 Lejun Tong
Lejun Tong Li Li
Li Li Jiehua Chen
Jiehua Chen- Department of Respiratory Diseases, Shenzhen Children’s Hospital Affiliated to Shantou University Medical College, Shenzhen, China
Primary ciliary dyskinesia (PCD) is a hereditary disorder characterized by defects in cilia that impair mucociliary clearance. This study focuses on PCD caused by mutations in the Cyclin O (CCNO) gene and reports on three cases involving Chinese children. Case 1 was an 8-year-and-3-month-old boy who presented with respiratory distress after birth and later developed a recurrent productive cough and purulent nasal discharge. He was initially diagnosed with diffuse panbronchiolitis (DPB) due to the presence of diffuse micronodules in lung CT scans. Case 2 was the younger sister of case 1. She also presented with respiratory distress after birth, with a chest radiograph revealing atelectasis. She required oxygen supplementation until the age of 2 months. Case 3 was a 4-year-and-4-month-old girl with a history of neonatal pneumonia, persistent pulmonary atelectasis, and recurrent lower respiratory tract infections. Her chest radiograph also showed diffuse micronodules. In all three cases, the final diagnosis of PCD was confirmed by genetic testing. Cases 1 and 2 exhibited homozygous c.248_252dup TGCCC (p.G85Cfs*11) mutations in the CCNO gene, while case 3 harbored a homozygous c.258_262dup GGCCC (p.Q88Rfs*8) mutation. A literature review indicated that the common clinical features of CCNO-PCD include neonatal respiratory distress (40/49, 81.6%), chronic cough (31/33, 93.9%), rhinosinusitis (30/35, 85.7%), bronchiectasis (26/35, 74.3%), and low nasal nitric oxide (nNO, 40/43, 93.0%). Notably, situs inversus has not been reported. In CCNO-PCD patients, cilia may appear structurally normal but were severely reduced in number or entirely absent. Lung CT scans in these patients may exhibit diffuse micronodules and “tree-in-bud” signs, which can lead to a clinical misdiagnosis of DPB. nNO screening combined with genetic testing is an optimized diagnostic strategy. Treatment options include the use of anti-infective and anti-inflammatory agent, along with daily airway clearance. This study underscores the importance of genetic testing in neonates and children with suspected PCD or those clinically diagnosed with DPB to enable an early diagnosis and prompt intervention, thereby enhancing the prognosis for these patients.
1 Introduction
Primary ciliary dyskinesia (PCD) is a genetically and clinically heterogeneous disorder, commonly manifesting as recurrent productive cough, respiratory infections, chronic sinusitis, and otitis media. Approximately half of the patients present with situs inversus totalis (1), which could raise suspicion of PCD if combined with chronic respiratory symptoms and bronchiectasis. To date, over 50 genes associated with PCD have been identified (2), with most mutations resulting in abnormalities in specific ultrastructural components of cilia, thereby impairing ciliary function.
The Cyclin O (CCNO) gene was first reported by Wallmeier et al. (3) in 2014. In this genotype of PCD, situs inversus has not been reported and cilia are significantly reduced or absent, while the ultrastructure of the axoneme may appear normal (3–6). This can lead to missed or delayed diagnosis when relying solely on conventional transmission electron microscopy (TEM) or high-speed videomicroscopy analysis (HSVA). Lung CT scans in patients with CCNO-PCD may exhibit diffuse micronodules and “tree-in-bud” signs, which can be misdiagnosed as diffuse panbronchiolitis (DPB). Despite ongoing reports of new cases, there remain a limited number of documented cases globally.
In this article, we report three cases of PCD caused by CCNO gene mutations and review the literature to elucidate the clinical features and diagnostic challenges and promote early recognition of this PCD genotype.
2 Case presentation
2.1 Case 1
An 8-year-and-3-month-old boy was admitted to the respiratory department of our hospital with a 6-day history of cough and fever. Born at term, the boy developed tachypnea 12 h after birth, which was diagnosed as neonatal pneumonia. He had no history of mechanical ventilation or prolonged oxygen supplementation and exhibited situs solitus. Since he was 1 year old, he had experienced a recurrent productive cough and nasal discharge, with a temporary improvement following antibiotic treatment. He showed no hydrocephalus or hearing problems. His cognitive, motor, and language development were normal. At 7 years of age, he was admitted to our hospital for an acute respiratory exacerbation. A chest CT scan revealed diffuse small nodules in the lower fields of both lungs and bronchiectasis. Bronchoscopy indicated suppurative bronchitis. A bacterial culture of bronchoalveolar lavage fluid (BALF) identified Haemophilus influenzae. Immunological tests, including T and B lymphocyte counts, serum immunoglobulin levels, and the respiratory burst test, were unremarkable. His levels of specific immunoglobulin E (IgE) for food and inhalant allergens, as well as total IgE levels, were within normal range. TEM revealed an absence of cilia in multiple sections (Figure 1A, KingMed Diagnostics, Guangzhou, China). His nasal nitric oxide (nNO) levels were consistently measured at 4 parts per billion (ppb) across multiple assessments. His fractional exhaled nitric oxide (FeNO) levels were recorded at 2 and 4 ppb in repeated measurements (using the SUNVOU nitric oxide device, Wuxi, China, with a flow rate of 10 ml/s for nNO and 50 ml/s for FeNO).
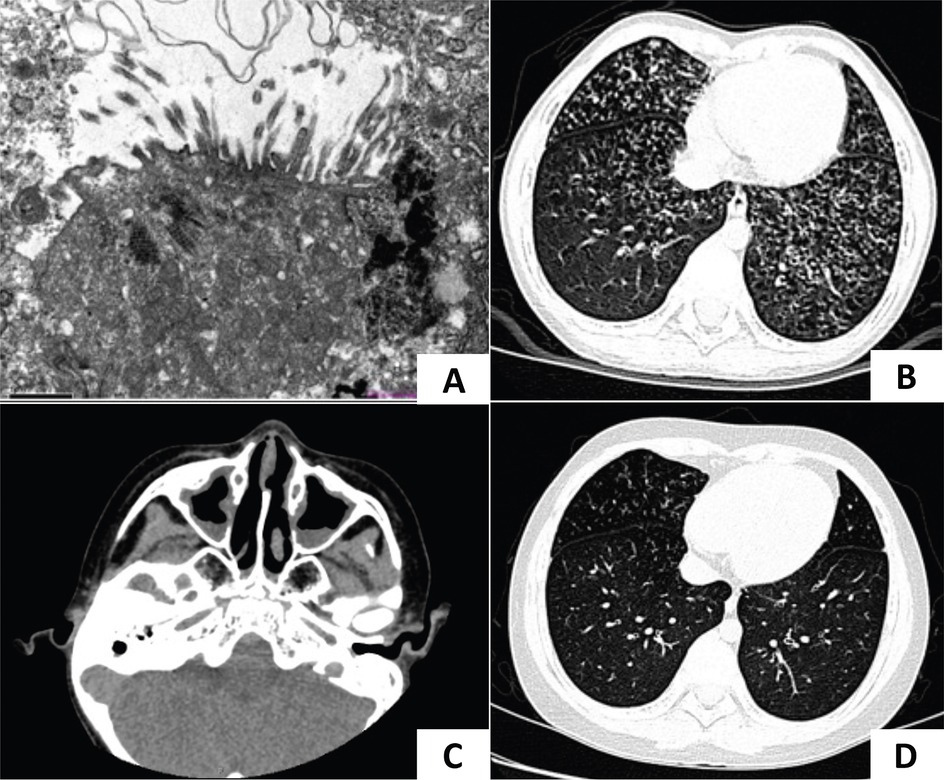
Figure 1. CT imaging and TEM of case 1. (A) TEM of case 1 showed microvilli on the epithelial surface, inflammatory cell infiltration, and absent cilia in multiple sections. (B) A lung CT scan identified bilateral diffuse micronodules and “tree-in-bud” signs. (C) A sinus CT scan showed mucosal thickening of bilateral nasal cavities and maxillary sinuses. (D) Lesions in the lower lungs improved after treatment.
On physical examination, the patient exhibited no dyspnea or cyanosis. Auscultation revealed coarse to moderate crackles in both lungs, with diminished breath sounds in the left lung.
Auxiliary examination: A lung CT scan demonstrated diffuse centrilobular micronodules in the left lingula segment, left lower lobe, and right middle and lower lobes. Bronchial wall thickening and lumen dilation were also observed (Figure 1B). A sinus CT scan revealed a thickening of the mucosa in the nasal cavity and the maxillary sinus (Figure 1C). A pulmonary function test indicated mild obstructive ventilatory impairment, with forced vital capacity (FVC) at 90.46% of the predicted value, forced expiratory capacity in 1 s (FEV1) at 81.27% of the predicted value, and an FEV1/FVC ratio at 75.51% of the predicted value. Bronchoscopy revealed inflammation of the bronchial mucosa. Bacterial culture, Mycoplasma pneumoniae DNA, and Mycobacterium tuberculosis DNA testing of BALF were negative. nNO was measured at 12 ppb (7.2 nl/min).
Diagnosis and treatment: Given the patient's history of neonatal respiratory distress, chronic respiratory symptoms, bronchiectasis, and markedly decreased nNO levels, PCD was considered. However, the previous TEM results showed absent cilia and the lung CT scan revealed diffuse centrilobular micronodules rather than the typical consolidation, atelectasis, or significant bronchiectasis commonly associated with PCD, leading to an inconclusive diagnosis. According to the Japanese diagnostic criteria for DPB (7), which include
1. Persistent cough, sputum, and exertional dyspnea;
2. History of chronic paranasal sinusitis;
3. Bilateral diffuse small nodular shadows on a plain chest radiography film or centrilobular micronodules on chest computed tomography images;
4. Coarse crackles;
5. FEV1/FVC <70% and PaO2 <80 mmHg; and
6. Titer of cold hemagglutinin ≥64.
The first four criteria were met in case 1, raising suspicion of DPB. The patient was treated with intravenous amoxicillin–sulbactam, low-dose oral erythromycin, a nasal glucocorticoid, and expectorants.
Follow-up: A review of the chest CT scan 1 year after discharge showed persistent diffuse centrilobular micronodules. Whole exome sequencing (WES, MyGenostics, Beijing, China) identified a homozygous c.248_252dup TGCCC (p.G85Cfs*11) mutation in the CCNO gene, confirming a definitive diagnosis of PCD. Following regular daily treatment with hypertonic saline nebulization (3%), airway clearance, and low-dose oral erythromycin, his productive cough and lung CT improved (Figure 1D). A pulmonary function test conducted at 13 years of age revealed normal ventilatory function, but diffuse micronodules persisted in the lower fields of both lungs.
2.2 Case 2
A female infant was admitted to the neonatal department of our hospital 12 h after birth. She was the younger sister of case 1, born at term, and exhibited grunting and tachypnea 8 h postnatally. On physical examination, crackles were auscultated in both lungs. A chest x-ray revealed atelectasis of the right upper lobe (Figure 2A). The patient exhibited situs solitus. A nasopharyngeal swab for Group B streptococcus DNA was negative. Multiplex polymerase chain reaction (PCR) tests for a panel of respiratory pathogens, including metapneumovirus, adenovirus, rhinovirus, parainfluenza virus, influenza B virus, chlamydia, M. pneumoniae, influenza A virus, bocavirus, coronavirus, and respiratory syncytial virus, were all negative. Toxoplasmosis, rubella, cytomegalovirus and herpes simplex virus (TORCH) screenings were unremarkable. Echocardiography revealed a patent foramen ovale. Bronchoscopy showed inflammation of the bronchial mucosa and laryngomalacia. The bacteria culture of BALF was negative. High-throughput sequencing of BALF identified Rothia mucilaginosa (11,684 sequences) and Streptococcus mitis (9,464 sequences), which were considered colonization. Sanger sequencing confirmed the same mutation in this patient as her brother, leading to a diagnosis of PCD. She was treated with heated, humidified high-flow nasal cannula oxygen therapy and intravenous penicillin combined with ceftazidime.
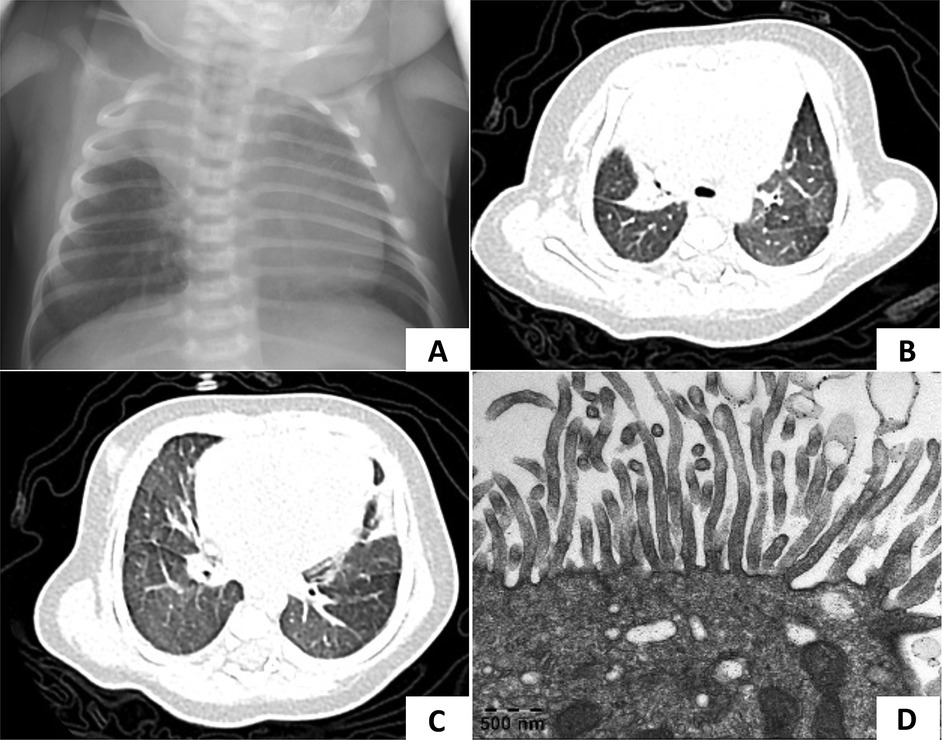
Figure 2. Chest radiography and TEM of case 2. (A) Chest x-ray showed atelectasis of the right upper lobe at 1 week after birth. (B,C) At 1 month of age, a lung CT scan showed a decreased volume of the right upper lobe and patchy opacities in both lungs which demonstrated pneumonia with right upper lobe atelectasis. (D) TEM showed microvilli with few cilia.
Follow-up: After discharge, the patient continued to require family nasal cannula oxygen supplementation at home. She experienced two episodes of acute respiratory exacerbations at 1 and 5 months of age, respectively. Lung CT scans revealed pneumonia with persistent right upper lobe atelectasis (Figures 2B,C). TEM showed normal microvilli with a few cilia (Figure 2D). With regular daily hypertonic saline nebulization and sputum suction, oxygen supplementation was discontinued by 2 months of age.
2.3 Case 3
A 4-year-and-4-month-old girl was admitted to our hospital with a 2-week history of cough. She had a history of pneumonia and atelectasis during the neonatal period. She required non-invasive ventilation in the neonatal period and continued to require oxygen supplementation until 6 months of age. The patient exhibited situs solitus and had a history of recurrent lower respiratory tract infections (LRTIs) and nasal discharge. A sputum culture identified H. influenzae. Bronchoscopy revealed suppurative bronchitis. Multiple chest x-rays showed persistent atelectasis of the right middle lobe (Figures 3A,B). On physical examination, crackles were auscultated in both lungs. Given the history of mechanical ventilation, oxygen supplementation, recurrent LRTIs, and persistent atelectasis, PCD was suspected. WES revealed a homozygous c.258_262dup GGCCC (p.Q88Rfs*8) mutation in the CCNO gene, confirming the diagnosis of PCD. During follow-up, lung CT scans showed diffuse micronodules in the lower fields of the lungs bilaterally, with persistent atelectasis in the right middle lobe (Figures 3C,D).
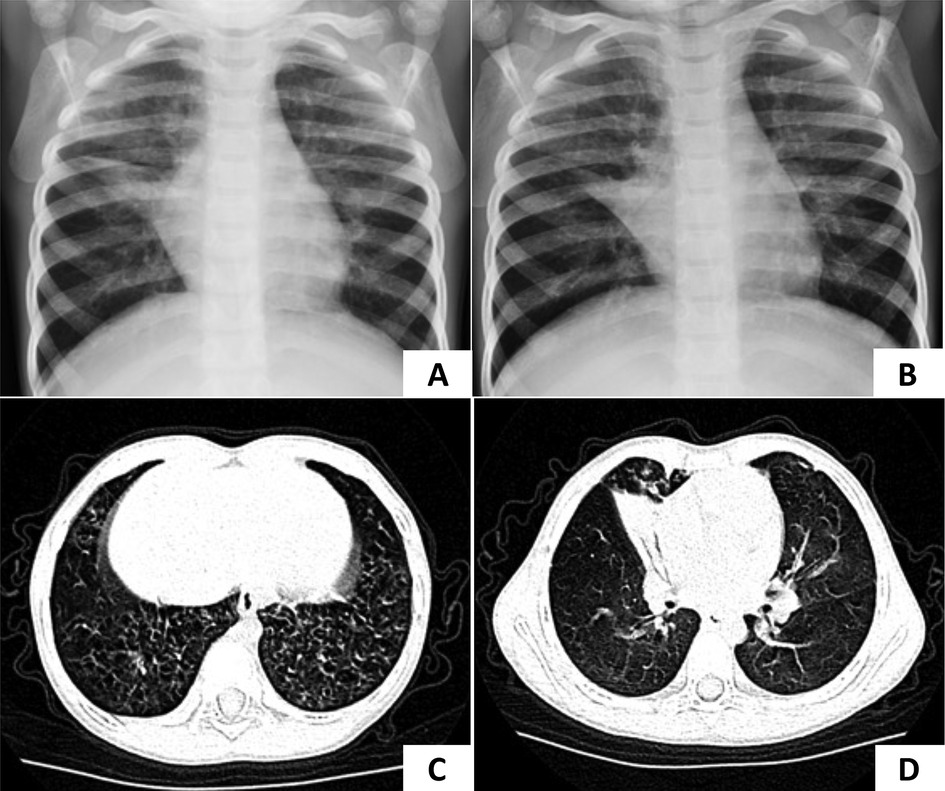
Figure 3. Chest radiography of case 3. (A,B) Atelectasis of the right middle lobe was persistent in two separate x-rays. (C,D) The lung CT scan showed diffuse micronodules in the lower fields of both lungs and decreased volume of the right middle lobe which were considered pneumonia with atelectasis of the right middle lobe.
3 Literature review
A literature search was conducted in PubMed using the keywords “primary ciliary dyskinesia”, “immotile-cilia syndrome” and “CCNO gene” to find studies published before April 2024. A total of 15 articles were reviewed, including 1 in Chinese and 14 in English (3, 4, 8–20). In total, including our 3 cases, 58 patients (26 males and 32 females) were included. The demographic and clinical features of these patients are summarized in Table 1, while studies on CCNO-PCD are summarized in Supplementary Table 2.
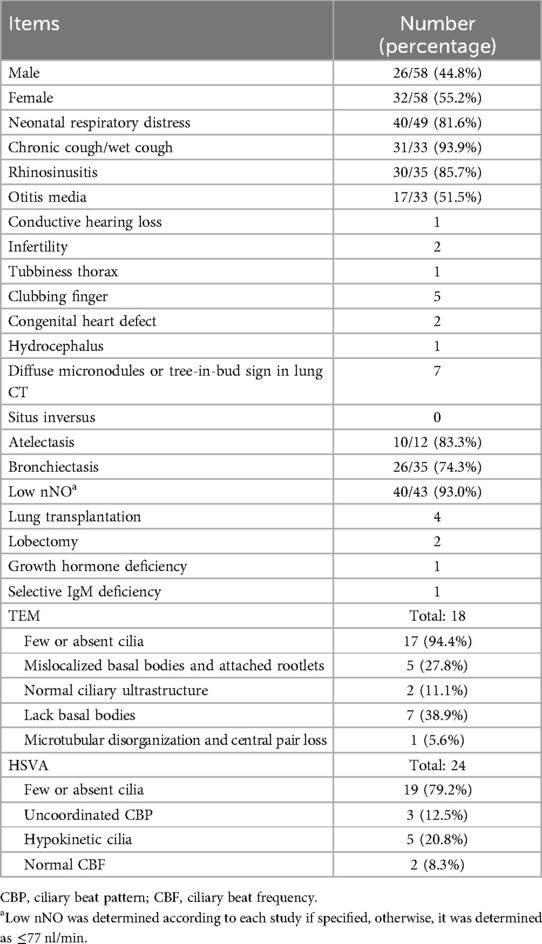
Table 1. Demographic and clinical features of CCNO-PCD.
4 Discussion
PCD is a highly heterogenous disorder resulting from ciliary dysfunction. To date, over 50 pathogenic genes have been identified (2). Most PCD genotypes lead to defects of specific ciliary ultrastructural components, such as the outer and inner dynamin arms, nexin links, and radial spokes (21), resulting in ciliary immotility or dyskinesia. PCD caused by mutations in the CCNO gene presents with reduced or absent cilia, a phenotype known as ciliary aplasia. However, the ciliary ultrastructure can be preserved (3–6), although a recent report has described microtubular disorganization and central pair loss (20). Residual cilia may beat in a normal pattern but are unable to beat coordinately due to their sparse distribution, ultimately impairing ciliary clearance. Consequently, this phenotype is also referred to as congenital mucociliary clearance disorder with reduced generation of multiple motile cilia (RGMC) (3).
Absent or decreased cilia might initially be mistaken for secondary loss due to brushing or infection. However, a previous study observed a severe reduction in motile cilia and basal bodies in the apical cell region of individuals with CCNO mutations, as well as defects in the migration of cytoplasmic basal bodies (3). An in vitro ciliogenesis study confirmed a ciliary generation defect in CCNO-PCD patients, and further research using Xenopus embryos demonstrated that CCNO is critical for the amplification and migration of mother centrioles (3). Therefore, the generation defect appears to be the primary cause of the ciliary aplasia phenotype, rather than secondary loss. In addition, DNAH5 and CCDC39, which are essential for ciliary beating, were found to be expressed in the residual cilia (3). These findings help explain the reduced number of cilia with preserved motility in the remaining cilia. Notably, no laterality defects have been reported in CCNO-PCD. This phenomenon has been investigated in mouse models, where it was shown that the ability to form multiple motile cilia is not entirely lost in CCNO-deficient embryos (22). Even as few as two rotating cilia are sufficient to generate nodal flow and establish left–right asymmetry (23), preventing the appearance of laterality defects due to the preservation of residual cilia.
Patients with CCNO-PCD commonly present with neonatal respiratory distress (40/49, 81.6%), chronic cough (31/33, 93.9%), rhinosinusitis (30/35, 85.7%), and otitis media (17/33, 51.5%). However, the clinical presentation and disease progression can be highly variable. For instance, one child was initially misdiagnosed with bronchiolitis obliterans due to recurrent cough and wheezing with exercise intolerance (16), while another was diagnosed with asthma due to recurrent wet cough without a history of sinusitis or otitis, though he did not respond to asthma medications (15). Two children were considered to have coexistent asthma (13). Alhalabi et al. (19) reported two siblings in whom bronchiectasis was detected at 14 years of age in the elder sister and 7 years of age in the younger sister. Similarly, in our case 1, bronchiectasis was identified at 7 years of age. However, the reported younger sibling exhibited diffuse cystic bronchiectasis at this age, whereas our case 1 demonstrated only mild bronchial wall thickening and lumen dilation. Moreover, our case 1 maintained normal pulmonary ventilatory function at 13 years of age, while the reported elder sibling progressed to end-stage lung disease requiring lung transplantation at 17 years old, highlighting the heterogeneity in the disease progression of CCNO-PCD. Previous studies have demonstrated that lung diseases in CCNO-PCD tend to manifest earlier and progress more severely compared to other PCD genotypes. Emiralioğlu et al. (12) demonstrated that patients with CCNO variants showed an early onset of symptoms, with the lowest median age of diagnosis at 3 years old. Raidt et al. (24) found that individuals with CCNO variants had the poorest median FEV1 z-score among a cohort of 1,072 genotyped PCD individuals with available lung function data. Extrapulmonary manifestations in CCNO-PCD patients include infertility, arrested hydrocephalus, hearing impairment, and congenital heart defects (3, 4, 9, 12). In addition, one child with a growth hormone deficiency and one with a selective IgM deficiency have been reported (15, 17).
In patients with CCNO-PCD, lung radiographs frequently reveal atelectasis (10/12, 83.3%) and bronchiectasis (26/35, 74.3%). In cases 2 and 3, atelectasis was observed in the same lung field in repeated radiographic tests, suggesting that PCD should be suspected in children with persistent atelectasis. Interestingly, five previously reported cases of CCNO-PCD demonstrated diffuse micronodules and “tree-in-bud” signs in lung CT scans (9, 11, 14, 16, 19), similar to those seen in cases 1 and 3, which mimic DPB. Combined with the chronic respiratory manifestations overlapping with DPB, it is difficult to determine whether these imaging features are pathognomic for DPB, represent a common pattern in CCNO-PCD, or suggest that PCD may be an underlying etiology of DPB, as a previous study found that nNO is also low in patients with DPB (7). Chen et al. (25) reported a PCD case without situs inversus yet complicated with DPB (genetic testing was not available) and reviewed the literature, identifying only 17 reported cases of Kartagener's syndrome complicated with DPB between 1999 and 2014, all of which exhibited situs inversus. Although diffuse bronchiolitis-like changes have been reported in two PCD patients with genetically confirmed CCDC39 and CCDC40 mutations, both of these cases also exhibited situs inversus (26, 27). The relatively frequent occurrence of a diffuse bronchiolitis-like change in CCNO-PCD, as seen in our cases and the reviewed literature, suggests that clinically diagnosed DPB with situs solitus and equivocal ciliary ultrastructural findings should be differentiated from CCNO-PCD, even if the clinical criteria for DPB are met. As genetic diagnosis improves, the genetic bases of various diseases are increasingly illuminated. Whether DPB is an isolated idiopathic inflammatory disease or a clinical syndrome that at least results from CCNO-PCD needs to be further studied. In addition, H. influenzae is one of the most common bacteria in airways affected by bronchiectasis (28). In two of our cases that presented with diffuse micronodules in lung CT scans, cultures of respiratory specimens were positive for H. influenzae. Okada et al. (29) found that centrilobular nodules appeared in lung CT scans in 64.9% of patients with acute H. influenzae pulmonary infection, suggesting that infectious factors may also contribute to the diffuse bronchiolitis-like change observed in these patients.
The diagnosis of PCD relies on a combination of typical history and diagnostic tests. Both the European Respiratory Society (ERS) and the American Thoracic Society (ATS) recommend using nNO as the primary screening tool for cooperative patients with a typical history as nNO levels are consistently low in PCD patients (≤77 nl/min) (30). In our review, nNO was significantly reduced in 93.0% (40/43) of CCNO-PCD patients, with only three cases presenting normal levels, reinforcing its value as an initial screening test for CCNO-PCD. In the subsequent diagnostic program, the ERS guidelines recommend using HSVA to observe ciliary beating frequency or TEM to assess ciliary ultrastructure (21). However, HSVA and TEM may fail to visualize the cilia due to reduced or absent cilia, as observed in 94.4% (17/18) of TEM and 79.2% (19/24) of HSVA assessments. Even if the cilia are observed, the absence of hallmark ciliary ultrastructural defects may lead to an ambiguous or delayed diagnosis. Recent research has demonstrated that the sensitivity of TEM varies depending on the regional prevalence of distinct PCD genotypes. In Turkey, where CCNO and DNAH11 variants are prevalent, TEM failed to diagnose more than half of the PCD cases (24). In China, a previous cohort study has identified CCNO variants as the fifth most prevalent genotype, a finding consistent with a multinational cohort study that included patients from Europe, Asia, and South America (13, 24). Given the relatively high prevalence of CCNO variants, skipping conventional modalities such as TEM and HSVA, nNO screening combined with genetic testing may enhance the detection of PCD and lead to more accurate diagnoses.
The treatment of PCD primarily focuses on managing acute exacerbations caused by infections and ensuring long-term airway clearance, which can compensate for the ciliary clearance function. Effective antibiotic therapy can delay the progression of bronchiectasis. Prophylactic macrolides may be used in patients aged 7 years and older who experience recurrent acute exacerbations. A multinational randomized trial including 90 PCD patients found that maintenance therapy with azithromycin was well tolerated and halved the rate of acute exacerbations (31). Case 1 showed clinical improvement and a significant reduction in small airway lesions in lung CT scans following regular oral low-dose erythromycin and airway clearance with hypertonic saline nebulization. Similarly, Zhang et al. (14) reported a case of CCNO-PCD in which the patient experienced a reduction in diffuse lung nodules after 2 months of oral azithromycin, highlighting the positive effects of macrolides and airway clearance. Surgical interventions are generally reserved for complications. Lobectomy may be considered in cases with significant bronchiectasis. Lung transplantation remains an option for patients with end-stage lung disease to extend survival (32). In the reviewed literature, one child underwent left lower lobectomy at 4 years of age due to persistent atelectasis and recurrent respiratory symptoms. Postoperatively, lower respiratory tract infections decreased, but the patient continued to experience a chronic wet cough and obstructive ventilatory impairment (4). Four patients underwent lung transplantation due to end-stage lung disease at the ages of 34, 34, and 43 years, and one before adulthood (3, 8, 19).
Our study highlights the importance of suspecting CCNO-PCD in term newborns with unexplained later-onset respiratory distress and in children with chronic respiratory symptoms, persistent atelectasis, or bronchiectasis in radiography. No laterality defects have been reported in CCNO-PCD. The clinical manifestations and disease progression are highly heterogeneous, with lung disease appearing earlier and more severely compared to other PCD genotypes. Diffuse micronodules and “tree-in-bud” signs in lung CT scans may lead to a clinical misdiagnosis of DPB. CCNO-PCD may be overlooked if the diagnosis is only based on an airway mucosal biopsy, due to reduced or absent cilia without hallmark ciliary ultrastructural defects. A combination of nNO screening and genetic testing offers an optimized diagnostic approach. In addition, parents of children with PCD should be offered prenatal screening and genetic counseling if planning to have more children.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
LT: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation. LL: Resources, Data curation, Writing – review & editing. WW: Funding acquisition, Resources, Validation, Writing – review & editing. JC: Resources, Supervision, Validation, Writing – review & editing, Methodology.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Shenzhen Key Medical Discipline Construction Fund (SZXK032) and the Guangdong Provincial High-level Hospital Construction Special Fund (YNKT2021-ZZ10).
Acknowledgments
We thank the institutions who performed the TEM and WES.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2024.1458660/full#supplementary-material
References
1. Kuehni CE, Frischer T, Strippoli MP, Maurer E, Bush A, Nielsen KG, et al. Factors influencing age at diagnosis of primary ciliary dyskinesia in European children. Eur Respir J. (2010) 36(6):1248–58. doi: 10.1183/09031936.00001010
2. Pioch CO, Connell DW, Shoemark A. Primary ciliary dyskinesia and bronchiectasis: new data and future challenges. Arch Bronconeumol. (2023) 59(3):134–6. doi: 10.1016/j.arbres.2022.12.001
3. Wallmeier J, Al-Mutairi DA, Chen CT, Loges NT, Pennekamp P, Menchen T, et al. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Genet. (2014) 46(6):646–51. doi: 10.1038/ng.2961
4. Henriques AR, Constant C, Descalço A, Pinto A, Moura Nunes J, Sampaio P, et al. Primary ciliary dyskinesia due to CCNO mutations—a genotype-phenotype correlation contribution. Pediatr Pulmonol. (2021) 56(8):2776–9. doi: 10.1002/ppul.25440
5. Shoemark A, Boon M, Brochhausen C, Bukowy-Bieryllo Z, De Santi MM, Goggin P, et al. International consensus guideline for reporting transmission electron microscopy results in the diagnosis of primary ciliary dyskinesia (Beat Pcd Tem Criteria). Eur Respir J. (2020) 55(4):1900725. doi: 10.1183/13993003.00725-2019
6. Shapiro AJ, Leigh MW. Value of transmission electron microscopy for primary ciliary dyskinesia diagnosis in the era of molecular medicine: genetic defects with normal and non-diagnostic ciliary ultrastructure. Ultrastruct Pathol. (2017) 41(6):373–85. doi: 10.1080/01913123.2017.1362088
7. Poletti V, Casoni G, Chilosi M, Zompatori M. Diffuse panbronchiolitis. Eur Respir J. (2006) 28(4):862–71. doi: 10.1183/09031936.06.00131805
8. Casey JP, McGettigan PA, Healy F, Hogg C, Reynolds A, Kennedy BN, et al. Unexpected genetic heterogeneity for primary ciliary dyskinesia in the Irish traveller population. Eur J Hum Genet. (2015) 23(2):210–7. doi: 10.1038/ejhg.2014.79
9. Amirav I, Wallmeier J, Loges NT, Menchen T, Pennekamp P, Mussaffi H, et al. Systematic analysis of CCNO variants in a defined population: implications for clinical phenotype and differential diagnosis. Hum Mutat. (2016) 37(4):396–405. doi: 10.1002/humu.22957
10. Guo T, Tan ZP, Chen HM, Zheng DY, Liu L, Huang XG, et al. An effective combination of whole-exome sequencing and runs of homozygosity for the diagnosis of primary ciliary dyskinesia in consanguineous families. Sci Rep. (2017) 7(1):7905. doi: 10.1038/s41598-017-08510-z
11. Shen N, Meng C, Liu Y, Gai Z. Genetic diagnosis of a case with primary ciliary dyskinesia type 29 by next generation sequencing. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2019) 36(3):225–8. doi: 10.3760/cma.j.issn.1003-9406.2019.03.008
12. Emiralioğlu N, Taşkıran EZ, Koşukcu C, Bilgiç E, Atilla P, Kaya B, et al. Genotype and phenotype evaluation of patients with primary ciliary dyskinesia: first results from Turkey. Pediatr Pulmonol. (2020) 55(2):383–93. doi: 10.1002/ppul.24583
13. Guan Y, Yang H, Yao X, Xu H, Liu H, Tang X, et al. Clinical and genetic spectrum of children with primary ciliary dyskinesia in China. Chest. (2021) 159(5):1768–81. doi: 10.1016/j.chest.2021.02.006
14. Zhang YY, Lou Y, Yan H, Tang H. CCNO mutation as a cause of primary ciliary dyskinesia: a case report. World J Clin Cases. (2022) 10(25):9148–55. doi: 10.12998/wjcc.v10.i25.9148
15. Celiksoy MH, Turan I, Gezdirici A, Kayalar O, Aydoğmuş Ç, Naiboglu S. A novel homozygous frameshift CCNO variant presenting with primary ciliary dyskinesia and selective IgM deficiency. Pediatr Pulmonol. (2023) 58(11):3333–6. doi: 10.1002/ppul.26607
16. Wang L-n, Gao L-w, Liu X-y, Xu B-p. Mutations in CCNO result in primary ciliary dyskinesia complicated with diffuse bronchiolitis. Indian J Pediatr. 90(5):522. doi: 10.1007/s12098-023-04543-7
17. Gong D, Tang Q, Yan LJ, Ye XM, Yang YC, Zou L, et al. Case report: a novel homozygous mutation of cyclin O gene mutation in primary ciliary dyskinesia with short stature. Pharmgenomics Pers Med. (2023) 16:443–8. doi: 10.2147/pgpm.S406445
18. Petrarca L, De Luca A, Nenna R, Hadchouel A, Mazza T, Conti MG, et al. Early genetic analysis by next-generation sequencing improves diagnosis of primary ciliary dyskinesia. Pediatr Pulmonol. (2023) 58(10):2950–3. doi: 10.1002/ppul.26604
19. Alhalabi O, Abdulwahab A, Thomas M. The first case of a homozygous CCNO NM 021147.4 mutation associated with primary ciliary dyskinesia in two Indian siblings. Cureus. (2024) 16(1):e52237. doi: 10.7759/cureus.52237
20. Xu Y, Ueda K, Nishikido T, Matsumoto T, Takeuchi K. Two Japanese pediatric patients with primary ciliary dyskinesia caused by loss-of-function variants in the CCNO gene. Cureus. (2024) 16(4):e58854. doi: 10.7759/cureus.58854
21. Lucas JS, Barbato A, Collins SA, Goutaki M, Behan L, Caudri D, et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J. (2017) 49(1):1601090. doi: 10.1183/13993003.01090-2016
22. Funk MC, Bera AN, Menchen T, Kuales G, Thriene K, Lienkamp SS, et al. Cyclin O (CCNO) functions during deuterosome-mediated centriole amplification of multiciliated cells. EMBO J. (2015) 34(8):1078–89. doi: 10.15252/embj.201490805
23. Shinohara K, Kawasumi A, Takamatsu A, Yoshiba S, Botilde Y, Motoyama N, et al. Two rotating cilia in the node cavity are sufficient to break left-right symmetry in the mouse embryo. Nat Commun. (2012) 3:622. doi: 10.1038/ncomms1624
24. Raidt J, Riepenhausen S, Pennekamp P, Olbrich H, Amirav I, Athanazio RA, et al. Analyses of 1236 genotyped primary ciliary dyskinesia individuals identify regional clusters of distinct DNA variants and significant genotype-phenotype correlations. Eur Respir J. (2024) 64(2):2301769. doi: 10.1183/13993003.01769-2023
25. Chen W, Shao C, Song Y, Bai C. Primary ciliary dyskinesia complicated with diffuse panbronchiolitis: a case report and literature review. Clin Respir J. (2014) 8(4):425–30. doi: 10.1111/crj.12089
26. Sui W, Hou X, Che W, Ou M, Sun G, Huang S, et al. CCDC40 mutation as a cause of primary ciliary dyskinesia: a case report and review of literature. Clin Respir J. (2016) 10(5):614–21. doi: 10.1111/crj.12268
27. Wang K, Chen X, Guo CY, Liu FQ, Wang JR, Sun LF. Cilia ultrastructural and gene variation of primary ciliary dyskinesia: report of three cases and literatures review. Zhonghua Er Ke Za Zhi. (2018) 56(2):134–7. doi: 10.3760/cma.j.issn.0578-1310.2018.02.012
28. Dimakou K, Triantafillidou C, Toumbis M, Tsikritsaki K, Malagari K, Bakakos P. Non CF-bronchiectasis: aetiologic approach, clinical, radiological, microbiological and functional profile in 277 patients. Respir Med. (2016) 116:1–7. doi: 10.1016/j.rmed.2016.05.001
29. Okada F, Ando Y, Tanoue S, Ishii R, Matsushita S, Ono A, et al. Radiological findings in acute Haemophilus influenzae pulmonary infection. Br J Radiol. (2012) 85(1010):121–6. doi: 10.1259/bjr/48077494
30. Shoemark A, Dell S, Shapiro A, Lucas JS. ERS and ATS diagnostic guidelines for primary ciliary dyskinesia: similarities and differences in approach to diagnosis. Eur Respir J. (2019) 54(3):1901066. doi: 10.1183/13993003.01066-2019
31. Kobbernagel HE, Buchvald FF, Haarman EG, Casaulta C, Collins SA, Hogg C, et al. Efficacy and safety of azithromycin maintenance therapy in primary ciliary dyskinesia (BESTCILIA): a multicentre, double-blind, randomised, placebo-controlled phase 3 trial. Lancet Respir Med. (2020) 8(5):493–505. doi: 10.1016/s2213-2600(20)30058-8
Keywords: primary ciliary dyskinesia, CCNO gene, genetic diagnosis, diffuse panbronchiolitis, bronchiectasis
Citation: Tong L, Li L, Wang W and Chen J (2024) Case Report: Primary ciliary dyskinesia due to CCNO mutations: a Chinese pediatric case series and literature review. Front. Pediatr. 12:1458660. doi: 10.3389/fped.2024.1458660
Received: 2 July 2024; Accepted: 4 September 2024;
Published: 24 September 2024.
Edited by:
Raffaella Nenna, Sapienza University of Rome, ItalyReviewed by:
Laura Petrarca, Sapienza University of Rome, ItalyOphir Bar-On, Schneider Children’s Medical Center, Israel
Copyright: © 2024 Tong, Li, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiehua Chen, bG9vZjExQDE2My5jb20=