 Guihua Lai
Guihua Lai Qiying Gu
Qiying Gu Haijun Chen
Haijun Chen- Central Laboratory, Ganzhou Maternal and Child Health Hospital, Ganzhou, Jiangxi, China
Background: Genetic diseases exhibit significant clinical and genetic diversity, leading to a complex and challenging diagnostic process. Exploiting novel approaches is imperative for the molecular diagnosis of genetic diseases. In this study, we utilized whole-exome sequencing (WES) to facilitate early diagnosis in patients suspected of genetic disorders.
Methods: This retrospective analysis included 144 patients diagnosed by singleton-WES Trio-WES between January 2021 and December 2023. We investigated the relevance of diagnosis rates with age, clinical presentation, and sample type.
Results: Among the 144 patients, 61 were diagnosed, yielding an overall diagnostic rate of 42.36%, with Trio-WES demonstrating a significantly higher diagnostic rate of 51.43% (36/70) compared to singleton-WES at 33.78% (25/74) (p < 0.05). Global developmental delay had a diagnosis rate of 67.39%, significantly higher than muscular hypotonia at 30.43% (p < 0.01) among different clinical phenotypic groups. Autosomal dominant disorders accounted for 70.49% (43/61) of positive cases, with autosomal abnormalities being fivefold more prevalent than sex chromosome abnormalities. Notably, sex chromosome abnormalities were more prevalent in males (80%, 8/10). Furthermore, 80.56% (29/36) of pathogenic variants were identified as de novo mutations through Trio-WES.
Conclusions: These findings highlight the effectiveness of WES in identifying genetic variants, and elucidating the molecular basis of genetic diseases, ultimately enabling early diagnosis in affected children.
1 Introduction
Genetic diseases, caused by genetic alterations leading to abnormal function, result in diverse clinical phenotypes, which can occur at all ages and are often congenital. Therefore, it is particularly common in pediatric patients. With the advancement of life science and society, the spectrum of childhood diseases has undergone significant changes. According to the estimate of the World Health Organization in 2007, the current birth defect rate in China is 5.6%, significantly higher than the 4.72% in developed countries (1). Furthermore, birth defects account for 20.13% of child deaths in China (2). Therefore, the diagnosis and treatment of genetic diseases and the follow-up course management become crucial in the clinical work of pediatricians. However, children with potential genetic diseases often exhibit multiple systems, multiple organs, and diverse symptoms, which are more complex than ordinary pediatric patients and are easily misdiagnosed and missed. They also have a higher proportion of hospitalization, accounting for 9%–15%, and higher mortality rates (1.0%–1.3%) (3–5). Assessing the diagnosis of genetic diseases in children requires highly vigilant specialist physicians, often requiring complex clinical examinations and evaluations, which are time-consuming and costly. Even after detailed diagnostic evaluation, the majority of children still been diagnosed unclearly. Therefore, due to the often complex clinical manifestations of genetic disease, a comprehensive diagnostic technology is required to achieve early diagnosis. In recent years, with the continuous development of molecular diagnosis and the ability to reveal the genetic causes of genetic diseases, medical practice is undergoing a revolutionary transformation from traditional symptom-based diagnosis to modern cause-based diagnosis (6). Choosing the right molecular genetic diagnostic strategy can help shorten the diagnostic odyssey and avoid the economic burden of redundant diagnostic testing. This can help accurately intervene in diseases related to genetic causes with clearly defined locations, thereby improving disease prognosis (7). This study analyzed the clinical records and diagnostic data of 144 cases diagnosed by whole-exome sequencing (WES) to evaluate the diagnostic efficacy of WES in different ages and clinical phenotypes.
2 Materials and methods
2.1 Study design and participants
This was a retrospective cohort study conducted at the Clinical Genetics Laboratory of the Ganzhou Maternal and Child Health Hospital. Pediatric patients were recruited between January 2021, and December 2023. The inclusion criteria were as follows: (1) children with an unclear clinical diagnosis for whom genetic disorders were considered; and (2) an order for Next-Generation Sequencing and complete medical history. Notably, Patients were excluded if they were undergoing emergency surgery or external blood transfusion. Whole exome sequencing was conducted as Trio-WES (both parents and their affected child sequenced simultaneously) to effectively detect de novo and compound heterozygous variants or as singleton-WES (only the affected individual sequenced) when parental samples were not available. Trio-WES were performed by 70 patients of this cohort with non-consanguineous healthy parents, and the remaining 74 probands underwent singleton-WES. We collected basic information about each patient through a retrospective review of medical records and examined the correlation between clinical information and diagnostic findings. The study was approved by the ethics committee of the Ganzhou Maternal and Child Health Hospital (202396). The legal guardians of the participating children gave their signed, informed consent for their children to be included in the study.
2.2 Next-generation sequencing
Blood samples of patients and any participating family members were collected, and genomic DNA was extracted using the QIAamp DNA Mini Kit (Hilden, Germany) following the manufacturer's protocol. The coding exons of target genes were captured using an Exome Panel v2.0 (Nanodigmbio, Nanjing), and libraries generated from enriched DNA were sequenced using the Illumina NovaSeq 6000 platform (Illumina Inc., San Diego, CA, USA) in paired-end mode. The average on-target sequencing depth for exome sequencing was 90X, and more than 98% of target bases had a coverage of over 20X. The sequencing reads were aligned to the human reference genome (UCSC GRCh37/hg19) using the Burrows-Wheeler Aligner. Subsequently, single nucleotide variant (SNV) and small insertion/deletion (Indel) detection were conducted. Finally, variant filtering was performed with the PhenoPro (8) phenotype-scoring algorithm. Copy number variants (CNV) analysis is primarily based on sequencing depth or read counts. The analysis process includes GC correction of the samples, normalization of sample data within batches, calculation of log2Ratio and Z-scores, and finally, hierarchical clustering analysis and segmentation process, resulting in a VCF file containing CNV information. The causative variants detected through Trio/singleton-WES were subsequently confirmed by PCR or Sanger sequencing in the proband and parents if available using a 3500XL Genetic Analyzer (Applied Biosystems) according to the manufacturer's specifications. The variant's pathogenicity was determined using the criteria established by the American College of Medical Genetics and Genomics (9).
2.3 Statistical analysis
The significance of the diagnostic rate of Trio-WES vs. singleton-WES (p values) was calculated by one-tailed Fisher's exact test. All other comparisons were done by a two-tailed Fisher's exact test. A p value of 0.05 was used as a significance threshold. The statistical analyses were conducted using the software package SPSS (version 26.0, IBM Corp., 2019).
3 Results
3.1 Demographics of clinical feature
From January 1, 2021, to December 31, 2023, a total of 144 unrelated patients were included in the study. The cohort consisted of 85 males and 59 females, with a median age of 4 years (range 0–17). All participants underwent WES for suspected genetic disorders. Among the 144 pediatric cases, there were 100 young children (aged 5 and under). Notably, 55% (55 out of 100) of the young children underwent Trio-WES analysis. In contrast, only 15 out of 44 older children (over 5 years old) underwent this analysis, representing 34.09% (p < 0.05). The clinical manifestations observed in the patients included global developmental delay (n = 46, 31.94%), intellectual disability (n = 44, 30.56%), abnormal metabolism (n = 26, 18.06%), dysmorphic facial features (n = 25, 17.36%), seizures (n = 24, 16.67%), muscular hypotonia (n = 23, 15.97%), and autistic behavior (n = 19, 13.19%), as detailed in Table 1, Supplementary Table S1 and Supplementary Table S2.
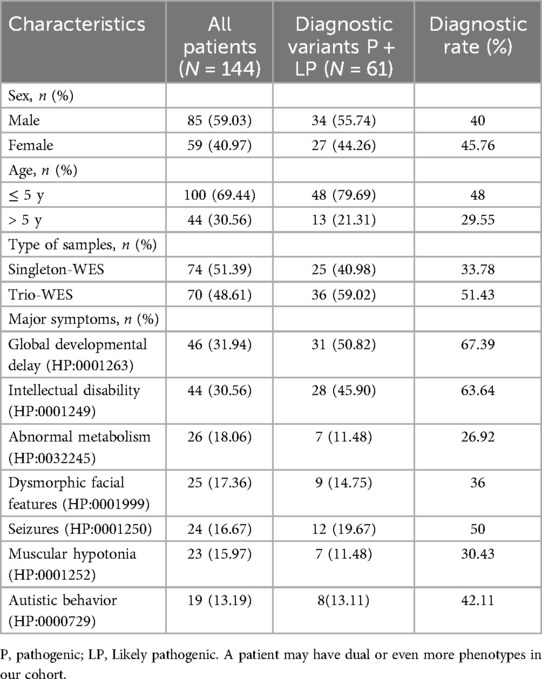
Table 1. Molecular diagnostic rates of WES by assay type, age and clinical phenotype.
3.2 Diagnostic yield of WES
The diagnostic results were obtained in 61 out of 144 patients and the overall diagnosis rate of WES was 42.36%, with a significantly higher diagnosis rate of 51.43% (36/70) in Trio-WES than 33.78% (25/74) in singleton-WES (p < 0.05). Furthermore, we grouped children according to age at different stages and found that the diagnosis rate of young children was significantly higher at 48% (48/100) than that of older children at 29.55% (13/44, p < 0.05). To explore the diagnostic rate of WES in terms of clinical phenotype, we analyzed the molecular diagnostic rate of clinical phenotype subgroups, as shown in Table 1. We found that the diagnostic rate in the global developmental delay patient group was (67.39%, 31/46), which was significantly higher than the diagnostic rate in the muscular hypotonia patient group (30.43%, 7/23, p < 0.01). Moreover, the diagnosis rate in the intellectual disability patient group was (63.64%, 28/44), which was significantly higher than the diagnosis rate in the abnormal metabolism patient group (26.92%, 7/26, p < 0.01). These findings suggested significant differences in diagnostic rates across clinical phenotypes.
3.3 Analysis of genetic variation
An overall diagnostic yield of 42.36% (61/144) was obtained through the combined analysis of Indels, CNVs, and SNVs. This included 63.93% (39/61) from SNV/Indel analysis and 36.06% (22/61) from exome-based CNV analysis. CNV analysis identified 22 pathogenic CNVs, with 27.87% (17/61) being deletions and 8.2% (5/61) duplications, ranging from 406 Kb to 25.3 Mb. SNV/Indel analysis identified 39 pathogenic or likely pathogenic variants in 33 different genes, based on ACMG guidelines. Among these, 24.59% (15/61) were frameshift variants, 16.39% (10/61) were missense variants, 14.75% (9/61) were nonsense variants, 6.56% (4/61) were splice variants, and 1.64% (1/61) were inframe variant (Figure 1). Specifically, among these positive patients, 70.49% (43/61) had autosomal dominant (AD) disorders, with global developmental delay (60.47%, 26/43) and intellectual disability (53.49%, 23/43) as the most common clinical phenotypes. Furthermore, 13.12% (8/61) had autosomal recessive (AR) disorders, with abnormal metabolism (87.5%, 7/8) being the most common clinical phenotype. Additionally, 13.12% (8/61) had X-linked recessive (XLR) disorders, which were exclusively in males, with muscular hypotonia (37.5%, 3/8) and intellectual disability (37.5%, 3/8) being the most common clinical phenotype. In 3.27% (2/61) of the children with X-linked dominant (XLD) disease, all of them were females, and the clinical phenotype of the two children was characterized primarily by intellectual disability. In the 61 positive children, the prevalence of autosomal abnormalities was above fivefold higher than sex chromosome abnormalities (51/10). Sex chromosome abnormalities were predominantly found in 80% (8/10) of males (Table 2). Furthermore, 80.56% (29/36) of pathogenic variants were identified as de novo mutations by Trio-WES, and the majority of these children had AD conditions 82.76% (24/29), while AR conditions was found in only 3.45% (1/17, Figure 2). The most common clinical phenotypes in patients with de novo mutations were intellectual disability (55.17%, 16/29), global developmental delay (44.83%, 13/29), and seizures (24.14%, 7/29).
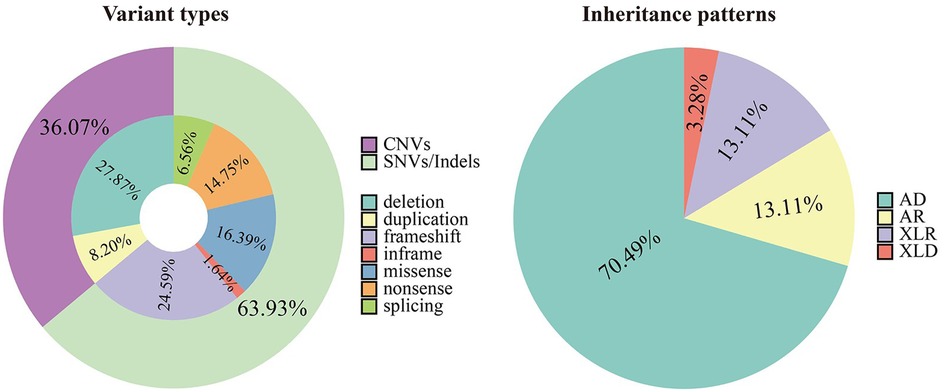
Figure 1. Variant types and inheritance patterns in 61 children with genetic diseases.
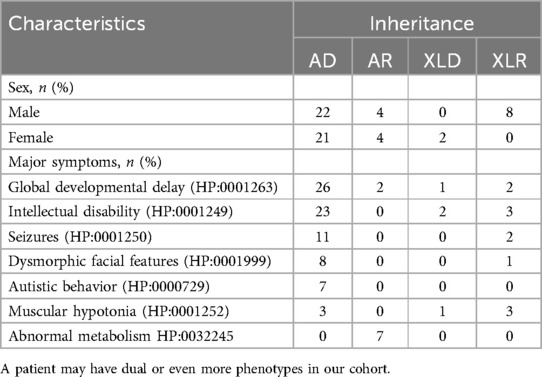
Table 2. Inheritance patterns of different clinical phenotype subgroups.
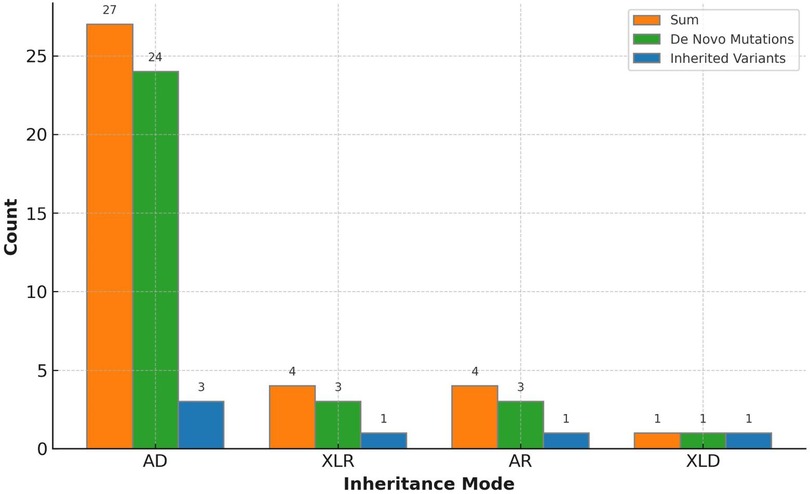
Figure 2. Variant inheritance of the 36 positive trio-WES cases.
4 Discussion
Genetic disorders, categorized as chromosomal, monogenic, polygenic, and mitochondrial disorders, involve congenital malformations, physiological defects, or metabolic abnormalities. The incidence of genetic disorders is estimated to be between 40 and 82 per 1,000 live births (10). For most patients with genetic disorders, a reliable diagnosis is essential to improve management and quality of life for patients and their families. Some studies have shown that molecular diagnosis of genetic diseases is an important and indispensable method for their accurate diagnosis (11). Genome sequencing technology has been shown to be an important tool for the discovery of new disease-associated genetic loci and it is an effective alternative to gene panel testing (12, 13). It is known that exonic regions comprise only about 1% of the entire human genome but contain 85% of the disease-causing variants (14). In this study, we used WES to directly detect the causative genes of genetic diseases for early diagnosis of diseases. Our results showed that 42.36 percent (61/144) of patients received a molecular diagnosis. Although there is no cure for most genetic disorders, symptomatic treatment of patients with a clear molecular etiology can significantly reduce the burden of events and improve subsequent clinical management (15). Furthermore, in our study, 79% of the patients with genetic disorders were young children, and our results are consistent with the early onset of genetic disorders and the fact that they are mostly congenital. Furthermore, among 144 children suspected of having genetic diseases, we found a higher percentage of male than female patients, which we considered to be due to the presence of sex chromosome variants. Our results showed that there are far more males than females with X-linked genetic disorders in the population, and the results could be more significant if the sample size is increased.
Furthermore, the results indicated that the highest percentage of patients were associated with AD conditions (70.49%). Using Trio-WES, we found that the variants in these patients were predominantly de novo mutations. It is possible due to the fact that AD conditions have a certain rate of disability and mortality, most families reject having children or undergo prenatal diagnosis (16, 17), and thus do not have a similar family history. Our study found that the patients associated with AR conditions accounted for 13.12%, and the disease phenotype was predominantly characterized by aberrant metabolism. Most variants associated with AR conditions were inherited from parents, with a higher prevalence observed in consanguineous marriage families (18). In this study, no Y-linked genetic diseases were detected.
Previous studies have shown that the diagnostic sensitivity of WES varies depending on the organ system involved (19). In this study, we grouped 144 patients with major clinical phenotypes and found significant differences in diagnostic rates between phenotype groups. Global developmental delay had the highest positive rate of 67.39%, followed by Intellectual disability (63.64%). It has been reported that the diagnostic rate of WES in children with neurological abnormalities was significantly higher than that of non-neurological abnormalities (20, 21). In our study, the diagnostic rates of global developmental delay, intellectual disability, autistic behavior, and seizures were higher than dysmorphic facial features, muscular hypotonia, and abnormal metabolism, which is consistent with the trend of previous findings (20). The low diagnostic rate of non-neurologic abnormalities suggested that the proportion of unknown genes, complex structural variation, or non-genetic underlying mechanisms may be greater. Compared to WES, Whole genome sequencing (WGS), with its superior and comprehensive genome coverage, enhances the detection of deep intronic regions and complex structural variants. However, considering efficacy and difficulty of interpretation, WES is more applicable for clinical testing currently. In recent years, as the cost of WGS decreases and the interpretation level of genetic testing improves, future studies could be considered to employ this technique on undiagnosed patients but highly suspected with genetic diseases to discover novel genes or pathogenic variants (22, 23). In addition, we found that the diagnostic rate of Trio-WES was significantly higher than that of singleton-WES. Trio-WES can determine whether the variants detected in proband samples are inherited from the parents, which is extremely essential in analyzing de novo mutations. Previous studies have found that patients with intellectual disability (24), global developmental delay (25), and epilepsy (26) are the most common groups affected by de novo mutations. Our results showed that de novo mutations were present in up to 80% (29/36) of all pathogenic variants, with the most common clinical phenotype being Intellectual disability 55.17% (16/29), followed by global developmental delay 44.83% (13/29). We found that the percentage of de novo pathogenic variants inherited as AR was only 3.45% (1/29), which may be due to the fact that de novo mutations are limited by the frequency of mutations in the species (27). In this study, the higher detection rate of de novo mutations was attributed to the Trio-WES testing strategy, which enhanced the clinical interpretation of the pathogenicity of the variants through co-testing of the proband and its parental samples (28). Therefore, the testing protocol of Trio-WES should be prioritized to improve the diagnostic rate when parents are available (29). However, there are limitations of using the WES approach, as WES targets only the coding region of the genome and may not reliably detect CNVs at the single-gene or exon level, which means WES may not accurately detect non-coding, deep intronic regions, or copy number changes, as well as complex genomic structural variants, such as gene rearrangement or inversion. In addition, variant of uncertain significance (VUS) results were detected and reported. Incorrect interpretations of these variants can cause patient anxiety and lead to inappropriate patient management.
In recent years, with the improvement of tools for classifying genetic variants and the continuous updating of genetic databases and clinical phenotypes (30), reanalysis of WES may improve the diagnosis of the disease (13, 31). Therefore, enhanced follow-up and reanalysis of patients with non-diagnostic findings is important for our subsequent studies. In addition, likely pathogenic variants only mean 90% chance of pathogenicity and may not be diagnostic. WES is a phenotype-driven analysis, providing accurate clinical phenotypes is essential for the lab to filter out the potential diagnostic findings. Therefore, the combination of clinical phenotype with genetic finding to make a clinical judgment is crucial in the diagnosis.
5 Conclusions
In this study, 144 patients suspected of genetic diseases were analyzed by WES, which revealed significant differences in diagnostic efficiency among different clinical phenotypes. Besides, we found that AD disorders dominated by de novo mutations were the most prevalent, variants associated with AR and XLR were inherited from parents. In conclusion, we proposed that Trio-WES can efficiently identify the genetic causes of the diseases, and improve the diagnostic rate of the genetic diseases in clinical work.
Data availability statement
The original data is available online (https://figshare.com/s/431c345a768b15dee77a). Requests to access the raw data should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethics committee of the Ganzhou Maternal and Child Health Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
GL: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. QG: Writing – original draft, Data curation, Methodology, Software. ZL: Data curation, Methodology, Software, Writing – original draft, Investigation. HC: Conceptualization, Data curation, Software, Supervision, Writing – review & editing, Methodology. JC: Conceptualization, Methodology, Software, Supervision, Writing – original draft, Writing – review & editing. JH: Conceptualization, Data curation, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project was funded by Science and Technology Programme of Jiangxi Provincial Health and Wellness Commission, Grant/Award Number: 202410833.
Acknowledgments
We thank the patients for their willingness to participate in this study, Zezhang Liu provided great help in polishing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2024.1448895/full#supplementary-material
Supplementary Table S1 | Detailed information on each case.
Supplementary Table S2 | Numbers of cases with each symptom.
References
1. Yu M, Ping Z, Zhang S, He Y, Dong R, Guo X. The survey of birth defects rate based on birth registration system. Chin Med J. (2015) 128(1):7–14. doi: 10.4103/0366-6999.147785
2. Cai L, Zheng LA, He L. The forty years of medical genetics in China. J Genet Genomics. (2018) 45(11):569–82. doi: 10.1016/j.jgg.2018.10.001
3. Gonzaludo N, Belmont JW, Gainullin VG, Taft RJ. Estimating the burden and economic impact of pediatric genetic disease. Genet Med. (2019) 21(8):1781–9. doi: 10.1038/s41436-018-0398-5
4. Wojcik MH, Schwartz TS, Yamin I, Edward HL, Genetti CA, Towne MC, et al. Genetic disorders and mortality in infancy and early childhood: delayed diagnoses and missed opportunities. Genet Med. (2018) 20(11):1396–404. doi: 10.1038/gim.2018.17
5. Shashi V, McConkie-Rosell A, Rosell B, Schoch K, Vellore K, McDonald M, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med. (2014) 16(2):176–82. doi: 10.1038/gim.2013.99
6. Zhang Q, He L, Shen YP. [The arrival of the clinical whole genome era]. Zhonghua er ke za zhi. (2019) 57(6):401–4. doi: 10.3760/cma.j.issn.0578-1310.2019.06.001
7. Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. (2017) 171(12):e173438. doi: 10.1001/jamapediatrics.2017.3438
8. Li Z, Zhang F, Wang Y, Qiu Y, Wu Y, Lu Y, et al. Phenopro: a novel toolkit for assisting in the diagnosis of Mendelian disease. Bioinformatics. (2019) 35(19):3559–66. doi: 10.1093/bioinformatics/btz100
9. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
10. Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet. (2016) 89(3):275–84. doi: 10.1111/cge.12654
11. Wright CF, FitzPatrick DR, Firth HV. Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet. (2018) 19(5):253–68. doi: 10.1038/nrg.2017.116
12. Platt CD, Zaman F, Bainter W, Stafstrom K, Almutairi A, Reigle M, et al. Efficacy and economics of targeted panel versus whole-exome sequencing in 878 patients with suspected primary immunodeficiency. J Allergy Clin Immunol. (2021) 147(2):723–6. doi: 10.1016/j.jaci.2020.08.022
13. Salfati EL, Spencer EG, Topol SE, Muse ED, Rueda M, Lucas JR, et al. Re-analysis of whole-exome sequencing data uncovers novel diagnostic variants and improves molecular diagnostic yields for sudden death and idiopathic diseases. Genome Med. (2019) 11(1):83. doi: 10.1186/s13073-019-0702-2
14. Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. (2009) 106(45):19096–101. doi: 10.1073/pnas.0910672106
15. Bhatia NS, Lim JY, Bonnard C, Kuan JL, Brett M, Wei H, et al. Singapore undiagnosed disease program: genomic analysis aids diagnosis and clinical management. Arch Dis Child. (2021) 106(1):31–7. doi: 10.1136/archdischild-2020-319180
16. de Die-Smulders CE, de Wert GM, Liebaers I, Tibben A, Evers-Kiebooms G. Reproductive options for prospective parents in families with Huntington’s disease: clinical, psychological and ethical reflections. Hum Reprod Update. (2013) 19(3):304–15. doi: 10.1093/humupd/dms058
17. Fahy N, Rice C, Lahiri N, Desai R, Stott J. Genetic risk for Huntington disease and reproductive decision-making: a systematic review. Clin Genet. (2023) 104(2):147–62. doi: 10.1111/cge.14345
18. Musante L, Ropers HH. Genetics of recessive cognitive disorders. Trends Genet. (2014) 30(1):32–9. doi: 10.1016/j.tig.2013.09.008
19. Adams DR, Eng CM. Next-generation sequencing to diagnose suspected genetic disorders. N Engl J Med. (2018) 379(14):1353–62. doi: 10.1056/NEJMra1711801
20. Splinter K, Adams DR, Bacino CA, Bellen HJ, Bernstein JA, Cheatle-Jarvela AM, et al. Effect of genetic diagnosis on patients with previously undiagnosed disease. N Engl J Med. (2018) 379(22):2131–9. doi: 10.1056/NEJMoa1714458
21. Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. (2019) 21(11):2413–21. doi: 10.1038/s41436-019-0554-6
22. Meienberg J, Bruggmann R, Oexle K, Matyas G. Clinical sequencing: is WGS the better WES? Hum Genet. (2016) 135(3):359–62. doi: 10.1007/s00439-015-1631-9
23. Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A. (2015) 112(17):5473–8. doi: 10.1073/pnas.1418631112
24. Hamdan FF, Srour M, Capo-Chichi JM, Daoud H, Nassif C, Patry L, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. (2014) 10(10):e1004772. doi: 10.1371/journal.pgen.1004772
25. Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. (2017) 9(1):43. doi: 10.1186/s13073-017-0433-1
26. McCormack M, McGinty RN, Zhu X, Slattery L, Heinzen EL, Costello DJ, et al. De-novo mutations in patients with chronic ultra-refractory epilepsy with onset after age five years. Eur J Med Genet. (2020) 63(1):103625. doi: 10.1016/j.ejmg.2019.01.015
27. Chintalapati M, Moorjani P. Evolution of the mutation rate across primates. Curr Opin Genet Dev. (2020) 62:58–64. doi: 10.1016/j.gde.2020.05.028
28. Tan TY, Lunke S, Chong B, Phelan D, Fanjul-Fernandez M, Marum JE, et al. A head-to-head evaluation of the diagnostic efficacy and costs of trio versus singleton exome sequencing analysis. Eur J Hum Genet. (2019) 27(12):1791–9. doi: 10.1038/s41431-019-0471-9
29. Du X, Gao X, Liu X, Shen L, Wang K, Fan Y, et al. Genetic diagnostic evaluation of trio-based whole exome sequencing among children with diagnosed or suspected autism spectrum disorder. Front Genet. (2018) 9:594. doi: 10.3389/fgene.2018.00594
30. Al-Nabhani M, Al-Rashdi S, Al-Murshedi F, Al-Kindi A, Al-Thihli K, Al-Saegh A, et al. Reanalysis of exome sequencing data of intellectual disability samples: yields and benefits. Clin Genet. (2018) 94(6):495–501. doi: 10.1111/cge.13438
Keywords: whole-exome sequencing, genetic diseases, genetic diagnosis, children, rare disease
Citation: Lai G, Gu Q, Lai Z, Chen H, Chen J and Huang J (2024) The application of whole-exome sequencing in the early diagnosis of rare genetic diseases in children: a study from Southeastern China. Front. Pediatr. 12:1448895. doi: 10.3389/fped.2024.1448895
Received: 14 June 2024; Accepted: 23 September 2024;
Published: 8 October 2024.
Edited by:
Yongchu Pan, Nanjing Medical University, ChinaReviewed by:
Amy Brower, American College of Medical Genetics and Genomics (ACMG), United StatesWenying Zhang, Cincinnati Children’s Hospital Medical Center, United States
Copyright: © 2024 Lai, Gu, Lai, Chen, Chen and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jungao Huang, MTgxNDY3OTUxMjlAMTYzLmNvbQ==