 Hua Zhou
Hua Zhou Jun Lu
Jun Lu Tao Wang
Tao Wang Xiaoyan Gu1
Xiaoyan Gu1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 05 November 2024
Sec. Pediatric Hematology and Hematological Malignancies
Volume 12 - 2024 | https://doi.org/10.3389/fped.2024.1493362
Background: Asparaginase is a critical component of chemotherapy for pediatric acute lymphoblastic leukemia (ALL), but its use is often complicated by asparaginase-associated pancreatitis (AAP). Genetic predispositions, such as variants in the SPINK1 gene, have been linked to an increased risk of pancreatitis. However, the role of genetic factors in relation to asparaginase treatment remains incompletely understood, partly because mutations in pancreatitis-causing genes are rarely found in pediatric ALL.
Case description: A four-year and three-month-old Chinese girl was admitted to our hospital due to fever for half a day, with no history of significant prior medical history. Initial blood tests revealed hematological abnormalities, including leukopenia, anemia, and thrombocytosis. Bone marrow aspiration identified 81.5% blast cells with B-lymphocyte morphology and immunophenotype, leading to a diagnosis of B-cell acute lymphoblastic leukemia (B-ALL). The patient began treatment under the CCCG-ALL-2015 protocol, which included PEG-asparaginase (PEG-asp). On day 10 of induction, she developed AAP, which was primarily characterized by severe epigastric pain and elevated serum amylase. Despite effective symptom management with analgesics and anti-inflammatory therapy, AAP recurred following administration of L-asparaginase (L-asp). Genetic analysis revealed a heterozygous SPINK1 c.194 + 2T>C variant (rs148954387), a well-known pathogenic variant associated with increased susceptibility to pancreatitis. Sanger sequencing confirmed that the SPINK1 variant was inherited from her asymptomatic mother. The patient's AAP was managed conservatively, and an asparaginase-free regimen ultimately achieved complete remission without recurrence of pancreatitis.
Conclusions: The identification of the SPINK1 c.194 + 2T>C variant, which is recognized as pathogenic, provides valuable information for understanding the heightened risk of AAP in our pediatric ALL patient. Our case underscores the potential role of genetic predisposition in the development of AAP and highlights the importance of considering genetic screening prior to asparaginase therapy in pediatric ALL patients to identify those at increased risk.
Acute pancreatitis (AP) is characterized by the abrupt onset of pancreatic inflammation that arises when digestive enzymes activate within the pancreas itself, causing the organ to begin self-digestion (1–3). As a highly heterogeneous disease, AP presents a wide range of clinical manifestations, ranging from mild discomfort to more severe signs such as severe abdominal pain, nausea, and vomiting (1–3). Despite a survival rate about 95%, severe AP continues to be a major cause of death for those hospitalized patients with gastrointestinal diseases (1, 4, 5).
While both children and adults can develop AP, it is less common in children, with about 3–13 cases per 100,000 children per year, compared to 5–60 cases per 100,000 adults per year (6–8). In adults, gallstones and alcohol abuse are the predominant causes of AP, whereas in children, the etiology is diverse and often multifactorial (1, 2, 8). Common triggers in pediatric cases include medications, genetic factors, metabolic disorders, and infections (8). The intricate nature of AP, combined with the potential for severe complications and the necessity to address underlying causes, makes this condition particularly challenging to treat and prone to recurrence in children. It has been reported that nearly a third of children who experience AP will have at least one additional episode (9). Therefore, effective management of AP, especially in children, requires a tailored approach that considers the unique aspects of each patient.
AP is a significant and potentially serious complication in pediatric patients with acute lymphoblastic leukemia (ALL) undergoing chemotherapy, particularly when asparaginase such as L-asparaginase (L-asp) or PEG-asparaginase (PEG-asp), a pegylated form of L-asp, is included in the treatment regimen (10–12). Asparaginase catalyzes the hydrolysis of the amino acid asparagine into aspartic acid and ammonia. By depleting asparagine, asparaginase inhibits protein synthesis in leukemic cells, leading to their death. Although asparaginase is a critical component in the treatment of ALL, it comes with a range of potential adverse effects (13). Notably, asparaginase can cause the premature activation of pancreatic digestive enzymes within the pancreas (14, 15). This results in autodigestion of pancreatic tissue and ultimately asparaginase-associated pancreatitis (AAP), a distinct type of AP marked by the similar clinical signs of AP but with a specific etiology related to asparaginase therapy (10). The incidence of AAP varies, but it is estimated to occur in approximately 2%–18% of ALL patients, with a higher occurrence in older children and adults (10–12, 16, 17). AAP not only complicates pediatric ALL treatment but also increases the risk of mortality, necessitating vigilant monitoring and early intervention strategies.
What is noteworthy is that genetic factors also play a significant role in predisposing individuals to pancreatitis, with mutations in the PRSS1 (protease serine 1), SPINK1 (serine peptidase inhibitor Kazal type 1), CFTR (cystic fibrosis transmembrane conductance regulator), CTRC (chymotrypsin C), TRPV6 (transient receptor potential cation channel subfamily V member 6), and CPA1 (carboxypeptidase A1) genes being predominant contributors (18, 19). Recent machine learning studies by Nielsen et al. also identified several single nucleotide polymorphisms (SNPs) as potential risk factors for AAP, including rs10273639 (PRSS1-PRSS2), rs10436957 (CTRC), rs13228878 (PRSS1-PRSS2), rs1505495 (GALNTL6), and rs4655107 (EPHB2) (20). However, genetic screening for pancreatitis-causing genes in ALL patients is not currently standard practice, which limits our understanding of the impact of specific genetic variants on the development of AAP in pediatric ALL receiving asparaginase therapy. In this case report, we present a very rare instance of a pediatric B-cell ALL (B-ALL) patient with a SPINK1 NM 003122: c.194 + 2T>C heterozygous variant (rs148954387) and AAP, highlighting the intricate interplay between genetic predisposition and asparaginase therapy in AAP development. By examining this case, we underscore the importance of genetic screening before initiating asparaginase therapy in pediatric ALL to identify at-risk patients and tailor treatment approaches accordingly.
In October 2020, a four-year and three-month-old Chinese girl was admitted to the Affiliated Changzhou Children's Hospital of Nantong University due to fever for half a day. Her parents reported that the patient's maximum temperature reached 39.8°C at home, which could be temporarily relieved by physical cooling and ibuprofen. Transient chest pain and one episode of nonprojectile vomiting occurred during the fever, but there were no cold symptoms such as chills, cough, or a stuffy nose. Her parents were non-consanguineous, and her mother was gravida 1, para 1 (G1P1). The patient was born via cesarean section at term, with a birth weight of 3,100 g and a length of 50 cm. After birth, the patient had normal growth and development without significant medical history. A family history of hereditary, cancerous, and contagious diseases, as well as any epidemiologic connection to other COVID-19 cases or impacted regions, was denied by her parents.
On admission, the patient's temperature was 36.4°C, pulse rate was 108 beats/min, respiration rate was 24 breaths/min, and blood pressure was measured at 120/75 mmHg. Physical examination revealed enlarged tonsils and hyperemia of the pharynx. All other general and neurological examinations showed no abnormalities. Blood laboratory test revealed that the white blood cells count was 1.1 × 109/L (normal range, 4–10 × 109/L), monocytes count was 0.04 × 109/L (normal range, 0.12–0.8 × 109/L), and neutrophils count was 0.01 × 109/L (normal range, 2–7.7 × 109/L), all of which were remarkably reduced. The red blood cells count was 3.15 × 1012/L (normal range, 3.5–5.5 × 1012/L) and hemoglobin was 89 g/L (normal range, 110–160 g/L), both of which were also reduced. In addition, the platelet count showed an increase to 510 × 109/L (normal range, 100–300 × 109/L), and the C-reactive protein level was elevated to 30.03 mg/L (normal range, 0–10 mg/ml).
To investigate the cause of abnormalities in blood cell counts, we further performed bone marrow (BM) aspiration, which showed hyperplastic BM with 81.5% of blast cells exhibiting lymphoid morphology. The blasts were generally large with a high nucleus-to-cytoplasm ratio, finely dispersed chromatin, and prominent nucleoli (Figure 1A). All blasts were negative for myeloperoxidase (MPO) staining. Multiparameter flow cytometry of BM aspirate detected 89.6% blasts and showed an immunophenotype that was positive for CD10, CD19, CD34, CD38, and HLA-DR, and negative for CD2, CD7, CD13, CD14, CD15, CD20, CD33, and CD117, corresponding to B-ALL features. G-band analysis of BM metaphase cells was further performed to analyze chromosomal abnormalities. The karyotypes of ten analyzed cells were 46,XX[6]/54,XX,dup(1)(q21q31),+4,+6,+10,+14,+17,+18,+21,+21[1]/55,idem,+8[1]/56,idem,+6,+8[2] (Figure 1B). Although common fusion genes typically found in leukemia were not detected in this patient, further transcriptome analysis using bulk RNA sequencing (RNA-seq) detected a rare RNF213-SLC26A11 fusion gene. Taken together, the patient eventually was diagnosed with B-ALL.
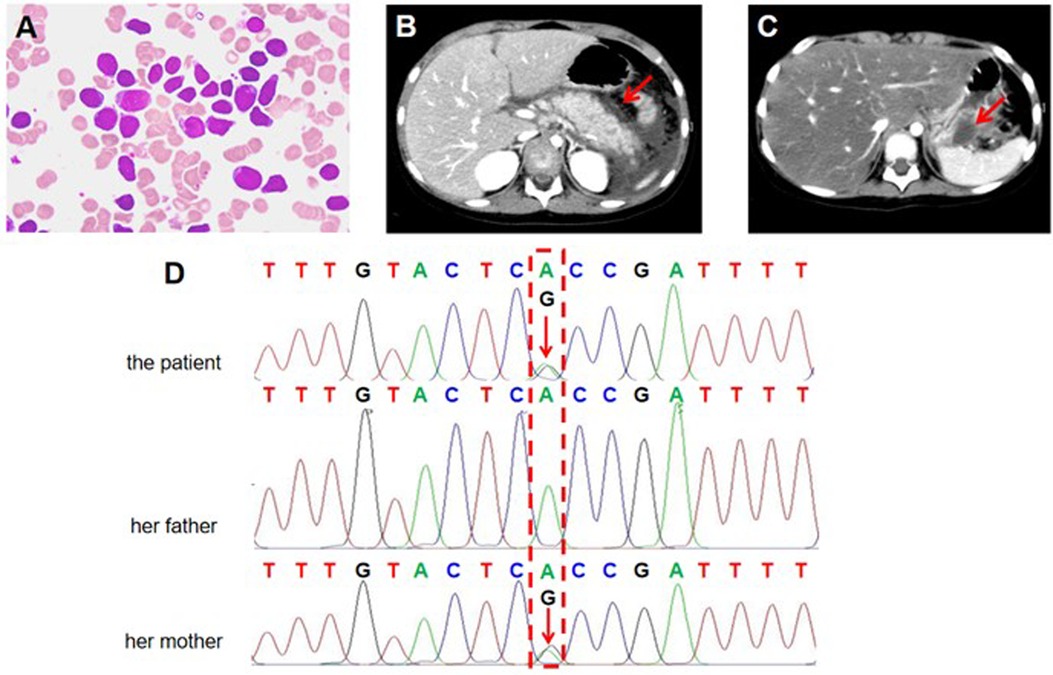
Figure 1. Clinical and genetic analysis of the investigated family. (A) Representative image of a bone marrow (BM) aspirate stained with Wright-Giemsa stain from the patient with B-cell acute lymphoblastic leukemia (B-ALL). The image shows a predominance of lymphoblasts, characterized by large, immature cells with scant cytoplasm, round to irregular nuclei, fine chromatin, and prominent nucleoli. (B,C) Contrast-enhanced axial abdominal CT scan of the pediatric patient showing features consistent with acute pancreatitis (AP). (B) The first episode of AP. The red arrow indicates that the pancreas is enlarged with diffuse peripancreatic fat stranding and peri-pancreatic fluid accumulation. The surrounding organs, including the liver and kidneys, appear unremarkable. (C) The second episode of AP. The red arrow indicates pancreatic pseudocyst. (D) Sanger sequencing (reverse strands) analysis of the SPINK1 c.194 + 2T>C variant in the investigated family. The results show the B-ALL patient and her mother carries a heterozygous SPINK1 c.194 + 2T>C variant. The patient's father has normal SPINK1 gene. The red arrow indicates the identified variant position.
According to Chinese Children's Cancer Group study ALL-2015 (CCCG-ALL-2015) protocol, the patient was classified as low-risk ALL and treated with standard induction regimen, which included vincristine, daunorubicin, PEG-asp, and dexamethasone. On day 10, the patient developed vomiting and severe epigastric pain. Serum amylase was elevated to 508.0 U/L, which was more than three times the upper limit of normal. Abdominal computed tomography (CT) showed pancreas edema, suggesting the development of AAP (Figure 1B). Therefore, the patient fasted for one week and was treated with analgesic therapy (octreotide) and anti-inflammatory therapy (ulinastatin) for ten days, which significantly relieved her epigastric pain. On day 19, the minimal residual disease (MRD) was 1.2 × 10−2 by flow cytometry. Therefore, the patient continued to take the same regimen as re-induction, albeit PEG-asp was replaced by L-asp. Notably, symptoms of pancreatitis reoccurred immediately after L-asp administration. On abdominal CT scan, pancreatic pseudocyst was detected (Figure 1C). Thus, both PEG-asp and L-asp were discontinued. After symptomatic treatment such as fasting, fluid intake, octreotide, and ulinastatin, the patient's symptoms were gradually improved. On day 46, end-of-induction BM aspiration revealed 0% blasts by flow cytometry MRD. Following the CCCG-ALL-2015 protocol, the patient underwent consolidation therapy with high-dose methotrexate (MTX) and maintenance therapy with low-dose MTX and mercaptopurine, leading to complete remission of ALL.
The patient's parents were both healthy with no history of pancreatitis or hematological diseases. To accurately determine the cause of our patient's AAP, we collected her peripheral blood genomic DNA and performed whole-exome sequencing (WES) analysis using standard protocols. Data quality assurance, bioinformatics analysis, and interpretation were conducted following the American College of Medical Genetics and Genomics (ACMG) guidelines and established approaches. In our analysis of the exome data, we specifically examined genes associated with hematological and metabolic diseases, as well as those related to pancreatitis, including PRSS1, SPINK1, CFTR, CTRC, TRPV6, and CPA1. After filtering common (allele frequency of >5%) and non-coding variants, we found a heterozygous variant NM_003122.2: c.194 + 2T>C in intron 3 of SPINK1 gene (rs148954387), which may contribute to the development of AAP. Sanger sequencing confirmed the presence of SPINK1 c.194 + 2T>C intronic variant in the patient (Figure 1D). Further analysis revealed that the patient's healthy mother was also heterozygous for SPINK1 c.194 + 2T>C, while the father carried the normal SPINK1 gene (Figure 1D). According to the ACMG guidelines, SPINK1 c.194 + 2T>C is defined as a pathogenic variant (PVS1, PS1). Based on clinical and genetic evidence, we conclude that the SPINK1 c.194 + 2T>C variant increases the susceptibility to AAP during asparaginase treatment in our B-ALL patient.
AAP is a significant and common adverse effect observed during asparaginase-based therapy for ALL, particularly in pediatric patients. Although the precise mechanism of AAP is multifactorial and not fully elucidated, it likely involves a combination of inappropriate pancreatic enzymes activation, immune responses, and metabolic disturbances that collectively increase the risk of pancreatic inflammation and injury (21). One of the mechanisms by which asparaginase induces pancreatic injury is through toxic calcium (Ca2+) signaling, which eventually leads to necrosis of pancreatic acinar cells (22). For accurate diagnosis of AAP in pediatric ALL patients, a thorough evaluation is essential. This includes a detailed assessment of clinical history, symptoms, laboratory tests, and imaging studies (1–3). For our patient, the key evidence for diagnosing AAP included abdominal pain that occurred rapidly following asparaginase administration, elevated serum amylase levels (more than three times the upper limit of normal), and appearance of pancreatic edema on CT scan.
Although genetic factors may influence the development of AAP, routine screening for mutations in pancreatitis-related genes is not commonly performed before treatment in pediatric ALL. As a result, the specific role of these gene mutations in AAP occurrence in pediatric ALL remains poorly documented and is considered rare. In this report, we identified the SPINK1 c.194 + 2T>C variant (rs148954387) as key pathogenic factor of the AAP in a B-ALL patient. To the best of our knowledge, this is the first report identifying this SPINK1 variant in pediatric ALL. The SPINK1 gene encodes a single-chain polypeptide known as pancreatic secretory trypsin inhibitor (PSTI), a protein approximately 6.3 kDa in size that plays a crucial role in preventing the activation of trypsinogen, the inactive precursor of trypsin, within the pancreas (18, 19, 23). PSTI is estimated to inhibit around 20% of the pancreas's total potential trypsin activity. Mutations in the SPINK1 gene are widely perceived to reduce the activity of PSTI, allowing trypsinogen to be prematurely converted into trypsin within the pancreatic tissue (24). This excessive proteolytic activity may lead to autodigestion of the pancreas and initiates inflammation, contributing to the pathogenesis of pancreatitis. Although PSTI is primarily known for its ability to inhibit trypsin, its broader biological roles and interactions with other pancreatic proteins and enzymes remain incompletely understood, necessitating further research to clarify these functions and their implications for pancreatitis.
In fact, the SPINK1 c.194 + 2T>C variant (rs148954387), also known as IVS3 + 2T>C, is one of the most common genetic variants associated with pancreatitis, particularly in East-Asian populations (18). According gnomAD V4.1.0, the variant allele was found at a total frequency of 0.0001137 (25). c.194 + 2T>C variant occurs in the splice donor site of intron 3 of the SPINK1 gene. The pathogenic impact of this intronic variant is well-documented (26–28). The SPINK1 c.194 + 2T>C heterozygous variant has been independently demonstrated in multiple studies to disrupt pre-mRNA splicing, which significantly reduces the expression of full-length SPINK1 mRNA (26–28). Using pancreatitis mouse model, a recent study by Liu et al. demonstrated that the heterozygous Spink1 c.194 + 2T>C mutation promotes the development of pancreatitis by elevating interleukin-33 levels, inducing M2 macrophage polarization, and activating pancreatic stellate cells (29). However, like other pathogenic variants of pancreatitis, the SPINK1 c.194 + 2T>C variant is also found in healthy individuals, suggesting that additional factors such as other genetic variants or lifestyle influences likely play a significant role in disease expression (18). Indeed, our B-ALL patient had no symptoms of pancreatitis prior to this hospitalization but developed symptoms only after receiving asparaginase. Notably, her mother, who also carries the SPINK1 c.194 + 2T>C heterozygous variant, has not exhibited noticeable symptoms of pancreatitis in the past. Nevertheless, given that SPINK1 c.194 + 2T>C variant increases susceptibility to pancreatitis, the patient's mother has been advised to maintain a healthy lifestyle and to seek medical attention if any symptoms associated with pancreatitis develop.
A definitive consensus on managing ALL in patients with SPINK1 or other gene mutations that predispose them to AAP has not yet been established. In general, the management of AAP is a delicate balance between addressing the acute episode, preventing further complications, and ensuring that leukemia treatment remains effective, all while minimizing the risk of recurrent pancreatitis. Most patients with AAP require hospitalization, particularly if the episode is severe. When managing our patient's AAP, a typical supportive care approach includes fasting to rest the pancreas, intravenous fluids to maintain hydration, anti-inflammatory treatment to prevent infection, and pain management. This conservative approach is effective for most patients, although more severe cases might require additional interventions like nutritional support via enteral feeding or even surgical procedures if complications arise. Frequently, AAP leads to truncation of asparaginase therapy, which can increase the risk of leukemia relapse, making the reintroduction of asparaginase a critical consideration. Notably, studies indicate that reintroducing asparaginase after an initial episode of AAP carries a 40%–50% risk of a second pancreatitis episode (10, 11). Therefore, the decision to reintroduce asparaginase must be guided by a careful risk-benefit analysis. We must acknowledge that reintroducing asparaginase in our patient, primarily guided by the MRD level on day 19, without fully understanding her genetic background, carries significant risks. Fortunately, conservative treatment effectively controlled the symptoms of pancreatitis and prevent recurrence, and the asparaginase-free chemotherapy achieved an MRD-negative state without causing complications. As of the time of writing, there has been no recurrence of pancreatitis in the past four years.
This case report has several limitations that should be considered. First, as a single case study, the findings may not be generalizable to all pediatric ALL patients undergoing asparaginase therapy. The identification of the SPINK1 c.194 + 2T>C variant and its association with AAP in this patient highlights the potential importance of genetic screening, but the rarity of such findings limits the applicability of this approach in routine clinical practice. Additionally, although we performed comprehensive genetic analysis, other potential genetic, environmental, or treatment-related factors contributing to AAP were not fully explored. The absence of long-term follow-up data beyond four years also limits our understanding of the patient's risk of pancreatitis recurrence or other potential complications. Lastly, the decision to reintroduce asparaginase based on MRD levels, while achieving leukemia remission, poses a significant risk, which may not be acceptable in all clinical contexts. Further research is needed to establish standardized protocols for managing asparaginase therapy in patients with genetic predispositions to AAP.
To summarize, the identification of the SPINK1 c.194 + 2T>C variant, which is recognized as pathogenic, provides valuable information for understanding the heightened risk of AAP in our pediatric ALL patient. Our case underscores the potential role of genetic predisposition in the development of AAP and highlights the importance of considering genetic screening prior to asparaginase therapy in pediatric ALL patients to identify those at increased risk.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the ethics committee of the Affiliated Changzhou Children’s Hospital of Nantong University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
HZ: Data curation, Formal Analysis, Investigation, Writing – original draft. JL: Investigation, Writing – review & editing. TW: Formal Analysis, Investigation, Writing – review & editing. XG: Writing – review & editing. XL: Writing – review & editing. JZ: Conceptualization, Project administration, Resources, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We extend our gratitude to the patient and her family, as well as to all the physicians who assisted throughout the course of medical care.
TW was employed by Dian Diagnostics Group Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Boxhoorn L, Voermans RP, Bouwense SA, Bruno MJ, Verdonk RC, Boermeester MA, et al. Acute pancreatitis. Lancet. (2020) 396(10252):726–34. doi: 10.1016/S0140-6736(20)31310-6
2. Mederos MA, Reber HA, Girgis MD. Acute pancreatitis: a review. JAMA. (2021) 325(4):382–90. doi: 10.1001/jama.2020.20317
3. Szatmary P, Grammatikopoulos T, Cai W, Huang W, Mukherjee R, Halloran C, et al. Acute pancreatitis: diagnosis and treatment. Drugs. (2022) 82(12):1251–76. doi: 10.1007/s40265-022-01766-4
4. Czapári D, Váradi A, Farkas N, Nyári G, Márta K, Váncsa S, et al. Detailed characteristics of post-discharge mortality in acute pancreatitis. Gastroenterology. (2023) 165(3):682–95. doi: 10.1053/j.gastro.2023.05.028
5. Peery AF, Crockett SD, Murphy CC, Lund JL, Dellon ES, Williams JL, et al. Burden and cost of gastrointestinal, liver, and pancreatic diseases in the United States: update 2018. Gastroenterology. (2019) 156(1):254–272.e11. doi: 10.1053/j.gastro.2018.08.063
6. Nydegger A, Heine RG, Ranuh R, Gegati-Levy R, Crameri J, Oliver MR. Changing incidence of acute pancreatitis: 10-year experience at the royal children’s hospital, Melbourne. J Gastroenterol Hepatol. (2007) 22(8):1313–6. doi: 10.1111/j.1440-1746.2007.04936.x
7. Sellers ZM, MacIsaac D, Yu H, Dehghan M, Zhang KY, Bensen R, et al. Nationwide trends in acute and chronic pancreatitis among privately insured children and non-elderly adults in the United States, 2007–2014. Gastroenterology. (2018) 155(2):469–478.e1. doi: 10.1053/j.gastro.2018.04.013
8. Afzal S, Kleinhenz J. Acute pancreatitis in children. Pediatr Ann. (2021) 50(8):e330–5. doi: 10.3928/19382359-20210713-01
9. Gariepy CE, Ooi CY, Maqbool A, Ellery KM. Demographics and risk factors for pediatric recurrent acute pancreatitis. Curr Opin Gastroenterol. (2021) 37(5):491–7. doi: 10.1097/MOG.0000000000000764
10. Wolthers BO, Frandsen TL, Baruchel A, Attarbaschi A, Barzilai S, Colombini A, et al. Asparaginase-associated pancreatitis in childhood acute lymphoblastic leukaemia: an observational ponte di legno toxicity working group study. Lancet Oncol. (2017) 18(9):1238–48. doi: 10.1016/S1470-2045(17)30424-2
11. Liu C, Yang W, Devidas M, Cheng C, Pei D, Smith C, et al. Clinical and genetic risk factors for acute pancreatitis in patients with acute lymphoblastic leukemia. J Clin Oncol. (2016) 34(18):2133–40. doi: 10.1200/JCO.2015.64.5812
12. Rank CU, Wolthers BO, Grell K, Albertsen BK, Frandsen TL, Overgaard UM, et al. Asparaginase-Associated pancreatitis in acute lymphoblastic leukemia: results from the NOPHO ALL2008 treatment of patients 1–45 years of age. J Clin Oncol. (2020) 38(2):145–54. doi: 10.1200/JCO.19.02208
13. Hijiya N, van der Sluis IM. Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leuk Lymphoma. (2016) 57(4):748–57. doi: 10.3109/10428194.2015.1101098
14. Mukherjee A, Ahmed N, Rose FT, Ahmad AN, Javed TA, Wen L, et al. Asparagine synthetase is highly expressed at baseline in the pancreas through heightened PERK signaling. Cell Mol Gastroenterol Hepatol. (2020) 9(1):1–13. doi: 10.1016/j.jcmgh.2019.08.003
15. Tsai CY, Kilberg MS, Husain SZ. The role of asparagine synthetase on nutrient metabolism in pancreatic disease. Pancreatology. (2020) 20(6):1029–34. doi: 10.1016/j.pan.2020.08.002
16. Flores-Calderón J, Exiga-Gonzaléz E, Morán-Villota S, Martín-Trejo J, Yamamoto-Nagano A. Acute pancreatitis in children with acute lymphoblastic leukemia treated with L-asparaginase. J Pediatr Hematol Oncol. (2009) 31(10):790–3. doi: 10.1097/MPH.0b013e3181b794e8
17. Du X, Liu Z, Jia X, Wen Y, Tang Y, Xu X, et al. Clinical analysis of asparaginase-associated pancreatitis in children. Pancreatology. (2022) 22(6):706–12. doi: 10.1016/j.pan.2022.06.003
18. Suzuki M, Minowa K, Nakano S, Isayama H, Shimizu T. Genetic abnormalities in pancreatitis: an update on diagnosis, clinical features, and treatment. Diagnostics (Basel). (2020) 11(1):31. doi: 10.3390/diagnostics11010031
19. Wertheim-Tysarowska K, Oracz G, Rygiel AM. Genetic risk factors in early-onset nonalcoholic chronic pancreatitis: an update. Genes (Basel). (2021) 12(5):785. doi: 10.3390/genes12050785
20. Nielsen RL, Wolthers BO, Helenius M, Albertsen BK, Clemmensen L, Nielsen K, et al. Can machine learning models predict asparaginase-associated pancreatitis in childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol. (2022) 44(3):e628–36. doi: 10.1097/MPH.0000000000002292
21. Manohar M, Verma AK, Venkateshaiah SU, Sanders NL, Mishra A. Pathogenic mechanisms of pancreatitis. World J Gastrointest Pharmacol Ther. (2017) 8(1):10–25. doi: 10.4292/wjgpt.v8.i1.10
22. Peng S, Gerasimenko JV, Tsugorka T, Gryshchenko O, Samarasinghe S, Petersen OH, et al. Calcium and adenosine triphosphate control of cellular pathology: asparaginase-induced pancreatitis elicited via protease-activated receptor 2. Philos Trans R Soc Lond B Biol Sci. (2016) 371(1700):20150423. doi: 10.1098/rstb.2015.0423
23. Tukiainen E, Kylänpää ML, Kemppainen E, Nevanlinna H, Paju A, Repo H, et al. Pancreatic secretory trypsin inhibitor (SPINK1) gene mutations in patients with acute pancreatitis. Pancreas. (2005) 30(3):239–42. doi: 10.1097/01.mpa.0000157479.84036.ed
24. Szabó A, Toldi V, Gazda LD, Demcsák A, Tőzsér J, Sahin-Tóth M. Defective binding of SPINK1 variants is an uncommon mechanism for impaired trypsin inhibition in chronic pancreatitis. J Biol Chem. (2021) 296:100343. doi: 10.1016/j.jbc.2021.100343
25. GnomAD (Genome Aggregation Database). Available online at: https://gnomad.broadinstitute.org/ (accessed September 27, 2021).
26. Zou WB, Boulling A, Masson E, Cooper DN, Liao Z, Li ZS, et al. Clarifying the clinical relevance of SPINK1 intronic variants in chronic pancreatitis. Gut. (2016) 65(5):884–6. doi: 10.1136/gutjnl-2015-311168
27. Kume K, Masamune A, Kikuta K, Shimosegawa T. [-215G>A; IVS3+2T>C] mutation in the SPINK1 gene causes exon 3 skipping and loss of the trypsin binding site. Gut. (2005) 55(8):1214. doi: 10.1136/gut.2006.095752
28. Kereszturi E, Király O, Sahin-Tóth M. Minigene analysis of intronic variants in common SPINK1 haplotypes associated with chronic pancreatitis. Gut. (2009) 58(4):545–9. doi: 10.1136/gut.2008.164947
Keywords: pancreatitis, asparaginase, acute lymphoblastic leukemia, SPINK1, c.194 + 2T>C, case report
Citation: Zhou H, Lu J, Wang T, Gu X, Li X and Zhao J (2024) Acute pancreatitis following asparaginase treatment in pediatric acute lymphoblastic leukemia with a heterozygous SPINK1 c.194 + 2T>C intronic variant: a case report. Front. Pediatr. 12:1493362. doi: 10.3389/fped.2024.1493362
Received: 9 September 2024; Accepted: 24 October 2024;
Published: 5 November 2024.
Edited by:
Tomasz Szczepanski, Medical University of Silesia, PolandReviewed by:
Joanna Zawitkowska, Medical University of Lublin, PolandCopyright: © 2024 Zhou, Lu, Wang, Gu, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Zhao, MTMzMzk0MzZAcXEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.