 Kajetan Trojovsky1
Kajetan Trojovsky1 Maximilian Seidl2
Maximilian Seidl2 Florian Babor1
Florian Babor1 Stephan Ehl3,4
Stephan Ehl3,4 Min Ae Lee-Kirsch5Michael Friedt6
Min Ae Lee-Kirsch5Michael Friedt6 Hans-Juergen Laws1Nibras Naami7
Hans-Juergen Laws1Nibras Naami7 Prasad Thomas Oommen1,†
Prasad Thomas Oommen1,† Sujal Ghosh1*†
Sujal Ghosh1*†
- 1Department of Pediatric Oncology, Hematology and Clinical Immunology, Medical Faculty, Center of Child and Adolescent Health, Heinrich-Heine-University and University Hospital, Duesseldorf, Germany
- 2Institute of Pathology, Heinrich Heine University and University Hospital of Duesseldorf, Duesseldorf, Germany
- 3Center for Chronic Immunodeficiency, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 4Center for Pediatrics and Adolescent Medicine, Medical Center, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 5Department of Pediatrics, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
- 6Department of General Pediatrics, Neonatology and Pediatric Cardiology, Division of Pediatric Gastroenterology, Heinrich-Heine-University and University Hospital, Duesseldorf, Germany
- 7Department of Pediatric Oncology and Hematology Herdecke, University Hospital Witten/Herdecke, Herdecke, Germany
Suppressors of cytokine signaling (SOCS) proteins play a critical role in regulating immune signaling pathways. Deficiency of SOCS1 leads to various autoimmune pathologies. We present two unrelated patients with distinct clinical manifestations. Patient 1, a 16-year-old male from Guinea, presented with Evans Syndrome, musculoskeletal pain and elevated liver enzymes. Patient 2, a 6-year-old German boy, developed recurrent oral aphthous ulcers, mild inflammatory bowel disease and chronic recurrent multifocal osteomyelitis. Both patients were diagnosed with SOCS1 deficiency by genetic testing. Treatment strategies included steroids, JAK inhibition and colchicine. These cases emphasize the importance of considering SOCS1 deficiency in patients with autoimmune or autoinflammatory diseases but also in patients with unexplained elevated IgE levels. They highlight the need for further research in ongoing multicenter registries to better understand this condition.
Introduction
The family of suppressors of cytokine signaling (SOCS) proteins are considered as essential key regulators in IFN type I and II signaling. SOCS proteins act as negative regulators of intracellular Janus kinases (JAK), which lead to inhibition of the JAK/STAT pathway (1–3). SOCS1 in particular, has the ability, not only to help to catalyze the ubiquitination of signaling intermediates, but also to directly inhibit the JAK protein activity, predominantly the activity of JAK-2 (1, 4). Interestingly, SOCS1 also acts as a tumor suppressor and is therefore silenced in many different tumor entities (5, 6).
SOCS1 deficiency or haploinsufficiency is a rare condition, first described in human in 2020. It leads to a variety of autoimmune pathologies (7–9). The phenotypical appearance can vary considerably, even among patients harboring identical mutations (8).
In this report, we describe two unrelated cases with distinct clinical manifestations of SOCS1 deficiency.
Case descriptions
Patient 1, a 16-year-old young boy, newly migrated from Guinea, presented with neutropenia, low platelets, constipation and chronic musculoskeletal pain in shoulders and hips when he was first admitted to our hospital. Physical examination revealed splenomegaly. Whole body MRI and sonography did not reveal any features explaining the pain, however heterogeneous liver tissue suggested a history of hepatitis. Further investigations showed elevated liver enzymes and IgE levels, along with low memory B cells and mild T cell lymphopenia. Testing for hepatotropic viruses, autoimmune hepatitis, parasites and allergic history remained negative. “Evans syndrome” was diagnosed with positive anti-granulocyte and anti-platelet antibodies. Genetic panel sequencing covering inborn errors of immunity revealed a c.214_215delinsAA (p.A72 K) variant in SOCS1 indicating SOCS1 deficiency. Family testing was not possible as other family members were not available. T cell proliferation was normal (PHA, anti-CD3, anti-CD3/28), however we observed a slightly elevated interferon signature with a score of 27,61 (<12,49) as expression of interferon expressing genes (IFI27, IFI44, IFI44l, IFIT1, ISG15, RSAD2, SIGLEC1) and elevated STAT1 phosphorylation (Figure 1) (10). Due to insufficient pain control with metamizole and NSAID we started treatment with steroids, providing partial pain relief and normalization of ANCs. Due to adverse effects steroids had to be tapered. Subsequent treatment with ruxolitinib (up to 2 × 20 mg) improved pain control and overall well-being but had limited impact on blood counts. Paradoxically, ruxolitinib led to a strongly increased interferon signature with a score of 871,95 (<12,49).
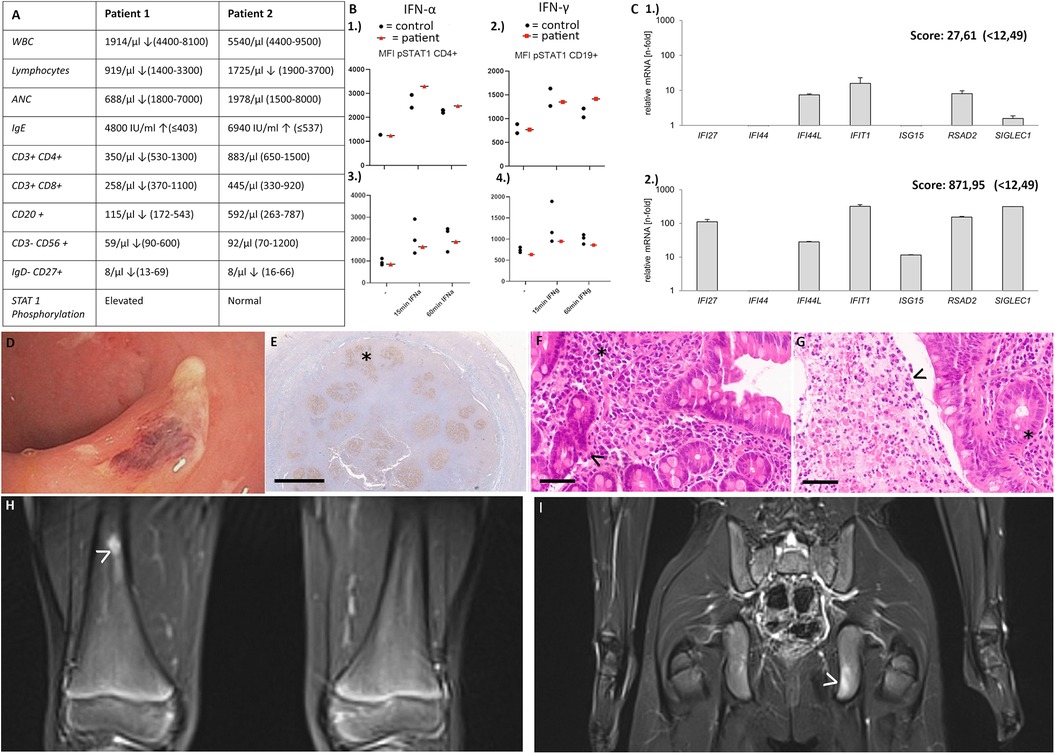
Figure 1. (A) Immunological features in patient 1 and patient 2 (B) STAT1 phosphorylation 1.) patient 1: elevated STAT1 phosphorylation after stimulation with IFN alpha and 2.) elevated STAT1 phosphorylation after 60 min of stimulation with IFN gamma 3.) patient 2: no elevated STAT1 phosphorylation after stimulation with IFN alpha and 4.) IFN gamma (C) 1.) interferon signature of patient 1 before start of ruxolitinib [slightly elevated, score 27,61 (<12,49)] 2.) interferon signature of patient 1 after 7 months of ruxolitinib treatment [strongly elevated, score 871,95 (<12,49)]. (D–G) Patient 2 (D) unspecific colitis with fibrin coated erosions in the rectum (E) Appendix at the age of 5 years showing irregular and progressively transformed germinal centers (BCL6 immunohistochemistry with DAB-based brown coloration, altered germinal center marked by asterisk) (F) Duodenum at the age of 8 years showing slightly elevated plasma cell densities in the lamina propria (asterisk), and slightly hyperplastic crypts (arrowhead) as signs of a minor, non-specific chronic inflammation. No villous atrophy and no intraepithelial lymphocytosis. (G) Rectum at the age of 8 years showing slightly hyperplastic mucosa on the right side, necrotic debris with neutrophils and macrophages on the left (pseudomembrane, arrowhead). Magnifications were as follows: (E) 1.4×, bar 2 mm; (F/G) 40×, bar 50 µm. Histological sections were digitized with the Aperio slide scanner (Leica, Nussloch, Germany), with an original magnification of 40× and an original resolution 0.253 µm per pixel. (H/I)Patient 2: Focal T2w signal alterations (arrowheads) in the distal third of the right femur (H), and in the ischium on the left (I) indicating lesions suggestive of CRMO.
Patient 2, a 6-year-old German boy, was referred to our department due to recurrent oral ulcers, diarrhea and recurrent upper respiratory tract infections. His medical history included suspected milk protein allergy, appendectomy at 5 years, atopic dermatitis since infancy, recurrent episodes of epistaxis and skin infections (11). Family history revealed a maternal grandmother with bronchial asthma and recurrent upper respiratory tract infections, arthrosis and type 2 diabetes. Oesophagogastroscopy and ileocolonoscopy in our patient showed unspecific colitis with fibrin coated erosions in the rectum, as well as distal gastritis with small, aphthous ulcers in the antrum and signs of proximal duodenitis (Figure 1). Histology showed a higher inflammation intensity in the large bowel. The inflammatory pattern shared some similarities with graft-vs.-host disease, and did not resemble ulcerative colitis or Crohn‘s disease. Histology revealed an inflammatory infiltrate dominated by (neutrophil) granulocytes in earlier, a lymphocytic pattern in the latest biopsy. There were no signs of a disturbed plasma cell maturation or homing. However, germinal centers were ill-defined throughout all biopsies. (Figure 1) Colchicine was initiated and led to complete resolution of oral aphthae. Furthermore, mesalazine led to improvement of diarrhea and lasting continuous remission, also histologically.
At the age of 9 years, the patient complained about progressive limb and neck pain, with MRI findings suggestive of chronic recurrent multifocal osteomyelitis (CRMO) in various bones. Treatment with NSAID significantly reduced pain and osseous inflammation.
Due to the unusual manifestations an inborn error of immunity was suspected. Further investigations revealed elevated IgE levels and low memory B cells (Figure 1), prompting whole exome sequencing. A heterozygous deletion in SOCS1 was identified. Further functional tests showed no abnormalities in STAT1 phosphorylation or T cell proliferation, and screening for auto-antibodies remained negative. Currently mesalazine and naproxen are well controlling the patients’ symptoms.
Discussion
Consistent with previous clinical cases of SOCS1 deficiency, our patients presented with a variety of autoimmune and autoinflammatory symptoms (8, 9, 11–14).
Notably, patient 1 experienced musculoskeletal pain without correlating MRI abnormalities, a novel observation in this context. Treatment with ruxolitinib improved pain control and quality of life, implicating underlying inflammation, despite no significant improvement in blood counts. Interestingly, treatment with ruxolitinib led to further increased interferon signature and liver enzymes, which might be conflicting for long term usage.
Patient 2 initially presented with gastrointestinal symptoms, without histological resembling of Crohn's disease, which has been observed in SOCS1 deficiency (8, 13). Additionally, this patient represents the first case of CRMO in SOCS1 deficiency. As treatment with colchicine, mesalazine, and naproxen was effective, we did not further intensify treatment. Interestingly, the complete deletion of SOCS1 as seen in this patient, does not appear to necessarily lead to more severe clinical features (13).
Our cases highlight the importance of screening patients with autoimmune or autoinflammatory diseases for SOCS1 deficiency (11, 13, 15). Furthermore, both patients showed highly elevated IgE levels exceeding the extent of atopy seen at the time of investigation. Further studies are needed to elucidate the potential link between autoimmune processes and elevated IgE levels in SOCS1 deficiency. Treatment strategies in our patients included symptom- and organ-orientated mediation. Given the paradox results of interferon signature in patient 1, targeted JAK-Inhibition with ruxolitinib needs further investigation.
It remains of paramount importance to include these patients in multicenter registries, as the ESID SOCS1 registry, to gain more structured data regarding complications, treatment strategies and long-term outcome.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The study of these participants is covered by the ESID approval issued by the Ethics committee at the University Hospital Düsseldorf (Study ID 5250). Written informed consent was obtained from the individual(s), and minor(s)' legal guardian for participation and publication of clinical data.
Author contributions
KT: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. MS: Writing – original draft, Writing – review & editing. FB: Writing – original draft, Writing – review & editing. SE: Writing – original draft, Writing – review & editing. ML-K: Writing – original draft, Writing – review & editing. MF: Writing – original draft, Writing – review & editing. H-JL: Writing – original draft, Writing – review & editing. NN: Writing – original draft, Writing – review & editing. PO: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. SG: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun. (2018) 9:1558. doi: 10.1038/s41467-018-04013-1
2. Yoshimura A, Ito M, Chikuma S, Akanuma T, Nakatsukasa H. Negative regulation of cytokine signaling in immunity. Cold Spring Harb Perspect Biol. (2018) 10:1–4. doi: 10.1101/cshperspect.a028571
3. Alexander WS. Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol. (2002) 2:410–6. doi: 10.1038/nri818
4. Kamizono S, Hanada T, Yasukawa H, Minoguchi S, Kato R, Minoguchi M, et al. The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J Biol Chem. (2001) 276:12530–8. doi: 10.1074/jbc.M010074200
5. Beaurivage C, Champagne A, Tobelaim WS, Pomerleau V, Menendez A, Saucier C. SOCS1 in cancer: an oncogene and a tumor suppressor. Cytokine. (2016) 82:87–94. doi: 10.1016/j.cyto.2016.01.005
6. Yeganeh M, Gui Y, Kandhi R, Bobbala D, Tobelaim WS, Saucier C, et al. Suppressor of cytokine signaling 1-dependent regulation of the expression and oncogenic functions of p21(CIP1/WAF1) in the liver. Oncogene. (2016) 35:4200–11. doi: 10.1038/onc.2015.485
7. Lee PY, Platt CD, Weeks S, Grace RF, Maher G, Gauthier K, et al. Immune dysregulation and multisystem inflammatory syndrome in children (MIS-C) in individuals with haploinsufficiency of SOCS1. J Allergy Clin Immunol. (2020) 146:1194–1200.e1. doi: 10.1016/j.jaci.2020.07.033
8. Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, et al. Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun. (2020) 11:5341. doi: 10.1038/s41467-020-18925-4
9. Rodari MM, Cazals-Hatem D, Uzzan M, Martin Silva N, Khiat A, Ta MC, et al. Insights into the expanding intestinal phenotypic spectrum of SOCS1 haploinsufficiency and therapeutic options. J Clin Immunol. (2023) 43:1403–13. doi: 10.1007/s10875-023-01495-7
10. Wolf C, Brück N, Koss S, Griep C, Kirschfink M, Palm-Beden K, et al. Janus kinase inhibition in complement component 1 deficiency. J Allergy Clin Immunol. (2020) 146:1439–1442.e5. doi: 10.1016/j.jaci.2020.04.002
11. Körholz J, Gabrielyan A, Sowerby JM, Boschann F, Chen LS, Paul D, et al. One gene, many facets: multiple immune pathway dysregulation in SOCS1 haploinsufficiency. Front Immunol. (2021) 12:680334. doi: 10.3389/fimmu.2021.680334
12. Hale RC, Owen N, Yuan B, Chinn IK. Phenotypic variability of SOCS1 haploinsufficiency. J Clin Immunol. (2023) 43:902–6. doi: 10.1007/s10875-023-01460-4
13. Michniacki TF, Walkovich K, DeMeyer L, Saad N, Hannibal M, Basiaga ML, et al. SOCS1 haploinsufficiency presenting as severe enthesitis, bone marrow hypocellularity, and refractory thrombocytopenia in a pediatric patient with subsequent response to JAK inhibition. J Clin Immunol. (2022) 42:1766–77. doi: 10.1007/s10875-022-01346-x
14. Gruber C, Lee A, Buta S, Khattri S, Gottlieb AB, Frost JM, et al. IL4Rα And IL17A blockade rescue autoinflammation in SOCS1 haploinsufficiency. J Clin Immunol. (2024) 44:36. doi: 10.1007/s10875-023-01635-z
Keywords: SOCS1, SOCS1 haploinsufficiency, SOCS1 deficiency, CRMO, CNO, autoinflammation, autoimmunity
Citation: Trojovsky K, Seidl M, Babor F, Ehl S, Lee-Kirsch MA, Friedt M, Laws H-J, Naami N, Oommen PT and Ghosh S (2025) SOCS1 deficiency—crossroads of autoimmunity and autoinflammation—two case reports. Front. Pediatr. 12:1516017. doi: 10.3389/fped.2024.1516017
Received: 23 October 2024; Accepted: 13 December 2024;
Published: 7 January 2025.
Edited by:
Guzide Aksu, Ege University, TürkiyeReviewed by:
Hermann Girschick, Vivantes Hospital, GermanyDewton Moraes Vasconcelos, University of São Paulo, Brazil
Copyright: © 2025 Trojovsky, Seidl, Babor, Ehl, Lee-Kirsch, Friedt, Laws, Naami, Oommen and Ghosh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sujal Ghosh, c3VqYWwuZ2hvc2hAbWVkLnVuaS1kdWVzc2VsZG9yZi5kZQ==
†These authors have contributed equally to this work