 Handan Kava
Handan Kava Ozlem Akgun-Dogan2,3,4,†
Ozlem Akgun-Dogan2,3,4,† Ahmet Yesilyurt
Ahmet Yesilyurt Yasemin Alanay
Yasemin Alanay Ugur Isik
Ugur Isik
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 27 February 2025
Sec. Pediatric Neurology
Volume 13 - 2025 | https://doi.org/10.3389/fped.2025.1471965
Background: We aimed to understand the genetic etiology in children presenting with epilepsy and/or developmental delay by using next-generation sequencing (NGS).
Materials and methods: We included children presenting to our pediatric neurology clinic with a diagnosis of epilepsy and/or developmental delay between January 2019 and December 2021. We evaluated the patients using the NGS equipment in our genetic laboratory.
Results: In total, 90 patients were included in the study. Twenty (34.4%) out of 58 patients who had undergone whole-exome sequencing (WES) had pathogenic or likely pathogenic (P/LP) variants and 11 (18.9%) had variants of unknown significance (VUS). Five (41.6%) out of 12 patients who had undergone whole-genome sequencing had P/LP variants and 5 (41.6%) had VUS. Eleven (55%) out of 20 patients who had undergone WES and chromosomal microarray had P/LP variants and 2 (10%) had VUS. Twenty-six novel variants were described. Twelve patients (13.3%) were diagnosed using a known specific treatment.
Conclusion: NGS aids in precisely diagnosing children with epilepsy and/or developmental delay. Furthermore, it provides a correct prognosis, specific treatment methods, and a multidisciplinary approach.
The incidence of epilepsy in children ranges from 0.5 to 8 per 1,000 individuals (1–4). Its estimated lifetime prevalence is 1%, and the highest incidence is observed during the first year of life (5). Various developmental disorders, such as developmental delay (DD), intellectual disability (ID), autism spectrum disorder (ASD), and attention deficit and hyperactivity disorder (ADHD) are more common in children with epilepsy (6).
In developed countries, the absence of expected psychomotor development is observed in approximately 1% of children (7). DD and ID may occur alone or in conjunction with congenital malformations or neurological findings such as epilepsy, hypotonia, ataxia, electroencephalogram (EEG) abnormalities, and behavioral problems such as ASD and ADHD. DD and ID are phenotypically and genetically heterogeneous, and a specific diagnosis cannot be reached in most cases. Therefore, genetic tests play a crucial role in determining the cause of abnormality in this patient group (8).
Next-generation sequencing (NGS) technologies enable the reliable identification of candidate genes responsible for epilepsy and DD in a single, cost-effective, and time-efficient process (9). There are compelling reasons to make a specific diagnosis in a child presenting with DD. Diagnosis permits families to understand the long-term prognosis, access special education services, and learn about potential treatment options. Moreover, it is crucial for counseling parents regarding recurrence risk, prenatal diagnosis, and preimplantation genetic diagnosis. Furthermore, a diagnosis can help parents access research studies and connect with other families facing similar challenges (10).
Chromosomal microarray (CMA) has long been utilized as a first-line genetic test in children with DD and ID. With the advancement of NGS techniques, whole-exome sequencing (WES) and whole-genome sequencing (WGS) have become pivotal diagnostic tools for children with DD and ID. The American College of Medical Genetics and Genomics (ACMG) guidelines strongly recommend considering WES/WGS as a first- or second-line test for patients with DD and ID (10).
NGS has unveiled new candidate genes implicated in the pathogenesis of epilepsy. Ion channels play a crucial role in maintaining proper neuronal excitability; these include voltage-gated sodium, potassium, calcium, and chloride ion channels, and ligand-gated acetylcholine receptor-mediated ion channels. Mutations in genes encoding components of these ion channels lead to ion channel dysfunction, known as channelopathy, and form the basis for the development of epilepsy syndromes. Genetic changes causing channelopathies may contribute to the pathogenesis of epilepsy via the gain or loss of ion channel function (11).
Early genetic testing and diagnosis are foundational for accurate treatment of epilepsy. However, they should be complemented with functional tests to explore the characteristics of the pathophysiological mechanisms and the functional impact underlying the detected variant. Specifically in ion channel disorders, understanding gain-of-function mutations and loss-of-function mutations is critical when making therapeutic decisions (12, 13).
Experience with most of the above mentioned treatments is limited to a relatively small number of patients. With a better understanding of the pathogenesis and cellular electrophysiology of genetic epilepsies, more treatment options are likely to emerge (14).
This retrospective cohort study included 90 patients aged 0–18 years, who were observed at the Acibadem Altunizade Hospital Pediatric Neurology Outpatient Clinic between January 2019 and December 2021 and had been diagnosed with epilepsy and/or DD. These patients had undetermined etiology despite undergoing biochemical and metabolic tests and magnetic resonance imaging (MRI). The genetic analyses were performed as part of the diagnostic process when patients first presented for evaluation. Informed consent for genetic testing was obtained by the accredited diagnostic laboratory prior to the tests. The WES and CMA results were processed at the Acibadem Labgen Genetic Diseases Evaluation Center, and the WGS results were obtained from the Centogene Laboratory (Germany).
Retrospective evaluation of the WES, CMA, and WGS results was conducted in collaboration with the Pediatric Genetics Department of Acibadem University. Patient medical records were retrospectively reviewed; these encompassed their medical history; background; family history; neurological symptoms; motor and mental developmental history; neurological examination findings; liver and kidney function test results; metabolic screening test, EEG, and MRI results; antiepileptic treatments; and genetic diagnoses.
Ethical approval for the retrospective study was obtained from the Scientific Research Ethics Committee of Acibadem University School of Medicine (Decision No: 2021/25/26).
Genomic DNA isolation was performed using peripheral blood leukocytes. Enrichment was performed using the Twist® Human Core Exome kits. Prepared libraries were sequenced using the Novaseq 6000 (Illumina) instrument. Using the Burrows–Wheeler Aligner (BWA-MEM; Sentieon), sequenced data were aligned with genome reference to GRCh37 (hg19) (15). Duplicate marking, indel realignment, baseline quality recalibration, and variant calling were performed using the Genome Analysis Toolkit (GATK) algorithms (Sentieon) (16). Variant annotation, filtering, and interpretation were performed with the VarSome Clinical platform (Spatter). Variants in the coding and splicing regions and known pathogenic variants in the noncoding regions were included in the analysis. Variants with minor allele frequencies (below 1%) in the gnomAD database were evaluated. Filtering and prioritization of potentially disease-causing variants were performed based on the inheritance pattern, information in the ClinVar database, Human Phenotype Ontology (HPO) terms associated with the patient's findings, and in silico pathogenicity estimates. Variants were classified according to the ACMG guidelines (17) and ClinGen recommendations (18), pathogenic (P), and likely pathogenic (LP), and clinically relevant variants of uncertain significance (VUS) associated with the patient's clinical findings were reported.
Genomic DNA was fragmented enzymatically, and libraries were generated using polymerase chain reaction-mediated addition of Illumina-compatible adapters. Libraries were sequenced with paired ends on an Illumina platform to achieve an average depth of coverage of ∼30×. Alignment of the GRCh37/hg19 genome and human mitochondrial DNA (NC_012920) to the revised Cambridge Reference Sequence (rCRS), variant calling, annotation, and extensive variant filtering were applied through an inhouse bioinformatics pipeline. The copy number variant (CNV) recall was based on Illumina's DRAGEN pipeline. All variants in the GnomAD database with minor allele frequency of <1% and disease-causing variants reported in the Human Gene Mutation Database, ClinVar, and CentoMD were evaluated. During the evaluation, the entire gene region was examined for candidate variants associated with the phenotype in exons and in ±20 intronic regions. All potential patterns were considered for the inheritance pattern. Furthermore, family history and clinical information were used to assess the pathogenicity and probability of the disease-causing effect of the detected variants. Variants were divided into five classes (pathogenic, possibly pathogenic, of uncertain clinical significance, possibly benign, and benign) according to the ACMG guidelines. All potential variants associated with the patient's phenotype were reported. CNVs of uncertain clinical significance were not reported. Variants with heteroplasmy levels of up to 15% were reported as mitochondrial variants. Centogene has established specific quality criteria and validation processes for variants detected by NGS. Variants with poor sequence quality and/or indeterminate zygosity were confirmed by orthogonal methods. As a result, a specificity of >99.9% was guaranteed for all reported variants.
Technical work was conducted using the Affymetrix Cytoscan 750 K Microarray Platform. Findings that fell within the resolution thresholds (at least 25 markers for decreases of 100 kb and greater and increases of 200 kb and greater in known genomic areas) were analyzed using filters in the “Chromosome Analysis Suite (CHAS)” analysis program. Findings outside the reporting standards; absence of heterozygosity regions; additional mosaic findings; findings below the resolution threshold overlapping with the Online Mendelian Inheritance in Man (OMIM) genes, above-threshold findings without the OMIM genes, the OMIM genes not detected but findings in loci associated with the phenotype; findings that cannot be directly associated with the reason for referral; and results other than the OMIM gene point mutations, small deletions or duplications, low rate mosaicism, and balanced chromosomal changes such as reciprocal and the Robertsonian translocations were not evaluated. Whether the findings were familial or not was determined only through the studies requested by the parents. When required, the segregation analysis of the CNVs detected in the mother and father via array analysis was investigated.
GRCh37/hg19 was used as the reference genome in the analysis. The ClinGen, Decipher, UNIQUE, OrphaNet, DGV, ClinVar, OMIM®, dbSNP, and HPO databases associated with the patient's clinical findings were used to assess the variants. Detected variants were assessed using the patient's clinical findings.
Detected variants were classified according to the ACMG-ClinGen Variant Classification Guidelines. Variants classified as pathogenic, possibly pathogenic, and of uncertain clinical significance associated with the patient's clinical findings were reported. Reported variants were named according to the ISCN 2020 nomenclature (19).
The demographic and clinical characteristics of the patients evaluated in the study were analyzed via descriptive statistical analyses (number, percentage, mean, standard deviation, etc.). The comparisons of age, clinical findings, perinatal history, demographic characteristics, MRI, and EEG findings according to the genetic analysis results of the patients evaluated in the study were conducted using chi-square analysis. The significance level for all analyses was set at p < 0.05. The conformity of the data to normal distribution was checked with the values of kurtosis and skewness (±1.5). IBM SPSS 22.0 was used to conduct the analyses.
In total, 90 patients were included in the study. Of the 90 patients, 53 (58.9%) were male, and 37 (41.1%) were female. The age of the patients at the time they were seen at our clinic was 0.04–16 years. The mean age of the patients was 3.99 ± 2.71 years, and the median age was 2.71 years. Forty-seven patients were from Turkey; the remaining 43 patients were international (Table 1). EEG abnormalities were detected in a total of 58 (64.4%) patients. EEG was not available from four patients (P68, P77, P79, P81) (Table 2).
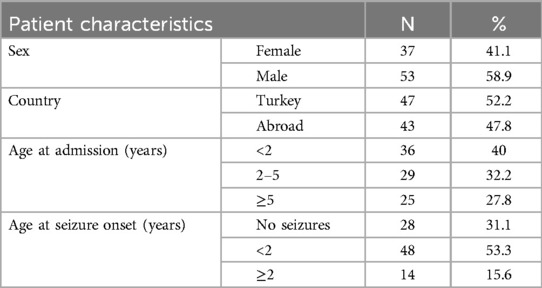
Table 1. Demographic characteristics of all patients.
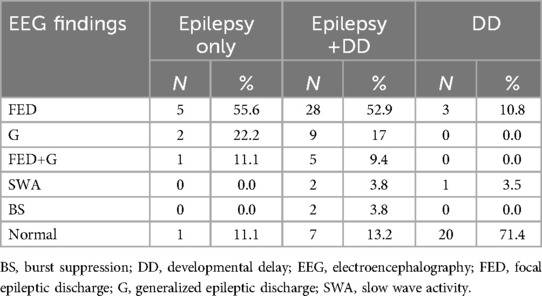
Table 2. Electroencephalography findings.
Cranial MRI was abnormal in 44 patients (48.8%). The MRI results were abnormal in 27 (50%) of 54 patients with P/LP variants and clinically relevant VUS variants. MRI was not available in two patients (P68, P82) (Table 3).
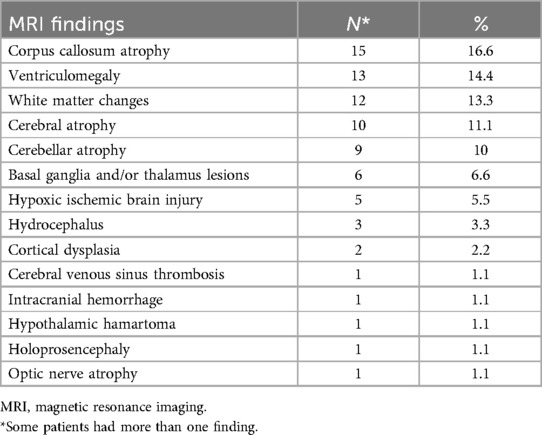
Table 3. Magnetic resonance imaging findings of all patients.
In 15 (16.6%) patients, parental consanguinity/origin from same village was reported. There was first-degree cousin marriage between the parents in seven patients (7.8%), and second-degree cousin marriage between the parents in five patients (5.6%). In total, 24 patients (26.7%) had a family history of similar diseases. Thirty-one patients (34.4%) had abnormal perinatal history, such as asphyxia, infection, and hospitalization in the neonatal intensive care unit (Supplementary Table S1).
The patients were classified into three main groups; epilepsy only (n = 9; 10%), DD and epilepsy (n = 53; 58.9%), and only DD (n = 28; 31.1%) (Figure 1).
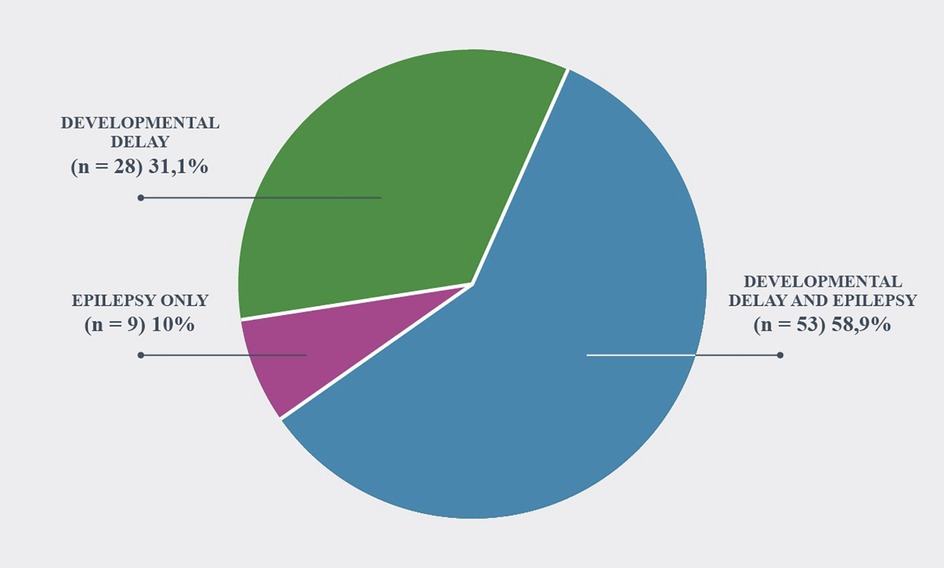
Figure 1. Phenotype of patients.
Among all patients (n = 90), WES was studied in 58 patients, WGS was studied in 12 patients, and WES and CMA were studied in 20 patients. Trio-WES was performed in four, and trio WGS was performed in two families. Among the 58 patients who had undergone WES, P/LP variants were detected in 20 (34.4%), and clinically relevant VUS was detected in 11 (18.9%). Among the 12 patients who had undergone WGS, P/LP variants were detected in 5 (41.6%), and VUS was detected in 5 (41.6%). Among the 20 patients who had undergone WES and CMA, P/LP variants were detected in 11 (55%), and VUS was detected in 2 (10%) (Figure 2). A molecular diagnosis enabling a potential treatment option was established 12 patients (13.3%), (Table 4).
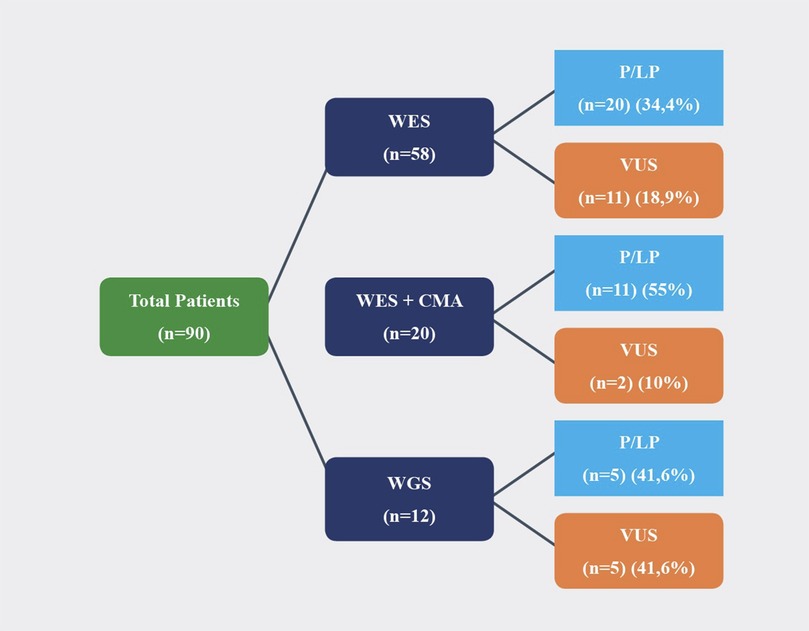
Figure 2. Molecular analysis results of patients.
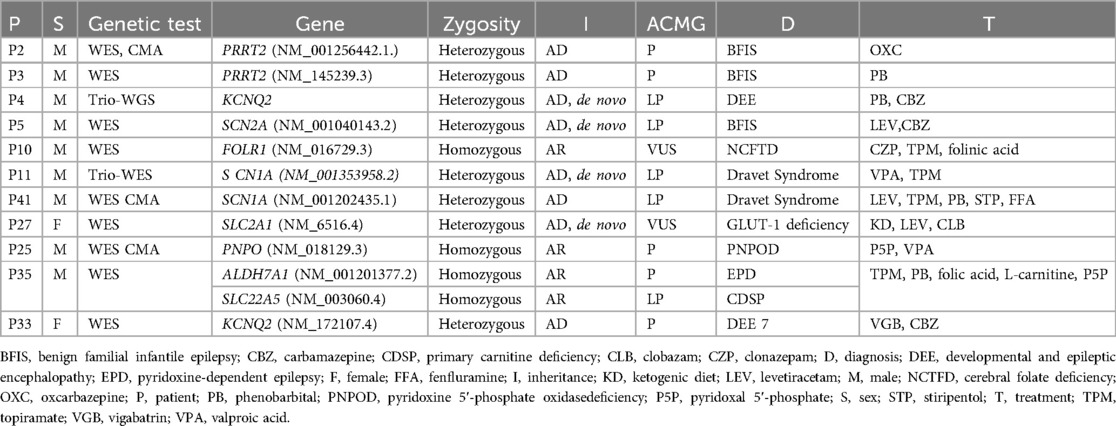
Table 4. Molecular diagnosis enabling potential treatment in epilepsy only and epilepsy+DD groups.
In our cohort, several genes were frequently implicated in cases of genetic epilepsy and developmental delay, including PRRT2 (n:2), KCNQ2 (n:2), SCN1A (n:2), PLA2G6 (n:2), and KDM6A (n:2). Among molecularly diagnosed cases SCN1A, SCN2A, KCNQ2, SCN8A, and ATP1A2 were identified as channelopathy-related genes, representing 7 patients. While channelopathies were less frequent compared to other conditions in the cohort, their distinct clinical characteristics warrant further attention.
Developmental delay was present in 71.4% of patients with channelopathies, slightly lower than the 93.6% observed in patients with other conditions. This lower prevalence may reflect the primary impact of channelopathies on seizure activity and excitability rather than broader neurodevelopmental processes. Similarly, dysmorphic features were observed in 14.2% of patients with channelopathies, significantly less frequent compared to 36.1% in other genetic conditions, highlighting the subtler phenotypic manifestations of channelopathies.
Abnormal MRI findings were detected in 28.5% of patients with channelopathies, which was markedly lower than the 53.1% observed in patients with other genetic conditions. This suggests that structural brain abnormalities are less commonly associated with channelopathies, reinforcing their functional nature. In contrast, EEG abnormalities were present in 71.4% of patients with channelopathies, slightly higher than the 61.7% seen in other genetic conditions. This reflects the prominent role of electrical disturbances in the pathophysiology of channelopathies.
Table 5 provides a detailed summary of the molecular findings in the diagnosed cohort, further illustrating the differences in clinical and genetic characteristics between channelopathies and other neurodevelopmental conditions.
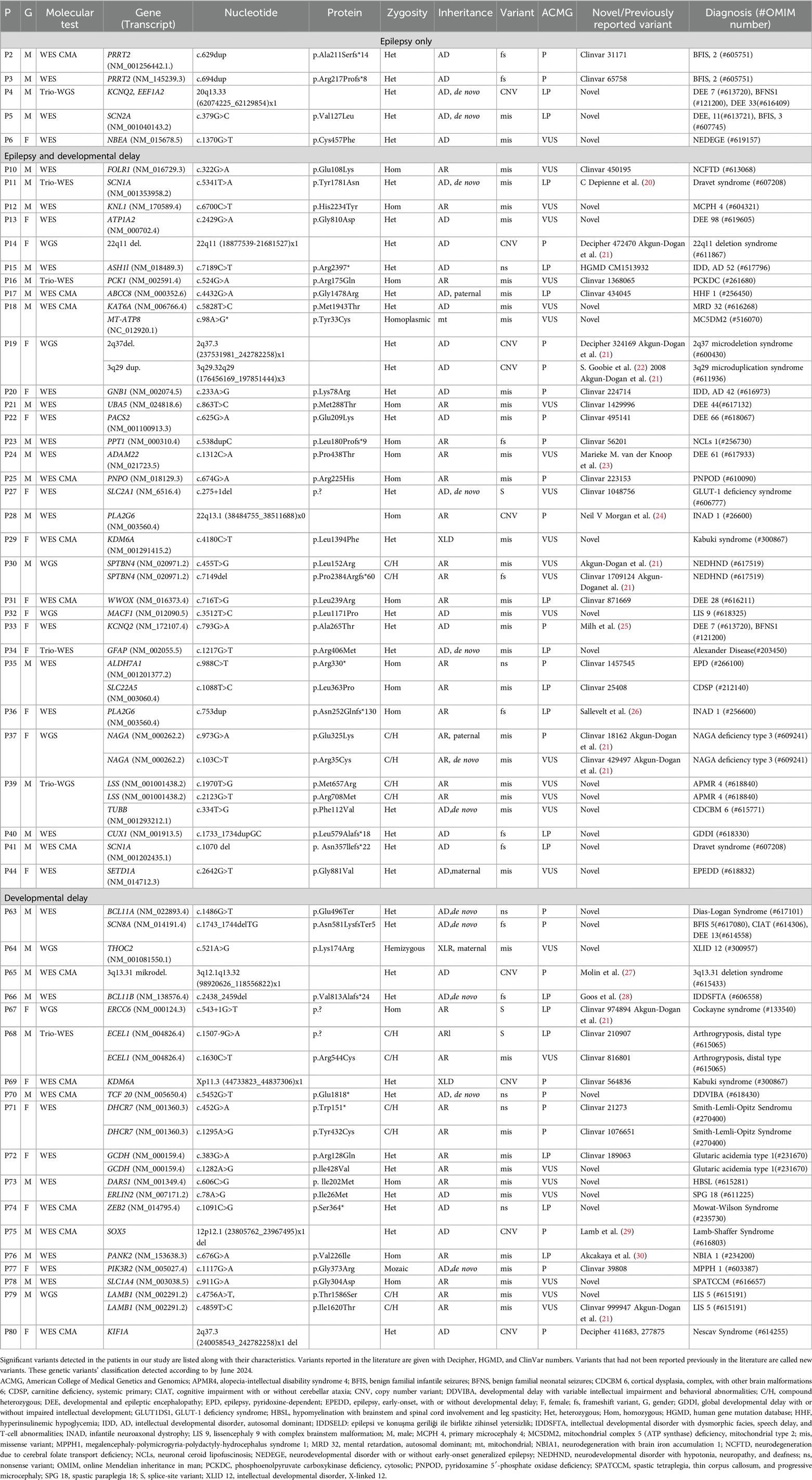
Table 5. Molecular findings in the diagnosed cohort.
Dysmorphic findings were significantly more frequent in the molecularly diagnosed group (P/LP/VUS variants), whereas other features, such as MRI abnormalities, neurobehavioral findings, and ocular findings, did not show a statistically significant difference (Table 6).

Table 6. Comparison of clinical characteristics according to patients' genetic results.
Several epilepsy phenotypes exhibit chromosomal imbalances or alterations in genes encoding ion channels and transcription factors. Genetic testing is crucial for elucidating the underlying cause in patients with epilepsy and DD (31). Despite advanced diagnostic technologies nearly half of the affected individuals remain undiagnosed.
In the literature, the rate of diagnosis of DD and epilepsy by utilizing WES is reported to be between 25% and 55% (32–36). Hiraide et al., have reported that WES, including CNV analysis, resulted in a diagnostic rate of 53.5% (37). In our study, the percentage of LP/P variants with WES was 34.4%. Clinically relevant VUS were observed in an additional 18.9%, providing a similar diagnostic rate (53.3%) despite absence of CNV analysis. Similarly, the diagnostic rate of WGS in DD and epilepsy cohorts is reported to be 21%–50% (38–42). Among the 12 patients who had undergone WGS, 5 (41.6%) had P/LP variants, and 5 (41.6%) had clinically relevant VUS. WGS was performed in patients without prior testing. Two out 5 P/LP WGS results showed CNV as the underlying cause which would have been missed by WES alone. WGS shortened the diagnostic duration in these patients as first-tier genetic test.
The findings of our study indicate that the most frequently observed genes are consistent with those reported in the literature (11–14). Furthermore, although a direct comparison is not feasible due to the significant differences in the numbers, channelopathy-related genes stood out prominently in our study. Similarly, in the literature, channelopathy-related genes have also been highlighted as a significant group, in line with our data. Compared to other genetic etiologies underlying epileptic encephalopathies, channelopathies are less frequently associated with dysmorphic features and abnormal MRI findings, while EEG abnormalities are more prominently observed. This highlights the critical importance of genetic analysis in establishing an accurate diagnosis. Furthermore, genetic findings play a pivotal role in informing and tailoring treatment strategies (11, 13).
In this study, a molecular genetic diagnosis with available treatment was established in 13.3% of the patients. Only one patient with an actionable variant was left untreated due to absence of clinical seizures. Mahler et al. reported a treatment change rate of 6%. The higher rate of specific treatment changes in our study may be attributed to our patients having a more heterogeneous phenotype, as well as the emergence of new treatment options involving genes related to ion channels (31). By re-evaluating the previously negative WES data of patient P27, a de novo VUS variant in the SLC2A1 gene was identified; this was followed by segregation analysis, which led to a diagnosis of GLUT-1 deficiency (43). Seizures were controlled using a ketogenic diet, demonstrating the importance of data reanalysis in treatment modifications.
Trio WES analysis was conducted in 4 families due to economic burden. Segregation analysis was performed following initial WES analysis. 20.7% of the diagnosed individuals showed de-novo monoallelic variants. Brunet et al. reported a much higher frequency of de- novo variants (40.3%) following trio WES in their study. The lower percentage of de-novo monoallelic variants in our cohort reflects the higher rate of consanguinity leading to a higher incidence of biallelic disorders (22.2%) in comparison to 16% reported by Brunet et al. (44).
Samati et al. reported abnormal MRI findings in one-third of individuals with epilepsy in a recent review. The most frequently observed abnormalities were encephalomalacia related to chronic infarcts, cerebral atrophy, disorders of neuronal migration, periventricular leukomalacia. Accompanying EEG finding was mainly focal discharges (78%) (45). In our study, corpus callosum atrophy (16.6%), ventriculomegaly (14.4%), and white matter changes (13.3%) were present in 48.8% of our patients and focal discharges were most common (40%) as well. Our cohort mainly consists of unexplained patients without evident focal lesions. The relatively lower percentage of focal discharges may be related to the lower number of focal lesions on MRI in our patient group who underwent genetic testing.
The main limitation of our study is the retrospective nature and heterogeneity of the genetic tests. Some patients had only WES, some had WES and CMA, and some had WGS. Therefore, the number of patients having each test was limited. However, our study adds novel genetic variants that have not been described previously in the literature. All patients received pre-test and post-test genetic counseling from two pediatric geneticists who also contributed to deep-phenotyping and reverse phenotyping of the diagnosed cohort. Patients with negative genetic results will be recalled for reanalysis yearly. Genetic counseling is provided by clinical geneticists in Turkey. Families with a diagnosis were counseled regarding recurrence risks and future reproductive options. This is the largest single center study with a multidisciplinary approach from our country.
In conclusión, NGS helps to determine the underlying molecular etiology of neurogenetic conditions. Moreover, an increasing percentage of patients receive personalized treatment after molecular diagnosis. WES and WGS increase the diagnosis rate and shorten the diagnostic odyssey for families when applied as first-tier genetic tests. Determining the genetic diagnosis of patients with epilepsy and/or DD using NGS is crucial in terms of an accurate prognosis, potential treatment methods, and a multidisciplinary approach involving molecular and clinical genetic expertise.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Scientific Research Ethics Committee of Acibadem University School of Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin.
HK: Data curation, Formal Analysis, Investigation, Writing – original draft, Methodology, Project administration. OA-D: Conceptualization, Data curation, Writing – review & editing, Supervision, Validation. AY: Methodology, Writing – review & editing, Data curation. YA: Data curation, Writing – review & editing, Supervision, Conceptualization, Validation. UI: Data curation, Investigation, Project administration, Supervision, Writing – review & editing, Conceptualization.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2025.1471965/full#supplementary-material
1. Hauser WA, Annegers JF, Kurland LT. Prevalence of epilepsy in Rochester, Minnesota: 1940–1980. Epilepsia. (1991) 32:429–45. doi: 10.1111/j.1528-1157.1991.tb04675.x
2. Oka E, Ohtsuka Y, Yoshinaga H, Murakami N, Kobayashi K, Ogino T. Prevalence of childhood epilepsy and distribution of epileptic syndromes: a population-based survey in Okayama, Japan. Epilepsia. (2006) 47(3):626–30. doi: 10.1111/j.1528-1167.2006.00477.x
3. Russ SA, Larson K, Halfon N. A national profile of childhood epilepsy and seizure disorder. Pediatrics. (2012) 129:256–64. doi: 10.1542/peds.2010-1371
4. Aaberg KM, Gunnes N, Bakken IJ, Lund Søraas C, Berntsen A, Magnus P, et al. Incidence and prevalence of childhood epilepsy: a nationwide cohort study. Pediatrics. (2017) 139(5):e20163908. doi: 10.1542/peds.2016-3908
5. Dang LT, Silverstein FS. Drug treatment of seizures and epilepsy in newborns and children. Pediatr Clin North Am. (2017) 64:1291–308. doi: 10.1016/j.pcl.2017.08.007
6. Zavadenko NN. Neurodevelopmental disorders in children with epilepsy intellectual disability, and autism spectrum disorders. Epilepsy and Paroxysmal Conditions. (2017) 9(4):64–71. doi: 10.17749/2077-8333.2017.9.4.064-071
7. McKenzie K, Milton M, Smith G, Ouellette-Kuntz H. Systematic review of the prevalence and incidence of intellectual disabilities: current trends and issues. Curr Dev Disord Rep. (2016) 3:104–15. doi: 10.1007/s40474-016-0085-7
8. McLaren J, Bryson SE. Review of recent epidemiological studies of mental retardation: prevalence, associated disorders, and etiology. Am J Ment Retard. (1987) 92(3):243–54.3322329
9. Rahman MM, Fatema K. Genetic diagnosis in children with epilepsy and developmental disorders by targeted gene panel analysis in a developing country. J Epilepsy Res. (2021) 11(1):22–31. doi: 10.14581/jer.21004
10. Manickam K, McClain MR, Demmer LA, Biswas S, Kearney HM, Malinowski J, et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2021) 23(11):2029–37. doi: 10.1038/s41436-021-01242-6
11. Zhang D, Liu X, Deng X. Genetic basis of pediatric epilepsy syndromes. Exp Ther Med. (2017) 13(5):2129–33. doi: 10.3892/etm.2017.4267
12. Striano P, Minassian BA. From genetic testing to precision medicine in epilepsy. Neurotherapeutics. (2020) 17(2):609–15. doi: 10.1007/s13311-020-00835-4
13. Bayat A, Bayat M, Rubboli G, Møller RS. Epilepsy syndromes in the first year of life and usefulness of genetic testing for precision therapy. Genes (Basel). (2021) 12(7):1051. doi: 10.3390/genes12071051
14. Moosa ANV. Antiepileptic drug treatment of epilepsy in children. Continuum (Minneap Minn). (2019) 25(2):381–407. doi: 10.1212/CON.0000000000000712
15. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. (2009) 25(14):1754–60. doi: 10.1093/bioinformatics/btp324
16. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
17. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17(5):405–23. doi: 10.1038/gim.2015.30
18. Clinical Genome Resource. Sequence variant interpretation. Working group (2021). Available online at: https://www.clinicalgenome.org/working-groups/sequence-variant-interpretation/ (Accessed January 3, 2024).
19. Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. (2020) 22(2):245–57. doi: 10.1038/s41436-019-0686-8
20. Depienne C, Trouillard O, Saint-Martin C, Gourfinkel-An I, Bouteiller D, Carpentier W, et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet. (2009) 46(3):183–91. doi: 10.1136/jmg.2008.062323
21. Akgun-Dogan O, Tuc Bengur E, Ay B, Ozkose GS, Kar E, Bengur FB, et al. Impact of deep phenotyping: high diagnostic yield in a diverse pediatric population of 172 patients through clinical whole-genome sequencing at a single center. Front Genet. (2024) 15:1347474. doi: 10.3389/fgene.2024.1347474
22. Goobie S, Knijnenburg J, Fitzpatrick D, Sharkey FH, Lionel AC, Marshall CR, et al. Molecular and clinical characterization of de novo and familial cases with microduplication 3q29: guidelines for copy number variation case reporting. Cytogenet Genome Res. (2008) 123(1–4):65–78. doi: 10.1159/000184693
23. van der Knoop MM, Maroofian R, Fukata Y, van Ierland Y, Karimiani EG, Lehesjoki AE, et al. Biallelic ADAM22 pathogenic variants cause progressive encephalopathy and infantile-onset refractory epilepsy. Brain. (2022) 145(7):2301–12. doi: 10.1093/brain/awac116
24. Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet. (2006) 38(7):752–4. doi: 10.1038/ng1826
25. Milh M, Boutry-Kryza N, Sutera-Sardo J, Mignot C, Auvin S, Lacoste C, et al. Similar early characteristics but variable neurological outcome of patients with a de novo mutation of KCNQ2. Orphanet J Rare Dis. (2013) 8:80. doi: 10.1186/1750-1172-8-80
26. Sallevelt SCEH, Stegmann APA, de Koning B, Velter C, Steyls A, van Esch M, et al. Diagnostic exome-based preconception carrier testing in consanguineous couples: results from the first 100 couples in clinical practice. Genet Med. (2021) 23(6):1125–36. doi: 10.1038/s41436-021-01116-x
27. Molin AM, Andrieux J, Koolen DA, Malan V, Carella M, Colleaux L, et al. A novel microdeletion syndrome at 3q13.31 characterised by developmental delay, postnatal overgrowth, hypoplastic male genitals, and characteristic facial features. J Med Genet. (2012) 49(2):104–9. doi: 10.1136/jmedgenet-2011-100534
28. Goos JAC, Vogel WK, Mlcochova H, Millard CJ, Esfandiari E, Selman WH, et al. A de novo substitution in BCL11B leads to loss of interaction with transcriptional complexes and craniosynostosis. Hum Mol Genet. (2019) 28(15):2501–13. doi: 10.1093/hmg/ddz072
29. Lamb AN, Rosenfeld JA, Neill NJ, Talkowski ME, Blumenthal I, Girirajan S, et al. Haploinsufficiency of SOX5 at 12p12.1 is associated with developmental delays with prominent language delay, behavior problems, and mild dysmorphic features. Hum Mutat. (2012) 33(4):728–40. doi: 10.1002/humu.22037
30. Akcakaya NH, Iseri SU, Bilir B, Battaloglu E, Tekturk P, Gultekin M, et al. Clinical and genetic features of PKAN patients in a tertiary centre in Turkey. Clin Neurol Neurosurg. (2017) 154:34–42. doi: 10.1016/j.clineuro.2017.01.011
31. Mahler EA, Johannsen J, Tsiakas K, Kloth K, Lüttgen S, Mühlhausen C, et al. Exome sequencing in children. Dtsch Arztebl Int. (2019) 116(12):197–204. doi: 10.3238/arztebl.2019.0197
32. Nouri N, Bahreini A, Nasiri J, Salehi M. Clinical and genetic profile of children with unexplained intellectual disability/developmental delay and epilepsy. Epilepsy Res. (2021) 177:106782. doi: 10.1016/j.eplepsyres.2021.106782
33. Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. (2015) 385(9975):1305–14. doi: 10.1016/S0140-6736(14)61705-0
34. Kuperberg M, Lev D, Blumkin L, Zerem A, Ginsberg M, Linder I, et al. Utility of whole exome sequencing for genetic diagnosis of previously undiagnosed pediatric neurology patients. J Child Neurol. (2016) 31(14):1534–9. doi: 10.1177/0883073816664836
35. Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. (2014) 312(18):1870–9. doi: 10.1001/jama.2014.14601
36. Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, et al. Enhanced utility of family- centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. (2015) 17(7):578–86. doi: 10.1038/gim.2014.154
37. Hiraide T, Yamoto K, Masunaga Y, Asahina M, Endoh Y, Ohkubo Y, et al. Genetic and phenotypic analysis of 101 patients with developmental delay or intellectual disability using whole-exome sequencing. Clin Genet. (2021) 100(1):40–50. doi: 10.1111/cge.13951
38. Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, et al. Genomic diagnosis for children with intellectual disability and or developmental delay. Genome Med. (2017) 9:43. doi: 10.1186/s13073-017-0433-1
39. Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. (2014) 511:344–7. doi: 10.1038/nature13394
40. Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by wholegenome sequencing. Am J Hum Genet. (2013) 93:249–63. doi: 10.1016/j.ajhg.2013.06.012
41. French CE, Delon I, Dolling H, Sanchis-Juan A, Shamardina O, Mégy K, et al. Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensive Care Med. (2019) 45(5):627–36. doi: 10.1007/s00134-019-05552-x
42. Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet. (2017) 101(5):664–85. doi: 10.1016/j.ajhg.2017.09.008
43. Bozkurt T, Alanay Y, Isik U, Sezerman U. Re-analysis of whole-exome sequencing data reveals a novel splicing variant in the SLC2A1 in a patient with GLUT1 deficiency syndrome 1 accompanied by hemangioma: a case report. BMC Med Genomics. (2021) 14(1):197. doi: 10.1186/s12920-021-01045-3
44. Brunet T, Jech R, Brugger M, Kovacs R, Alhaddad B, Leszinski G, et al. De novo variants in neurodevelopmental disorders-experiences from a tertiary care center. Clin Genet. (2021) 100(1):14–28. doi: 10.1111/cge.13946
Keywords: epilepsy, developmental delay, next-generation sequencing, pediatric neurology, pediatric geneticist
Citation: Kava H, Akgun-Dogan O, Yesilyurt A, Alanay Y and Isik U (2025) Evaluation of the etiology of epilepsy and/or developmental delay in children via next-generation sequencing: a single-center experience. Front. Pediatr. 13:1471965. doi: 10.3389/fped.2025.1471965
Received: 28 July 2024; Accepted: 28 January 2025;
Published: 27 February 2025.
Edited by:
Piero Pavone, University of Catania, ItalyReviewed by:
Raffaele Falsaperla, Policlinico San Marco, ItalyCopyright: © 2025 Kava, Akgun-Dogan, Yesilyurt, Alanay and Isik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yasemin Alanay, eWFzZW1pbi5hbGFuYXlAYWNpYmFkZW0uZWR1LnRy
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.