 Aqsa Ahmad
Aqsa Ahmad Liang Lijun2*
Liang Lijun2* Zhang Yan
Zhang Yan- 1The First Clinical Medical College of Ningxia Medical University, Yinchuan, China
- 2Department of Pediatrics, General Hospital of Ningxia Medical University, Yinchuan, China
- 3Department of Pediatrics, Graduate School, Dalian Medical University, Liaoning, China
Background: Alport syndrome (AS) is a multifaceted condition that primarily affects the basement membranes of the kidneys, ears, and eyes. AS is considered the second most common cause of hereditary renal failure, exhibiting varied clinical manifestations across different lifespans. The aim of this study is to investigate the clinical features and genetic profile of AS and to elucidate the genotype-phenotype correlation of AS.
Method: The clinical and genetic data of ten children with AS treated at the General Hospital of Ningxia Medical University between January 2021 and May 2024 were retrospectively analyzed.
Results: Ten children with AS, six male and four female patients, with a mean age of 9 years (ranging from 3 to 15 years) were reported. Hematuria was observed in all individuals, with six cases exhibiting microscopic hematuria and four cases exhibiting macroscopic hematuria. Furthermore, extra-renal manifestations were noted in five cases, encompassing ocular abnormalities (n = 2) and hearing impairment (n = 3). In total, eight cases displayed mutations in COL4A5 indicating XLAS, while two cases manifested mutations in COL4A4 indicating ADAS. Nine different variants were detected, with 3 mutations identified as novel. Two cases underwent histopathological analysis, revealing a thin basement membrane and mild to moderate mesangial proliferation. Three cases were lost to follow-up, while the remaining seven maintained regular visits to our hospital. As of August 1st, 2024, the median follow-up time was 30 (range 24–36) months, and the renal function of the children under observation remained within normal parameters.
Conclusion: In this study, the most commonly observed mutation was glycine substitution. Additionally, patients exhibiting severe mutations showed an increased vulnerability to complications, including proteinuria, ocular lesions, and hearing impairment. Genetic testing emerged as a critical resource for diagnosing AS. Furthermore, early diagnosis is crucial for implementing an appropriate management plan and assessing the prognosis.
Introduction
AS is a genetically and phenotypically heterogeneous disease that affects the glomerular basement membrane (GBM), cochlea, and ocular basement membrane. Mutations in the collagen IV genes COL4A3, COL4A4, and COL4A5 lead to this condition (1). XLAS is estimated to affect about one in every 2,300 of the normal population (2). AS accounts for 0.5% of recently diagnosed end-stage renal disease (ESRD) patients in adults (3) and 12.9% in children (4). It is distinguished by hematuria, gradual renal failure, unique ultrastructural abnormalities in the GBM, hearing loss, and ocular abnormalities (5, 6). There are four genetic forms of inheritance for AS: X-linked AS (XLAS), autosomal recessive AS (ARAS), autosomal dominant AS (ADAS) and digenic AS.
AS is often diagnosed through pathological examination, often using an electron microscope. But, the results of renal biopsy may not be typical for young patients, leading to misdiagnosis. As a result, genetic testing is the most accurate method of determining a diagnosis, as it also illustrates the method of inheritance and, in some cases, the possibility of early-onset kidney failure and extra-renal complications (7). Diagnosis of AS is crucial since effective and inexpensive therapy with renin-angiotensin-aldosterone system (RAAS) inhibition delays renal failure (8, 9).
The early diagnosis of AS poses challenges and often necessitates a kidney biopsy or genetic testing. Despite significant advancements in the research on the genotype and clinical phenotype of AS-related genes, ongoing technological developments continue to unveil novel genotypes. However, there remains a need for enhanced comprehension of the intricate relationship between the genotype and phenotype of AS. This study undertakes a retrospective analysis of the genetic and clinical characteristics of ten pediatric AS patients treated at our hospital in recent years, aiming to elucidate the genotype-phenotype correlations.
Method
Diagnostic criteria for AS
According to the Expert Consensus on Diagnosis and Treatment of Alport Syndrome (Chinese version 2023) (10), ①+ ② or ③ or ④ can be diagnosed as AS if any one of the following conditions are met.
①. The main clinical manifestations include persistent glomerular hematuria or hematuria with proteinuria.
②. The histological examination of renal biopsy shows a diffusely thickened basement membrane or has a varied thickness, the lamina densa is torn and stratified, showing a basket-weave pattern under an electronic microscope.
③. Immunofluorescence examination of type IV collagen α chain shows abnormal immunofluorescence staining at α3, α4, and α5 chains or abnormal staining of α5 chain in the skin during skin biopsy.
④. Positive genetic test showing a mutation in COL4A3-5 gene.
This criteria ensured that all patients diagnosed with AS in this study satisfied the above-mentioned conditions.
Clinical analysis
Individuals <18 years of age with AS were retrospectively analyzed at the General Hospital of Ningxia Medical University from January 2021 to May 2024. The clinical data, encompassing general information such as gender, age of onset, personal history, birth history, and family history, was gathered from the medical records. Additionally, clinical manifestations, including initial symptoms, special signs, extra renal symptoms, and relevant tests such as blood routine, urinalysis, and renal function tests, were compiled.
Mutation analysis
Through Berry Genomics, comprehensive whole-exome sequencing technology (WES) analysis were conducted for genetic evaluation. Venous whole blood samples (2–3 ml) were collected from the child and parents using EDTA anticoagulant tubes as per established protocols. Genomic DNA extraction was performed using a Blood Genome Column Medium Extraction Kit on the provided DNA. Subsequent to the qualitative examination of the samples, library preparation, sequencing, and bioinformatic analysis were executed. The interpretation of genetic mutations adhered to the American College of Medical Genetics and Genomics (ACMG) guidelines (11). In accordance with these guidelines, pathogenicity was categorized into the following classes: pathogenic (P), likely pathogenic (LP), and variants of unknown significance (VUS). The detection of COL4A5 mutations was achieved via the MPLA technique.
Ethical consideration
This study was reviewed and approved by the Institutional Review Board of the General Hospital of Ningxia Medical University. The parents/guardians of the study subjects have given informed consent to this study and signed a written informed consent.
Results
Clinical features
The clinical manifestations of ten patients with genetically confirmed AS are detailed in Table 1, while the family tree of the patients is illustrated in Figure 1. The study encompassed six male and four female patients, with a mean age of 9 years (ranging from 3 to 15 years). The laboratory results and auxiliary findings of the participants are outlined in Table 2. Hematuria was observed in all individuals, with six cases exhibiting microscopic hematuria and four cases exhibiting macroscopic hematuria. Additionally, proteinuria was identified in six cases, with three cases demonstrating trace protein levels in urine. Notably, all cases maintained normal renal function throughout the study. Furthermore, extra-renal manifestations were noted in five cases, encompassing ocular abnormalities (n = 2) and hearing impairment (n = 3). P1, diagnosed with strabismus at age 4 and treated with corrective eyewear, presented with decreased visual acuity but showed no other significant ocular abnormalities upon examination. P2 exhibited a shallow anterior chamber in both eyes on slit-lamp examination, although intraocular pressure (IOP) remained normal at 11 mmHg bilaterally. The cornea was clear, and lens shape and transparency were preserved bilaterally. Fundus examination of both eyes revealed a normal cup-to-disc (C/D) ratio of 0.3 and an artery-to-vein (A/V) ratio of 2:3, suggesting no evidence of glaucomatous damage. Considering the progressive nature of AS, a regular follow-up of 6 months was recommended for both patients. A family history of renal disorders was reported in eight cases, among which P6's mother had been diagnosed with stage 5 chronic kidney disease and had received peritoneal dialysis eight years prior. Renal ultrasound identified anomalies in three cases, indicating enhanced parenchymal echogenicity and unclear delimitation of the cortex and medulla. Only two cases (P7 and P10) underwent renal biopsy, revealing diffuse mesangial proliferation on a light microscope and a thin basement membrane on an electron microscope. P7 also revealed abnormal α5-staining upon immunofluorescence staining.
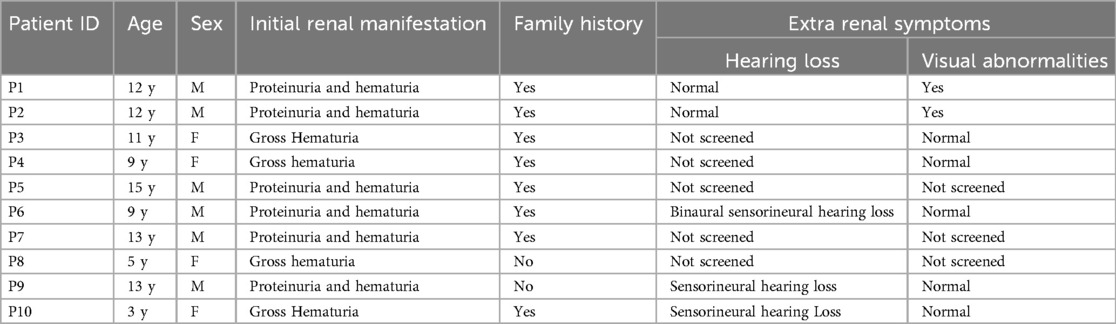
Table 1. Clinical features of the children.
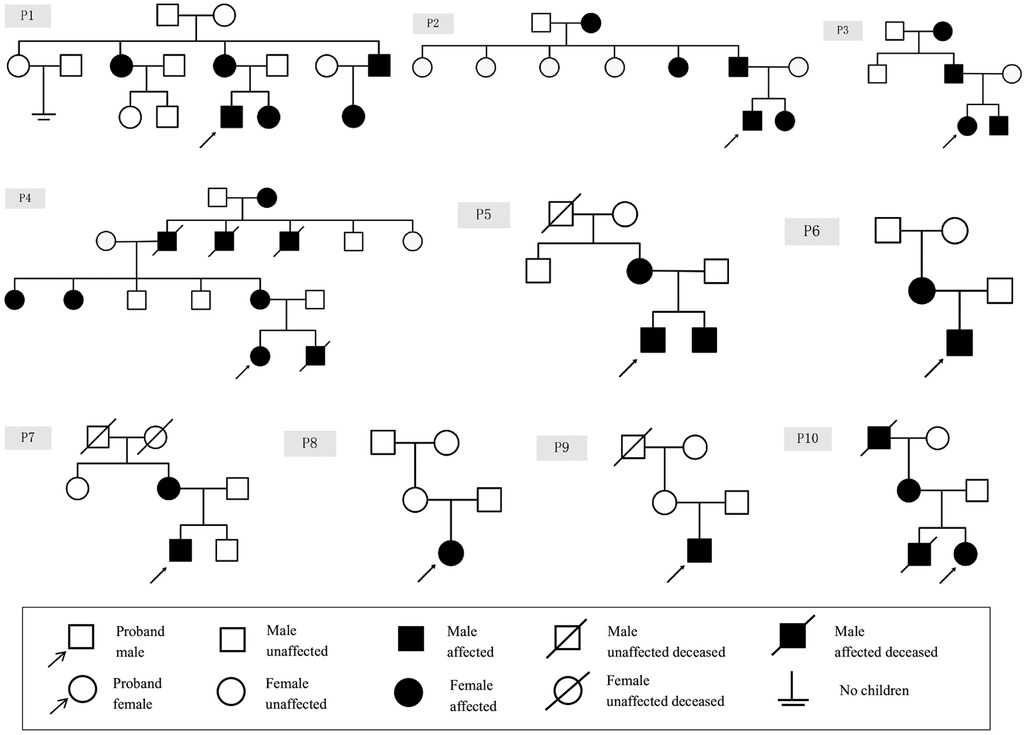
Figure 1. Family tree of children with AS.
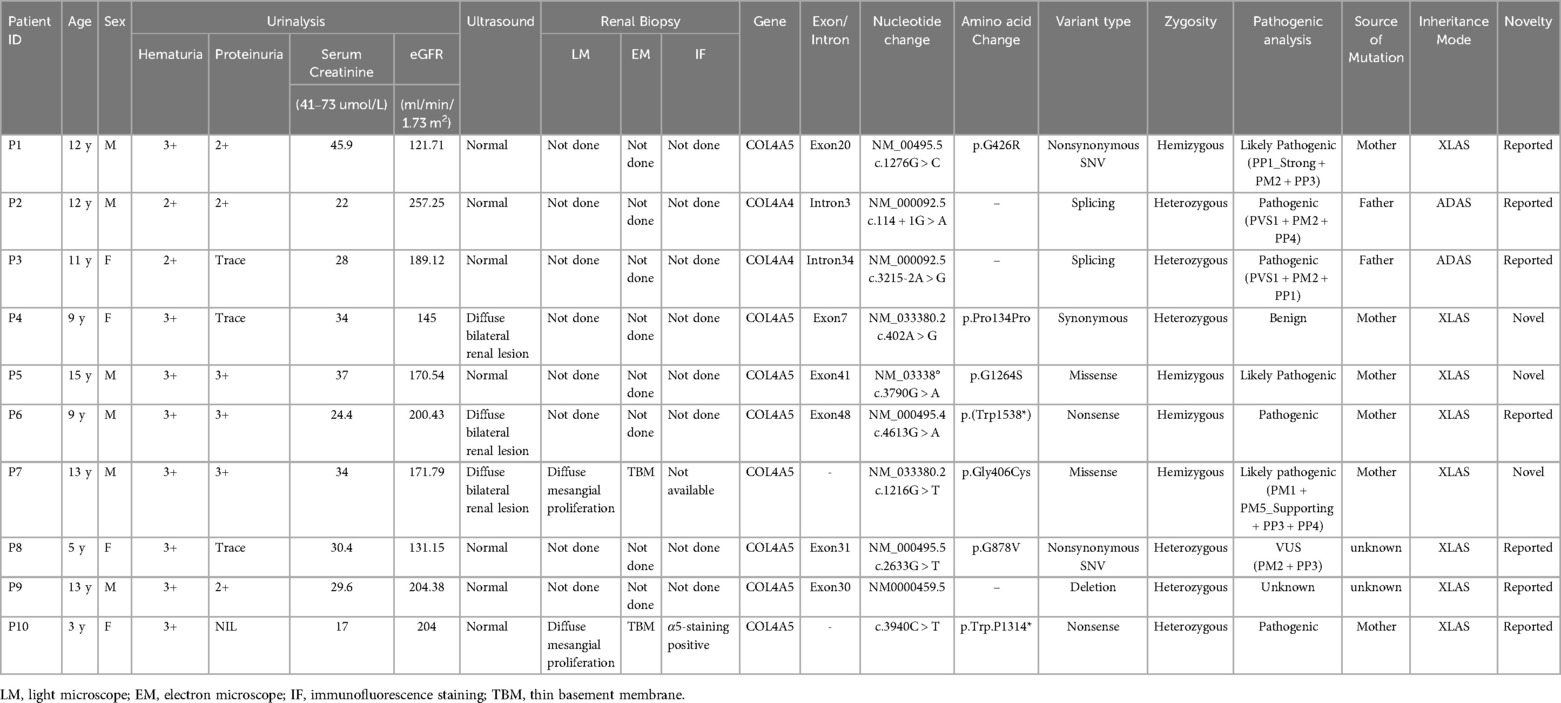
Table 2. Laboratory, pathological and genetic features of the children.
Genetic features
Whole exome sequencing was performed to detect probable disease-causing mutations in each family. After filtering against several databases, including the 1,000 Genome Project, Shenzhou Genome Database, Genome Aggregation Database (gnomAD), and Exome Aggregation Consortium (ExAC), patients' mutations were successfully identified in each family. The detected mutations are shown in Table 2, out of which 5 variants were confirmed by Sanger sequencing as shown in Figure 2. Among the cohort of ten children diagnosed with AS, eight cases displayed mutations in COL4A5 indicating XLAS, while two cases manifested mutations in COL4A4 exhibiting ADAS. In total, 9 different variants were identified, including 2 nonsynonymous variants (SNV), 2 splicing variants, 1 synonymous variant, 2 nonsense variants, and 2 missense variants. In addition to these variants, a heterozygous deletion of the COL4A5 gene located on exon 30 was identified in P9. Notably, seven mutations had been previously documented as the causative mutations for AS in prior studies, with the remaining three mutations recognized as novel findings in this study. Glycine substitution was the most prevalent form of mutation (n = 6).
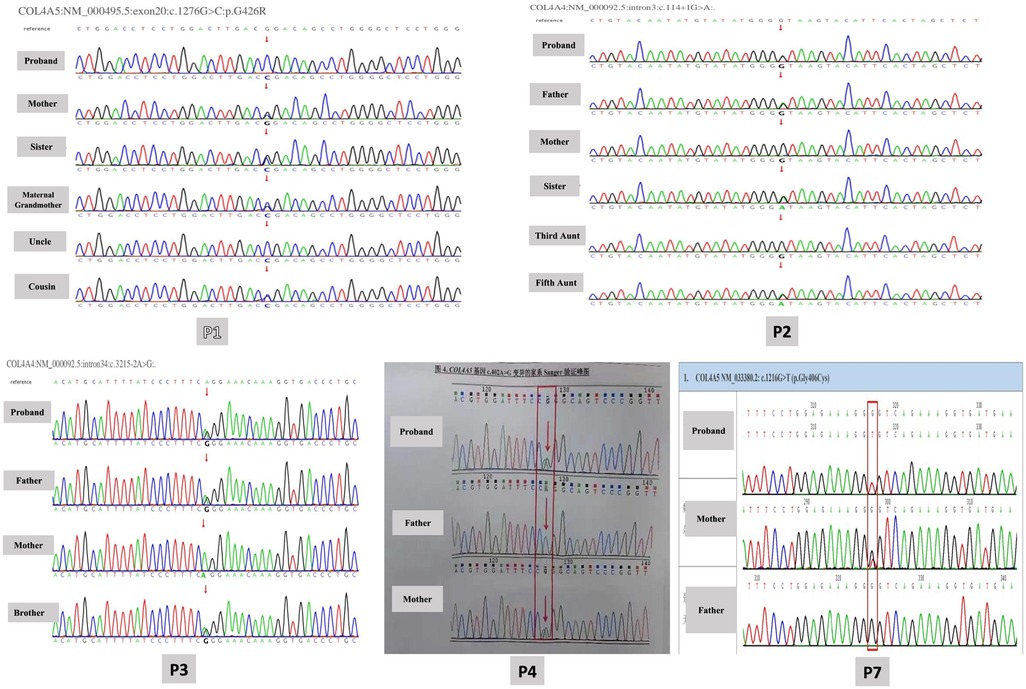
Figure 2. Variants confirmed by Sanger sequencing.
In total, eight cases had X-linked inheritance (six inherited from the mother and two had unknown origin), and two cases had autosomal dominant inheritance (inherited from the father). All the patients had hematuria at diagnosis with stable eGFR. Sensorineural hearing loss was noted in three of the XLAS patients with maternal inheritance, whereas the ADAS patients did not exhibit hearing loss. In the X-linked cohort, severe renal symptoms were observed in several affected relatives on the maternal side, suggesting a higher familial clustering of severe renal involvement. Affected individuals in this cohort, such as P1, exhibited renal manifestations in multiple maternal family members, including his mother, maternal grandmother, aunt, and uncle. Specifically, while P1's maternal uncle developed renal failure and required dialysis, his mother and maternal aunt both presented with microscopic hematuria. Similarly, P4's maternal relatives, including his mother and maternal aunts, also displayed microscopic hematuria. Furthermore, P5's mother had been diagnosed with AS years prior to his own diagnosis. Notably, P6's mother had been diagnosed with stage 5 chronic kidney disease and had undergone peritoneal dialysis eight years ago. Additionally, P7's mother and maternal grandmother also displayed microscopic hematuria. This was notably absent in the autosomal dominant cohort, where family members did not exhibit similar renal manifestations. Specifically, whereas P2's father exhibited no renal symptoms, P3's father presented only with mild microscopic hematuria (2+), highlighting a lack of significant renal involvement in this group.
Follow-up
Subsequent management of AS patients was tailored to their individual clinical presentations, with the majority (70%) receiving renin-angiotensin-aldosterone system (RAAS) inhibitors, while a portion (30%) received traditional Chinese medicine (TCM). The most commonly used TCM for the treatment of hematuria is Xue'an capsule (血尿胺胶囊), which is made from 4 Chinese herbs: white grass root, small thistle, yellow cypress, and orthosiphon aristatus. It has been reported in various Chinese literature that Xue'an capsule combined with western medicine has achieved commendable results in the treatment of glomerular hematuria (12). Out of the ten children studied, three were lost to follow-up, while the remaining seven maintained regular visits to our hospital, as shown in Table 3. As of August 1st, 2024, the median follow-up time was 30 months (range 24–36), and the renal function of the children under observation remained within normal parameters.
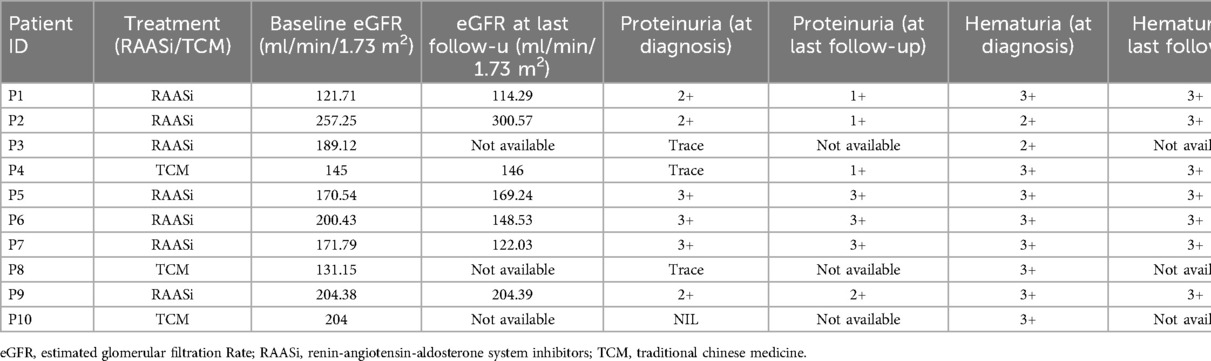
Table 3. Follow-up of children with AS.
Discussion
Arthur C. Alport first identified the disorder in 1927, and it was given the name “Alport syndrome” (AS) in 1961 (13). While AS is considered the most prevalent hereditary kidney disorder. However, there are instances where AS is diagnosed in individuals without any familial history of renal diseases. This underscores the importance of conducting genetic tests in patients exhibiting persistent abnormal urinalysis, regardless of their family history. Notably, here two cases were genetically confirmed to have AS despite the absences of a familial background. The clinical manifestations observed in patients with AS can vary significantly, ranging from isolated hematuria to the onset of renal failure, along with potential extra-renal manifestations, which emphasizes the significance of inter- and intra-familial variability in renal presentations among AS patients (14). Among the families affected by AS, impaired renal function was predominantly noted in patients with mutations in the COL4A5 gene. In contrast, individuals carrying heterozygous variants of COL4A4 exhibited more favorable renal outcomes, a finding that aligns with prior studies (15). Hearing loss associated with AS has not been characterized as congenital, and affected individuals typically pass newborn hearing screenings. It is generally identified through audiometric evaluation during late childhood or early adolescence, often manifesting as a bilateral decrease in sensitivity to mid and high frequencies (16–19). Research indicates that the likelihood of experiencing hearing loss before the age of 30 is approximately 60% for patients with missense mutations and can reach up to 90% for those with severe mutations (nonsense mutation, frameshift mutation, mutations at donor or acceptor site, large deletions) related to XLAS (20).
AS is characterized as a renal disorder with rapid progression, emphasizing the need for early diagnosis. A study revealed detection rates for immunofluorescence staining of type IV collagen, renal biopsy via electron microscopy, and genetic testing for AS at 77.8%, 92.6%, and 96.6%, respectively (21). Consequently, kidney biopsy and genetic testing are essential for the accurate diagnosis of this condition. In accordance with the recommendations related to genetic testing and the management of AS, specialists now recommend genetic testing for individuals with persistent hematuria, particularly when there is a suspicion of a heterozygous pathogenic variant in COL4A3 or COL4A4 (15). Additionally, cascade testing for first-degree relatives is encouraged due to their increased risk of compromised kidney function. In contrast to conventional gene-panel methodologies, whole exome sequencing (WES) offers enhanced capabilities for identifying potential genetic variants associated with phenotypic manifestations (22). WES also plays a pivotal role in uncovering the modifier genes associated with AS. A recent investigation revealed multiple loci linked to the variations in albuminuria and glomerular filtration rate (GFR) within a mouse model, which includes a locus on the X chromosome connected to X inactivation and another locus on chromosome 2 that encompasses the Fmn1 gene (23). Consequently, performing genetic analyses through WES on patients with AS will provide valuable insights into the intricate relationship between genotypes and phenotypes. It also plays a guiding role for the patient's family members in reproduction.
The current study identifies three variants that have not been previously reported, indicating a wide mutation spectrum of AS. Among the variants identified, two intronic mutations (P2 and P3) occurred at splicing sites, where alterations in the GT-AG box are likely to lead to splicing errors. It can be challenging to differentiate between intronic variants that cause splicing errors and those that are harmless polymorphisms. While various in silico methods exist to evaluate the function of intronic variants, functional analyses are essential to validate the in silico findings (24). Therefore, a thorough investigation of intronic variants is warranted, particularly for those located near conserved splicing sites.
In summary, the identification of these pathogenic variants contributes to a better understanding of the correlation between the phenotypic and genotypic characteristics of AS.
Limitations
However, this study had some inherent limitations due to its retrospective design. Firstly, the sample size of our study was small and was conducted at a single center, which could have led to biased findings. Therefore, further studies with a larger sample size are highly desirable to validate these findings. The second important limitation is the absence of a clear case explanation of extra renal manifestation, especially that of ocular abnormalities among AS patients. The third limitation of this study is that targeted assessments for ocular findings and hearing impairment were performed in only a subset of patients, therefore, the observed paucity of ocular and hearing abnormalities may not accurately reflect their true prevalence in the cohort. Lastly, lack of renal biopsy data limits our ability to provide direct histopathological correlation with the observed clinical and genetic findings.
Conclusion
This study presents a cohort of ten Chinese children diagnosed with AS. It is noteworthy that pediatric cases rarely exhibit extra-renal manifestations. The most frequently identified mutation involved glycine substitution. Furthermore, patients with more severe mutations demonstrated a higher susceptibility to complications such as proteinuria, ocular abnormalities, and hearing loss. Genetic testing proved to be an essential tool for the diagnosis of AS. It not only assists in diagnosis but also identifies the location and type of variant, thereby aiding in selecting appropriate treatment plan and predicting prognosis. Early diagnosis and an appropriate management possess significant potential for preventing complications among children.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Institutional Review Board of General Hospital of Ningxia Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
AA: Conceptualization, Data curation, Formal Analysis, Investigation, Writing – original draft, Writing – review & editing. LL: Supervision, Validation, Writing – review & editing. ZY: Visualization, Writing – review & editing. MY: Data curation, Writing – review & editing. ZS: Writing – review & editing. DW: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by Ningxia Hui Autonomous Region Key Research and Development Program under Grant No. 2022BEG03119.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AS, alport syndrome; GBM, glomerular basement membrane; ESRD, end-stage renal disease; XLAS, X-linked Alport syndrome; ARAS, autosomal recessive alport syndrome; ADAS, autosomal dominant alport syndrome; eGFR, estimated glomerular filtration rate; RAAS, renin-angiotensin-aldosterone system; IOP, intraocular pressure; TCM, traditional chinese medicine; WES, whole-exome sequencing.
References
1. Kashtan CE. Alport syndrome: achieving early diagnosis and treatment. Am J Kidney Dis. (2021) 77(2):272–9. doi: 10.1053/j.ajkd.2020.03.026
2. Gibson J, Fieldhouse R, Chan MMY, Sadeghi-Alavijeh O, Burnett L, Izzi V, et al. Prevalence estimates of predicted pathogenic COL4A3-COL4A5 variants in a population sequencing database and their implications for alport syndrome. J Am Soc Nephrol. (2021) 32(9):2273–90. doi: 10.1681/ASN.2020071065
3. Mallett A, Tang W, Clayton PA, Stevenson S, McDonald SP, Hawley CM, et al. End-stage kidney disease due to alport syndrome: outcomes in 296 consecutive Australia and New Zealand dialysis and transplant registry cases. Nephrol Dial Transplant. (2014) 29(12):2277–86. doi: 10.1093/ndt/gfu254
4. Hattori M, Sako M, Kaneko T, Ashida A, Matsunaga A, Igarashi T, et al. End-stage renal disease in Japanese children: a nationwide survey during 2006–2011. Clin Exp Nephrol. (2015) 19(5):933–8. doi: 10.1007/s10157-014-1077-8
5. Grünfeld JP, Joly D. Hereditary kidney diseases in adults. Rev Prat. (1997) 47(14):1566–9. https://www.ncbi.nlm.nih.gov/pubmed/9366116
6. Gubler M, Levy M, Broyer M, Naizot C, Gonzales G, Perrin D, et al. Alport’s syndrome. A report of 58 cases and a review of the literature. Am J Med. (1981) 70(3):493–505. doi: 10.1016/0002-9343(81)90571-4
7. Savige J, Storey H, Il Cheong H, Gyung Kang H, Park E, Hilbert P, et al. X-linked and autosomal recessive alport syndrome: pathogenic variant features and further genotype-phenotype correlations. PLoS One. (2016) 11(9):e0161802. doi: 10.1371/journal.pone.0161802
8. Gross O, Licht C, Anders HJ, Hoppe B, Beck B, Tönshoff B, et al. Study group members of the gesellschaft für pädiatrische nephrologie: early angiotensin-converting enzyme inhibition in alport syndrome delays renal failure and improves life expectancy. Kidney Int. (2012) 81:494–501. doi: 10.1038/ki.2011.407
9. Gross O, Tönshoff B, Weber LT, Pape L, Latta K, Fehrenbach H, et al. A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport’s syndrome. Kidney Int. (2020) 97:1275–86. doi: 10.1016/j.kint.2019.12.015
10. Alport Syndrome Collaborative Group; National Clinical Research Center of Kidney Diseases; Rare Diseases Branch of Beijing Medical Association. Expert consensus on the diagnosis and treatment of Alport syndrome (version 2023). Zhonghua Yi Xue Za Zhi. (2023) 103(20):1507–25. doi: 10.3760/cma.j.cn112137-20230203-00161
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the Association for Molecular Pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
12. Zhang C, Wang J, Ji Q, Wang K, Da H. 36 Cases of damp-heat bet primary glomerular hematuria treated with hematuria capsules[J]. Yunnan J Tradit Chinese Med. (2015) 36(10):22–4. doi: 10.16254/j.cnki.53-1120/r.2015.10.009
13. Alport AC. Hereditary familial congenital haemorrhagic nephritis. Br Med J. (1927) 1(3454):504–6. doi: 10.1136/bmj.1.3454.504
14. Xiao T, Zhang J, Liu L, Zhang B. Genetic diagnosis of alport syndrome in 16 Chinese families. Mol Genet Genomic Med. (2024) 12(3):e2406. doi: 10.1002/mgg3.2406
15. Savige J, Lipska-Zietkiewicz BS, Watson E, Hertz JM, Deltas C, Mari F, et al. Guidelines for genetic testing and management of alport syndrome. Clin J Am Soc Nephrol. (2022) 17(1):143–54. doi: 10.2215/CJN.04230321
16. Gleeson MJ. Alport’s syndrome: audiological manifestations and implications. J Laryngol Otol. (1984) 98(5):449–65. doi: 10.1017/S0022215100146894
17. Harvey SJ, Mount R, Sado Y, Naito I, Ninomiya Y, Harrison R, et al. The inner ear of dogs with X-linked nephritis provides clues to the pathogenesis of hearing loss in X-linked Alport syndrome. Am J Pathol. (2001) 159(3):1097–104. doi: 10.1016/S0002-9440(10)61785-3
18. Alves FRA, de A Quintanilha Ribeiro F. Revision about hearing loss in the alport’s syndrome, analyzing the clinical, genetic and bio-molecular aspects. Braz J Otorhinolaryngol. (2005) 71(6):813–9. doi: 10.1016/S1808-8694(15)31253-2
19. Zhang X, Zhang Y, Zhang Y, Gu H, Chen Z, Ren L, et al. X-linked Alport syndrome: pathogenic variant features and further auditory genotype-phenotype correlations in males. Orphanet J Rare Dis. (2018) 13(1):229. doi: 10.1186/s13023-018-0974-4
20. Jais JP, Knebelmann B, Giatras I, Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. (2000) 11(4):649–57. doi: 10.1681/ASN.V114649
21. An XG, Zhang YQ, Ding J, Wang F, Xiao HJ, Yao Y. Analysis of diagnosis and treatment of Alport syndrome. Zhonghua Er Ke Za Zhi. (2016) 54(9):669–73. doi: 10.3760/cma.j.issn.0578-1310.2016.09.008
22. Bullich G, Domingo-Gallego A, Vargas I, Ruiz P, Lorente-Grandoso L, Furlano M, et al. A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. (2018) 94(2):363–71. doi: 10.1016/j.kint.2018.02.027
23. Takemon Y, Wright V, Davenport B, Gatti DM, Sheehan SM, Letson K, et al. Uncovering modifier genes of X-linked Alport syndrome using a novel multiparent mouse model. J Am Soc Nephrol. (2021) 32(8):1961–73. doi: 10.1681/ASN.2020060777
Keywords: alport syndrome, clinical features, genetic testing, hematuria, children
Citation: Ahmad A, Lijun L, Yan Z, Yan M, Shuai Z and Wangnan D (2024) Clinical profile and molecular genetic analysis of alport syndrome in children: a single center experience. Front. Pediatr. 12:1487927. doi: 10.3389/fped.2024.1487927
Received: 29 August 2024; Accepted: 9 December 2024;
Published: 23 December 2024.
Edited by:
Arvind Bagga, All India Institute of Medical Sciences, IndiaReviewed by:
Judy Savige, The University of Melbourne, AustraliaMenka Yadav, All India Institute of Medical Sciences, India
Copyright: © 2024 Ahmad, Lijun, Yan, Yan, Shuai and Wangnan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang Lijun, bGlhbmdsaWp1bm54QHNpbmEuY29t