts AM, Vitkovic J, Lacey B, Andrew S, et al. The prone sleeping position impairs arousability in term infants. J Pediatr. (2001) 138(6):811–6. doi: 10.1067/mpd.2001.114475
140. Horne RS, Ferens D, Watts AM, Vitkovic J, Lacey B, Andrew S, et al. Effects of maternal tobacco smoking, sleeping position, and sleep state on arousal in healthy term infants. Arch Dis Child Fetal Neonatal Ed. (2002) 87(2):100F–5. doi: 10.1136/fn.87.2.F100
141. Kato I, Scaillet S, Groswasser J, Montemitro E, Togari H, Lin JS, et al. Spontaneous arousability in prone and supine position in healthy infants. Sleep. (2006) 29(6):785–90. doi: 10.1093/sleep/29.6.785
142. Hunt NJ, Waters KA, Rodriguez ML, Machaalani R. Decreased orexin (hypocretin) immunoreactivity in the hypothalamus and pontine nuclei in sudden infant death syndrome. Acta Neuropathol. (2015) 130(2):185–98. doi: 10.1007/s00401-015-1437-9
143. Saugstad OD. Hypoxanthine as an indicator of hypoxia: its role in health and disease through free radical production. Pediatr Res. (1988) 23(2):143–50. doi: 10.1203/00006450-198802000-00001
144. Vege A, Chen Y, Opdal SH, Saugstad OD, Rognum TO. Vitreous humor hypoxanthine levels in SIDS and infectious death. Acta Paediatr. (1994) 83(6):634–9. doi: 10.1111/j.1651-2227.1994.tb13096.x
145. Opdal SH, Rognum TO, Vege A, Saugstad OD. Hypoxanthine levels in vitreous humor: a study of influencing factors in sudden infant death syndrome. Pediatr Res. (1998) 44(2):192–6. doi: 10.1203/00006450-199808000-00009
146. Stoltenberg L, Rootwelt T, Oyasaeter S, Rognum TO, Saugstad OD. Hypoxanthine, xanthine, and uric acid concentrations in plasma, cerebrospinal fluid, vitreous humor, and urine in piglets subjected to intermittent versus continuous hypoxemia. Pediatr Res. (1993) 34(6):767–71. doi: 10.1203/00006450-199312000-00013
147. Storm H, Rognum TO, Saugstad OD, Reichelt KL. Elevated beta-endorphin immunoreactivity in the cerebrospinal fluid in victims of sudden infant death correlates with hypoxanthine in vitreous humour. Eur J Pediatr. (1993) 152(11):935–8. doi: 10.1007/BF01957536
148. Marti HH, Risau W. Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc Natl Acad Sci U S A. (1998) 95(26):15809–14. doi: 10.1073/pnas.95.26.15809
149. Jones KL, Krous HF, Nadeau J, Blackbourne B, Zielke HR, Gozal D. Vascular endothelial growth factor in the cerebrospinal fluid of infants who died of sudden infant death syndrome: evidence for antecedent hypoxia. Pediatrics. (2003) 111(2):358–63. doi: 10.1542/peds.111.2.358
150. Storm H, Rognum TO, Saugstad OD, Skullerud K, Reichelt KL. Beta-endorphin immunoreactivity in spinal fluid and hypoxanthine in vitreous humour related to brain stem gliosis in sudden infant death victims. Eur J Pediatr. (1994) 153(9):675–81. doi: 10.1007/BF02190691
151. Waters KA, Meehan B, Huang JQ, Gravel RA, Michaud J, Cote A. Neuronal apoptosis in sudden infant death syndrome. Pediatr Res. (1999) 45(2):166–72. doi: 10.1203/00006450-199902000-00002
152. Biondo B, Magagnin S, Bruni B, Cazzullo A, Tosi D, Matturri L. Glial and neuronal alterations in the nucleus tractus solitarii of sudden infant death syndrome victims. Acta Neuropathol. (2004) 108(4):309–18. doi: 10.1007/s00401-004-0895-2
153. Matturri L, Ottaviani G, Lavezzi AM. Maternal smoking and sudden infant death syndrome: epidemiological study related to pathology. Virchows Arch. (2006) 449(6):697–706. doi: 10.1007/s00428-006-0308-0
154. Luijerink L, Waters K, Rodriguez M, Machaalani R. GFAP expression in the BRAIN during human postnatal development. Neuropathol Appl Neurobiol. (2024) 50(5):e13007. doi: 10.1111/nan.13007
155. Oehmichen M, Woetzel F, Meissner C. Hypoxic-ischemic changes in SIDS brains as demonstrated by a reduction in MAP2-reactive neurons. Acta Neuropathol. (2009) 117(3):267–74. doi: 10.1007/s00401-008-0459-y
156. Jensen LL, Banner J, Byard RW. Does beta-APP staining of the brain in infant bed-sharing deaths differentiate these cases from sudden infant death syndrome? J Forensic Leg Med. (2014) 27:46–9. doi: 10.1016/j.jflm.2014.07.006
157. Jensen LL, Banner J, Ulhoi BP, Byard RW. Beta-amyloid precursor protein staining of the brain in sudden infant and early childhood death. Neuropathol Appl Neurobiol. (2014) 40(4):385–97. doi: 10.1111/nan.12109
158. Panigrahy A, Filiano J, Sleeper LA, Mandell F, Valdes-Dapena M, Krous HF, et al. Decreased serotonergic receptor binding in rhombic lip-derived regions of the medulla oblongata in the sudden infant death syndrome. J Neuropathol Exp Neurol. (2000) 59(5):377–84. doi: 10.1093/jnen/59.5.377
159. Kinney HC, Randall LL, Sleeper LA, Willinger M, Belliveau RA, Zec N, et al. Serotonergic brainstem abnormalities in northern plains Indians with the sudden infant death syndrome. J Neuropathol Exp Neurol. (2003) 62(11):1178–91. doi: 10.1093/jnen/62.11.1178
160. Ozawa Y, Okado N. Alteration of serotonergic receptors in the brain stems of human patients with respiratory disorders. Neuropediatrics. (2002) 33(3):142–9. doi: 10.1055/s-2002-33678
161. Machaalani R, Say M, Waters KA. Serotoninergic receptor 1A in the sudden infant death syndrome brainstem medulla and associations with clinical risk factors. Acta Neuropathol. (2009) 117(3):257–65. doi: 10.1007/s00401-008-0468-x
162. Bright FM, Byard RW, Vink R, Paterson DS. Medullary serotonin neuron abnormalities in an Australian cohort of sudden infant death syndrome. J Neuropathol Exp Neurol. (2017) 76(10):864–73. doi: 10.1093/jnen/nlx071
163. Haynes RL, Trachtenberg F, Darnall R, Haas EA, Goldstein RD, Mena OJ, et al. Altered 5-HT2A/C receptor binding in the medulla oblongata in the sudden infant death syndrome (SIDS): part I. Tissue-based evidence for serotonin receptor signaling abnormalities in cardiorespiratory- and arousal-related circuits. J Neuropathol Exp Neurol. (2023) 82(6):467–82. doi: 10.1093/jnen/nlad030
164. Audero E, Coppi E, Mlinar B, Rossetti T, Caprioli A, Banchaabouchi MA, et al. Sporadic autonomic dysregulation and death associated with excessive serotonin autoinhibition. Science. (2008) 321(5885):130–3. doi: 10.1126/science.1157871
165. Lavezzi AM. Altered development of mesencephalic dopaminergic neurons in SIDS: new insights into understanding sudden infant death pathogenesis. Biomedicines. (2021) 9(11):1–10. doi: 10.3390/biomedicines9111534
166. Nonnis Marzano F, Maldini M, Filonzi L, Lavezzi AM, Parmigiani S, Magnani C, et al. Genes regulating the serotonin metabolic pathway in the brain stem and their role in the etiopathogenesis of the sudden infant death syndrome. Genomics. (2008) 91(6):485–91. doi: 10.1016/j.ygeno.2008.01.010
167. Filonzi L, Magnani C, Lavezzi AM, Rindi G, Parmigiani S, Bevilacqua G, et al. Association of dopamine transporter and monoamine oxidase molecular polymorphisms with sudden infant death syndrome and stillbirth: new insights into the serotonin hypothesis. Neurogenetics. (2009) 10(1):65–72. doi: 10.1007/s10048-008-0149-x
168. Opdal SH, Vege A, Rognum TO. Genetic variation in the monoamine oxidase A and serotonin transporter genes in sudden infant death syndrome. Acta Paediatr. (2014) 103(4):393–7. doi: 10.1111/apa.12526
169. Broadbelt KG, Paterson DS, Belliveau RA, Trachtenberg FL, Haas EA, Stanley C, et al. Decreased GABAA receptor binding in the medullary serotonergic system in the sudden infant death syndrome. J Neuropathol Exp Neurol. (2011) 70(9):799–810. doi: 10.1097/NEN.0b013e31822c09bc
170. Dunn AJ. Effects of cytokines and infections on brain neurochemistry. Clin Neurosci Res. (2006) 6(1–2):52–68. doi: 10.1016/j.cnr.2006.04.002
171. Vivekanandarajah A, Nelson ME, Kinney HC, Elliott AJ, Folkerth RD, Tran H, et al. Nicotinic receptors in the brainstem ascending arousal system in SIDS with analysis of pre-natal exposures to maternal smoking and alcohol in high-risk populations of the safe passage study. Front Neurol. (2021) 12:636668. doi: 10.3389/fneur.2021.636668
172. Slotkin TA, Seidler FJ, Spindel ER. Prenatal nicotine exposure in rhesus monkeys compromises development of brainstem and cardiac monoamine pathways involved in perinatal adaptation and sudden infant death syndrome: amelioration by vitamin C. Neurotoxicol Teratol. (2011) 33(3):431–4. doi: 10.1016/j.ntt.2011.02.001
173. Lee SY, Sirieix CM, Nattie E, Li A. Pre- and early postnatal nicotine exposure exacerbates autoresuscitation failure in serotonin-deficient rat neonates. J Physiol. (2018) 596(23):5977–91. doi: 10.1113/JP275885
174. Amiry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nat Rev Neurosci. (2003) 4(12):991–1001. doi: 10.1038/nrn1252
175. Xu M, Xiao M, Li S, Yang B. Aquaporins in nervous system. Adv Exp Med Biol. (2017) 969:81–103. doi: 10.1007/978-94-024-1057-0_5
176. Manley GT, Binder DK, Papadopoulos MC, Verkman AS. New insights into water transport and edema in the central nervous system from phenotype analysis of aquaporin-4 null mice. Neuroscience. (2004) 129(4):981–91. doi: 10.1016/j.neuroscience.2004.06.088
177. Filippidis AS, Carozza RB, Rekate HL. Aquaporins in brain edema and neuropathological conditions. Int J Mol Sci. (2017) 18(1):1–13. doi: 10.1067/mpd.2001.114475. doi: 10.3390/ijms18010055
178. Nagelhus EA, Mathiisen TM, Ottersen OP. Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience. (2004) 129(4):905–13. doi: 10.1016/j.neuroscience.2004.08.053
179. Wang C, Yan M, Jiang H, Wang Q, He S, Chen J, et al. Mechanism of aquaporin 4 (AQP 4) up-regulation in rat cerebral edema under hypobaric hypoxia and the preventative effect of puerarin. Life Sci. (2018) 193:270–81. doi: 10.1016/j.lfs.2017.10.021
180. Lu H, Ai L, Zhang B. TNF-alpha induces AQP4 overexpression in astrocytes through the NF-kappaB pathway causing cellular edema and apoptosis. Biosci Rep. (2022) 42(3):1–13. doi: 10.1042/BSR20212224
181. Li L, Zhang H, Varrin-Doyer M, Zamvil SS, Verkman AS. Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB J. (2011) 25(5):1556–66. doi: 10.1096/fj.10-177279
182. Ito H, Yamamoto N, Arima H, Hirate H, Morishima T, Umenishi F, et al. Interleukin-1beta induces the expression of aquaporin-4 through a nuclear factor-kappaB pathway in rat astrocytes. J Neurochem. (2006) 99(1):107–18. doi: 10.1111/j.1471-4159.2006.04036.x
183. Eidahl JML, Stray-Pedersen A, Rognum TO, Opdal SH. Aquaporin 4 expression in the hippocampus in sudden infant death syndrome and sudden unexplained death in childhood. J Chem Neuroanat. (2021) 115:101962. doi: 10.1016/j.jchemneu.2021.101962
184. Fan Y, Zhang J, Sun XL, Gao L, Zeng XN, Ding JH, et al. Sex- and region-specific alterations of basal amino acid and monoamine metabolism in the brain of aquaporin-4 knockout mice. J Neurosci Res. (2005) 82(4):458–64. doi: 10.1002/jnr.20664
185. Ding JH, Sha LL, Chang J, Zhou XQ, Fan Y, Hu G. Alterations of striatal neurotransmitter release in aquaporin-4 deficient mice: an in vivo microdialysis study. Neurosci Lett. (2007) 422(3):175–80. doi: 10.1016/j.neulet.2007.06.018
186. Rite I, Machado A, Cano J, Venero JL. Intracerebral VEGF injection highly upregulates AQP4 mRNA and protein in the perivascular space and glia limitans externa. Neurochem Int. (2008) 52(4–5):897–903. doi: 10.1016/j.neuint.2007.10.004
187. Ding JY, Kreipke CW, Speirs SL, Schafer P, Schafer S, Rafols JA. Hypoxia-inducible factor-1alpha signaling in aquaporin upregulation after traumatic brain injury. Neurosci Lett. (2009) 453(1):68–72. doi: 10.1016/j.neulet.2009.01.077
188. Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P, et al. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. (2011) 114(1):92–101. doi: 10.3171/2010.6.JNS10207
189. Ding H, Liu X, Li X, Wen M, Li Y, Han Y, et al. Hypercapnia exacerbates the disruption of the blood-brain barrier by inducing interleukin-1beta overproduction in the blood of hypoxemic adult rats. Int J Mol Med. (2020) 46(2):762–72. doi: 10.3892/ijmm.2020.4604
190. Aranda FJ, Teixeira F, Becker LE. Assessment of growth in sudden infant death syndrome. Neuroepidemiology. (1990) 9(2):95–105. doi: 10.1159/000110756
191. Siebert JR, Haas JE. Organ weights in sudden infant death syndrome. Pediatr Pathol. (1994) 14(6):973–85. doi: 10.3109/15513819409037694
192. Kadhim H, Sebire G, Khalifa M, Evrard P, Groswasser J, Franco P, et al. Incongruent cerebral growth in sudden infant death syndrome. J Child Neurol. (2005) 20(3):244–6. doi: 10.1177/088307380502000303
193. Kinney HC, Brody BA, Finkelstein DM, Vawter GF, Mandell F, Gilles FH. Delayed central nervous system myelination in the sudden infant death syndrome. J Neuropathol Exp Neurol. (1991) 50(1):29–48. doi: 10.1097/00005072-199101000-00003
194. Eidahl JML, Rognum TO, Stray-Pedersen A, Opdal SH. Brain water content in sudden unexpected infant death. Forensic Sci Med Pathol. (2023) 19:507–16. doi: 10.1007/s12024-023-00584-8
195. Beckwith JB. SIDS and hypoxanthine. Pediatrics. (1991) 88(5):1076–7. doi: 10.1542/peds.88.5.1076b
196. Carpenter KH, Bonham JR, Worthy E, Variend S. Vitreous humour and cerebrospinal fluid hypoxanthine concentration as a marker of pre-mortem hypoxia in SIDS. J Clin Pathol. (1993) 46(7):650–3. doi: 10.1136/jcp.46.7.650
197. Rognum TO. SIDS or not SIDS? Classification problems of sudden infant death syndrome. Acta Paediatr. (1996) 85(4):401–3. doi: 10.1111/j.1651-2227.1996.tb14049.x
198. Byard RW, Shipstone RA, Young J. Continuing major inconsistencies in the classification of unexpected infant deaths. J Forensic Leg Med. (2019) 64:20–2. doi: 10.1016/j.jflm.2019.03.007
199. Tan L, Byard RW. An analysis of the use of standard SIDS definitions in the English language literature over a three-year period (2019–2021). Acta Paediatr. (2022) 111(5):1019–22. doi: 10.1111/apa.16266
Keywords: hypoxia, infection, risk factors, serotonergic network, SIDS, sudden infant death, triple risk hypothesis of SIDS, vicious spiral of SIDS
Citation: Opdal SH, Stray-Pedersen A, Eidahl JML, Vege Å, Ferrante L and Rognum TO (2025) The vicious spiral in Sudden Infant Death Syndrome. Front. Pediatr. 13:1487000. doi: 10.3389/fped.2025.1487000
Received: 27 August 2024; Accepted: 27 January 2025;
Published: 11 February 2025.
Edited by:
Luana Maria Nosetti, University of Insubria, ItalyReviewed by:
Rita Machaalani, The University of Sydney, AustraliaCopyright: © 2025 Opdal, Stray-Pedersen, Eidahl, Vege, Ferrante and Rognum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Siri Hauge Opdal, c2lyb3BkQG91cy1oZi5ubw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.