
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 21 August 2024
Sec. Pediatric Hematology and Hematological Malignancies
Volume 12 - 2024 | https://doi.org/10.3389/fped.2024.1448801
 Lara Chavaz1,2*†
Lara Chavaz1,2*† Laurent Cimasoni1,2*†
Laurent Cimasoni1,2*† Johanna A. Kremer Hovinga3,4,†
Johanna A. Kremer Hovinga3,4,† Paul Coppo5,6,7,†Marc Ansari1,2,†
Paul Coppo5,6,7,†Marc Ansari1,2,†
The cornerstone treatment for immune-mediated thrombotic thrombocytopenic purpura (iTTP) in children is a combination of therapeutic plasma exchange (TPE), corticosteroids, and rituximab. Caplacizumab is an anti-von Willebrand factor (VWF) NANOBODY molecule approved as a frontline therapy of iTTP for adults and children aged ≥12 years. Using caplacizumab in children aged <12 years remains a gray area based on recommendations but with no marketing authorization. We report the first case of a pediatric patient with iTTP successfully treated with a caplacizumab dose adjustment of 5 mg daily based on ADAMTS13 activity. We also review all published cases of iTTP in children aged <12 years treated with caplacizumab. This is a 7-year-old girl with clinical thrombotic microangiopathy, in the absence of diarrhea and kidney injury. With a French score of 2 and a PLASMIC score of 7 (high risk), the diagnosis of TTP was suspected and later confirmed by severely low ADAMTS13 activity (<5%). Immune-mediated TTP was distinguished from the congenital one due to the presence of a functional ADAMTS13 inhibitor. Daily TPE and intravenous corticosteroids were started on day 0 (D0). Rituximab was added on D4, and due to refractoriness under daily TPE, we considered off-label administration of caplacizumab from D12. A clinical answer, with a significant increase in the platelet count, was observed within 48 h. A complete ADAMTS13 recovery was reached on D62. No major adverse events were observed during the treatment. She was discharged from the hospital over 3 months ago with a platelet count still within normal ranges. In the literature, we identified a total of four case reports describing five iTTP patients aged <12 years treated with caplacizumab, with a 100% success and tolerability rate. These published data attest to the efficacy and safety of the systematic use of caplacizumab and rituximab as frontline therapy in pediatric iTTP under 12 years of age. Therefore, prospective data are needed to support commercial authorization of caplacizumab in this subpopulation. Close monitoring of ADAMTS13 activity is particularly of interest among children to limit the number of caplacizumab injections.
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is a specific form of thrombotic microangiopathy caused by a severe antibody-mediated deficiency of to a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13 (ADAMTS13), a plasma enzyme responsible for the physiological cleavage of von Willebrand factor (VWF) (1). In the absence of ADAMTS13, VWF remains a long, multimerized compound (2, 3) that triggers the spontaneous formation of microthrombi (1). The obstructed microcirculation consumes platelets, leads to microangiopathic hemolytic anemia with schistocyte formation, and contributes to ischemic organ injuries. In the case of a suspected acute episode of thrombotic microangiopathy, the pretest likelihood of a severe ADAMTS13 deficiency can be estimated using the French (4) and PLASMIC (5) scores, both validated for adults. Definitive diagnosis is established by documentation of an ADAMTS13 activity of <10% of the normal in the presence of anti-ADAMTS13 antibodies. Close follow-up for potentially life-threatening relapses is crucial after an iTTP episode (6, 7).
Historically, iTTP treatment was based on therapeutic plasma exchange (TPE) and corticosteroids, with rituximab only given as salvage therapy. Today, early administration of rituximab has become part of the first-line therapeutic strategy (1, 8). Caplacizumab is a humanized NANOBODY molecule inhibiting the interaction between platelets and VWF by shielding the VWF A1 domain. Based on two randomized control trials, TITAN and HERCULES, caplacizumab has been approved for initial therapy of iTTP in patients aged ≥12 years and weighing ≥40 kg (9, 10). The approved dosage is a 10 mg intravenous (IV) loading bolus, followed by 10 mg daily subcutaneously (9, 10). A 5 mg daily dose was suggested for children weighing <40 kg (11). In the Phase II trial, treatment duration was set at 30 days (9) but was found to be associated with an increased risk of iTTP recurrence after discontinuation (10, 12). ADAMTS13 activity is emerging as a crucial biomarker for guiding treatment duration and preventing recurrence; however, further studies are needed to confirm the discontinuation threshold (1), which currently ranges from 10% (13) to 20% (8, 14, 15). Caplacizumab is associated with faster platelet count recovery and lower risks of recurrence, refractoriness, and mortality due to iTTP (1, 9, 10). The most commonly described adverse events are bleeding, mainly mucocutaneous, as caplacizumab induces a von Willebrand disease type 2M-like state (10).
Already rare in adulthood, iTTP is even rarer in childhood (16), where hemolytic uremic syndrome and the congenital form of TTP are more prevalent (17). Consequently, guidelines for pediatric treatment of iTTP do not exist, and treatment is often based on recommendations for adults. Although caplacizumab is not authorized under 12 years of age, some pediatric iTTP cases treated with this drug have been reported. Here, we describe a patient treated in our institution who is the first published child with iTTP receiving caplacizumab dose adjustment based on ADAMTS13 activity and review published case reports.
A 7-year-old Asian girl presented at the emergency department with spontaneous bruising and three episodes of vomiting after a week of afebrile coughing. Her medical and family histories were unremarkable. Clinical examination revealed a petechial rash with hematomas of various sizes. Laboratory studies showed low platelet count (14 G/L), hemolytic anemia (hemoglobin, 73 g/L; reticulocyte count, 164 G/L; lactate dehydrogenase, 959 U/L; haptoglobin below the detection limit; indirect bilirubin, 39.2 µmol/L; and negative direct antiglobulin test), 2% schistocytes on the peripheral blood smear, and no apparent organ involvement. Anticardiolipin IgG was 23.8 U/ml (normal, <14 U/ml), and anticardiolipin IgM, beta2-glycoprotein1 IgG, IgM, and antinuclear antibodies were negative. Complement factor C3, C4, and CH50 plasma levels were within normal range.
The patient presented with a picture of thrombotic microangiopathy, but the absence of diarrhea and kidney injury ruled out hemolytic uremic syndrome. French score of 2 and a PLASMIC score of 7 (high risk) indicated probable TTP, soon confirmed by severely low ADAMTS13 activity (<5%), which was found to be acquired due to functional ADAMTS13 inhibitor of 1.5 BU/ml, thus ruling out congenital TTP. The patient was transferred to the pediatric intensive care unit on the day of admission (D0), receiving one packed red cell unit and fresh frozen plasma for central line placement, TPE, and intravenous (IV) methylprednisolone (1.5 mg/kg/day). Following daily TPE D0-D4, the patient showed clinical response with a platelet count of >150 G/L for 2 days, and TPE was paused. However, this was rapidly followed by an iTTP exacerbation, with the platelet count dropping to 40 G/L on D6 (Figure 1), increased hemolysis, and signs of cardiac and renal injury. TPE was restarted twice daily, with pulse methylprednisolone 15 mg/kg/day for 4 days. On D7, when the ADAMTS13 inhibitor result became available, she received the first of four doses of rituximab (375 mg/m2 on D7, D10, D15, and D21).
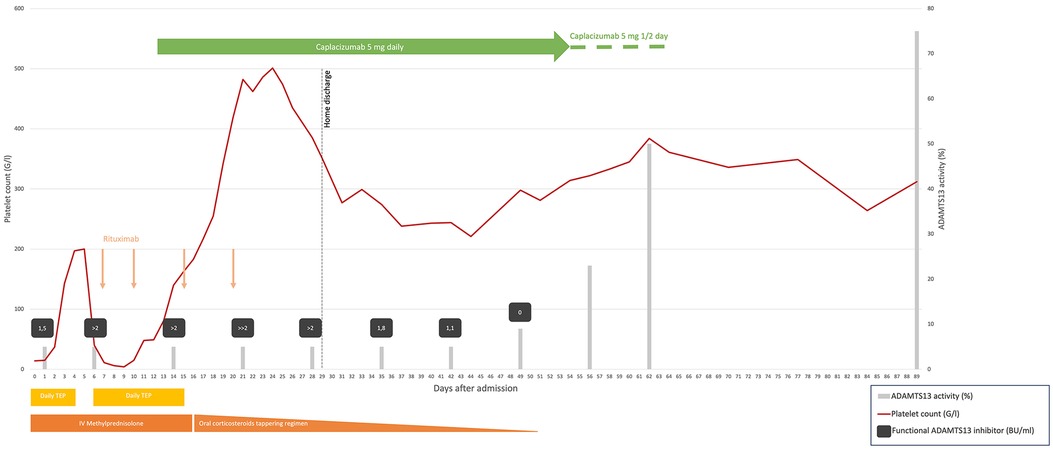
Figure 1. Summary of laboratory parameters and treatment administered in our case. TPE daily, days 0–4 and 6–15, temporarily increased bid days 9–12; intravenous methylprednisolone 1.5 mg/kg daily, days 0–16 temporarily increased to pulse 15 mg/kg/day, days 9–12; oral prednisone 1 mg/kg then slowly tapered, days 13–51; intravenous rituximab, 375 mg/m2, on days 7, 10, 15, and 20; caplacizumab 5 mg daily, intravenous day 12 and subcutaneous days 13–54, and then every second day, days 55–64. The functional ADAMTS13 inhibitor level is semiquantitative when the value is >2 BU/ml, ≫2 BU/ml, being the highest. TPE, therapeutic plasma exchange; IV, intravenous; G/L, giga/liter; BU/ml, Bethesda unit/milliliter. ADAMTS13 activity normal range, 51%–100%. Functional ADAMTS13 inhibitor normal range, <0.4 BU/ml.
Due to refractoriness under daily TPE, we considered off-label administration of caplacizumab and contacted Sanofi's managed access program. After obtaining the parents' informed consent, our institutional review board's approval, and advice from experts in the field, caplacizumab was started on D12 with a first dose of 5 mg IV followed by daily subcutaneous administration of the same dose. Her platelet count increased to 140 G/L after 2 days and remained in the normal range thereafter (Figure 1). Hospitalization was prolonged due to central line-associated bloodstream infection (CLABSI) by methicillin-sensitive Staphylococcus aureus on D14. The next day, we removed all central lines and stopped TPE. After completing the rituximab course, the patient was discharged home on D29 on a tapering steroid regimen. Daily caplacizumab was continued in the outpatient unit, until D54 when, because of painful injection site reaction, it was spaced to every other day, with monitoring of trough VWF activity using the VWF:GPIbM assay and platelet counts for 10 more days.
Anti-ADAMTS13 antibody titter started decreasing by D28, with partial ADAMTS13 recovery on D56 and complete recovery on D62, respectively, 7 and 6 weeks after TPE cessation and the last rituximab infusion. The only reported side effect reported was the painful injection site reaction with local redness and petechial rash. Our patient has been at home for 3 months now with normal platelet counts. Unfortunately, we have been unable to retest the patient for the antiphospholipid antibodies after 3 months, as she moved back to her home country.
This is the first pediatric patient successfully treated with dose adjustment of 5 mg daily and adaptation based on ADAMTS13 and VWF activities. The case underlines the risk of nosocomial CLABSI among iTTP patients dependent on a central line for their daily TPE. By reducing the number of TPE and length of stay, caplacizumab limits exposure to hospital-acquired infections. However, consideration should be given to how caplacizumab is administered to avoid real aversion to hospitalization in young children requiring chronic follow-up. Guiding treatment by measuring trough VWF activity levels allowed a reduction in the frequency of the injections.
We identified all published pediatric iTTP cases treated with caplacizumab in PubMed (“Purpura, Thrombotic, Thrombocytopenic” AND “caplacizumab” AND “Child*”) and EMBASE. Our search on 19 January 2024 found 15 and 34 results, respectively. Four case reports (18–21) described five iTTP patients aged <12 years treated with caplacizumab and are summarized in Table 1. Given the different anti-ADAMTS13 antibody and functional ADAMTS13 inhibitor tests used at the different sites, anti-ADAMTS13 antibody and functional ADAMTS13 inhibitor are not comparable and are given only as positive. Sex was evenly distributed; cutaneous, abdominal, and neurological symptoms were the most frequent clinical findings present at diagnosis. Doses of caplacizumab were either 5 mg or 10 mg daily for at least 30 days. All patients received TPE and immunosuppression with steroids, rituximab, and caplacizumab. All patients survived, and caplacizumab was well tolerated with only minor adverse events.
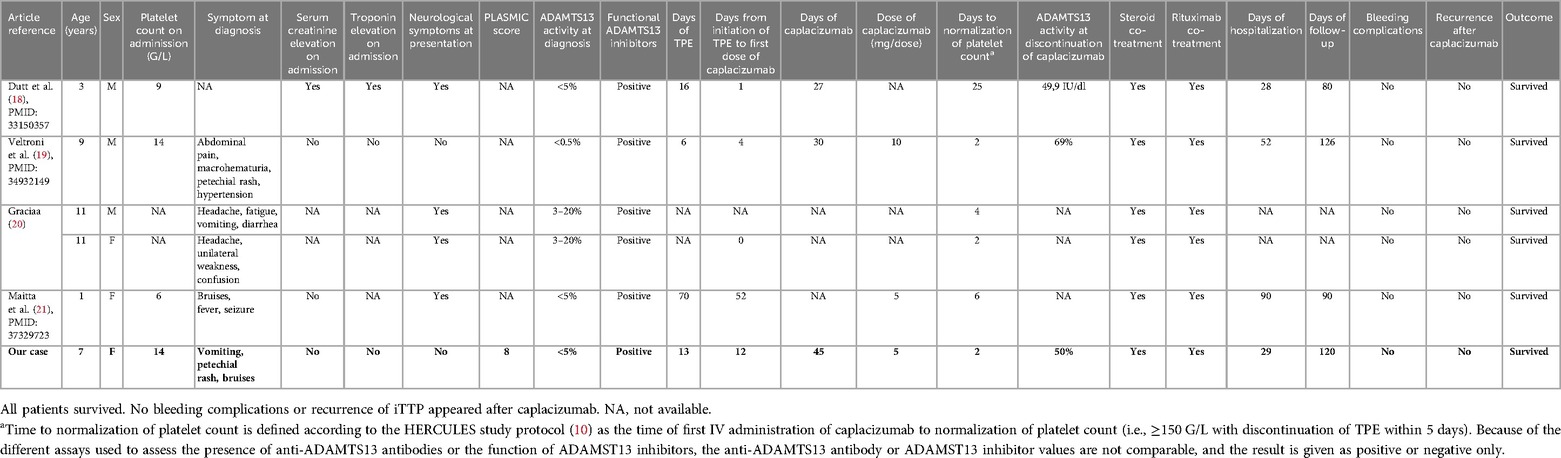
Table 1. Description of our case (in bold) and five pediatrics iTTP patients aged <12 years retrieved from the literature who were treated with caplacizumab.
These data, in line with reported experience from adult patients, support the systematic use of caplacizumab and rituximab as frontline therapy in pediatric iTTP patients. To date, there is only one retrospective clinical trial ongoing studying pediatric iTTP patients treated with caplacizumab (clinicaltrials.gov NCT05263193) with pending results (22). A randomized clinical trial on treatment with caplacizumab in children and adolescents aged <18 years or even 12 years suffering from acute iTTP would be highly appreciated, but given the low incidence rate of iTTP in children, it seems unlikely that such a study will be set up and recruit enough patients in due time. Therefore, case reports similar to the one described here or those already published are valuable in providing information and guiding physicians in treating iTTP in children.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Ethical approval was not required for this study involving only one unpublished patient. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
LC: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. LC: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. JK: Supervision, Validation, Visualization, Writing – review & editing. PC: Supervision, Validation, Visualization, Writing – review & editing. MA: Supervision, Validation, Visualization, Writing – review & editing.
The authors declare financial support was received for the research, authorship, and/or publication of this article. Open access funding by the University of Geneva.
The authors would like to thank Gaby Tanner, Medical Head of Rare Diseases and Rare Blood Disorders at Sanofi, for facilitating drug use. The authors also thank Darren Hart from Publish or Perish for his careful proofreading of the English language.
Caplacizumab treatment was financed by Sanofi. JK is a member of the advisory board of Takeda, a member of the Takeda group of companies, for the development of recombinant ADAMTS13, and of Ablynx, now part of Sanofi for the development of caplacizumab, and has received lecture fees from Takeda and Sanofi. All honoraria are paid to her employer, Insel Gruppe AG, Bern, Switzerland. PC is a member of Sanofi's advisory board and has received honoraria for participating in symposia.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Picod A, Veyradier A, Coppo P. Should all patients with immune-mediated thrombotic thrombocytopenic purpura receive caplacizumab? J Thromb Haemost. (2021) 19:58–67. doi: 10.1111/jth.15194
2. Moake JL, Rudy CK, Troll JH, Weinstein MJ, Colannino NM, Azocar J, et al. Unusually large plasma factor VIII: von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med. (1982) 307:1432–5. doi: 10.1056/NEJM198212023072306
3. Siddiqui A, Journeycake JM, Borogovac A, George JN. Recognizing and managing hereditary and acquired thrombotic thrombocytopenic purpura in infants and children. Pediatr Blood Cancer. (2021) 68:e28949. doi: 10.1002/pbc.28949
4. Coppo P, Schwarzinger M, Buffet M, Wynckel A, Clabault K, Presne C, et al. Predictive features of severe acquired ADAMTS13 deficiency in idiopathic thrombotic microangiopathies: the French TMA Reference Center experience. PLoS One. (2010) 5:e10208. doi: 10.1371/journal.pone.0010208
5. Bendapudi PK, Hurwitz S, Fry A, Marques MB, Waldo SW, Li A, et al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. Lancet Haematol. (2017) 4:e157–64. doi: 10.1016/S2352-3026(17)30026-1
6. Kremer Hovinga JA, Vesely SK, Terrell DR, Lämmle B, George JN. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood. (2010) 115:1500–11; quiz 1662. doi: 10.1182/blood-2009-09-243790
7. Thejeel B, Garg AX, Clark WF, Liu AR, Iansavichus AV, Hildebrand AM. Long-term outcomes of thrombotic microangiopathy treated with plasma exchange: a systematic review. Am J Hematol. (2016) 91:623–30. doi: 10.1002/ajh.24339
8. Coppo P, Bubenheim M, Azoulay E, Galicier L, Malot S, Bigé N, et al. A regimen with caplacizumab, immunosuppression, and plasma exchange prevents unfavorable outcomes in immune-mediated TTP. Blood. (2021) 137:733–42. doi: 10.1182/blood.2020008021
9. Peyvandi F, Scully M, Kremer Hovinga JA, Cataland S, Knöbl P, Wu H, et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med. (2016) 374:511–22. doi: 10.1056/NEJMoa1505533
10. Scully M, Cataland SR, Peyvandi F, Coppo P, Knöbl P, Kremer Hovinga JA, et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med. (2019) 380:335–46. doi: 10.1056/NEJMoa1806311
11. Bergstrand M, Hansson E, Delaey B, Callewaert F, De Passos Sousa R, Sargentini-Maier ML. Caplacizumab model-based dosing recommendations in pediatric patients with acquired thrombotic thrombocytopenic purpura. J Clin Pharmacol. (2022) 62:409–21. doi: 10.1002/jcph.1991
12. Völker LA, Kaufeld J, Miesbach W, Brähler S, Reinhardt M, Kühne L, et al. Real-world data confirm the effectiveness of caplacizumab in acquired thrombotic thrombocytopenic purpura. Blood Adv. (2020) 4:3085–92. doi: 10.1182/bloodadvances.2020001973
13. Völker LA, Kaufeld J, Miesbach W, Brähler S, Reinhardt M, Kühne L, et al. ADAMTS13 and VWF activities guide individualized caplacizumab treatment in patients with aTTP. Blood Adv. (2020) 4:3093–101. doi: 10.1182/bloodadvances.2020001987
14. Prasannan N, Thomas M, Stubbs M, Westwood J-P, de Groot R, Singh D, et al. Delayed normalization of ADAMTS13 activity in acute thrombotic thrombocytopenic purpura in the caplacizumab era. Blood. (2023) 141:2206–13. doi: 10.1182/blood.2022018847
15. Cuker A, Cataland SR, Coppo P, de la Rubia J, Friedman KD, George JN, et al. Redefining outcomes in immune TTP: an international working group consensus report. Blood. (2021) 137:1855–61. doi: 10.1182/blood.2020009150
16. Reese JA, Muthurajah DS, Kremer Hovinga JA, Vesely SK, Terrell DR, George JN. Children and adults with thrombotic thrombocytopenic purpura associated with severe, acquired Adamts13 deficiency: comparison of incidence, demographic and clinical features. Pediatr Blood Cancer. (2013) 60:1676–82. doi: 10.1002/pbc.24612
17. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. (2017) 129:2836–46. doi: 10.1182/blood-2016-10-709857
18. Dutt T, Shaw RJ, Stubbs M, Yong J, Bailiff B, Cranfield T, et al. Real-world experience with caplacizumab in the management of acute TTP. Blood. (2021) 137:1731–40. doi: 10.1182/blood.2020007599
19. Veltroni M, Pegoraro F, Scappini B, Brugnolo F, Allegro E, Ermini S, et al. Off-label caplacizumab as add-on therapy in a 9-year-old boy with refractory aTTP. Ann Hematol. (2022) 101:1369–71. doi: 10.1007/s00277-021-04740-4
20. Graciaa S. 2023 ASPHO conference paper and poster index. Pediatr Blood Cancer. (2023) 70:e30390. doi: 10.1002/pbc.30390
21. Maitta RW, Reeves HM, Downes KA, He X, Hackney LR, Ahuja SP. Immature platelet dynamics in management of protracted response to therapy of a young pediatric patient with immune-mediated thrombotic thrombocytopenic purpura. Thromb Res. (2023) 228:145–7. doi: 10.1016/j.thromres.2023.06.002
22. Researcher View | Retrospective Study on Caplacizumab-treated Pediatric Patients With Immune-mediated Thrombocytopenic Purpura (iTTP) | ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/study/NCT05263193?tab=table (accessed July 17, 2024)
Keywords: immune-mediated thrombotic thrombocytopenic purpura, iTTP, caplacizumab, pediatrics, benign hematological disorders
Citation: Chavaz L, Cimasoni L, Kremer Hovinga JA, Coppo P and Ansari M (2024) Caplacizumab as an add-on therapy in a 7-year-old girl with exacerbated immune-mediated thrombotic thrombocytopenic purpura, a case report and literature review. Front. Pediatr. 12:1448801. doi: 10.3389/fped.2024.1448801
Received: 13 June 2024; Accepted: 31 July 2024;
Published: 21 August 2024.
Edited by:
Tomasz Szczepanski, Medical University of Silesia, PolandReviewed by:
Szymon Janczar, Medical University of Lodz, PolandCopyright: © 2024 Chavaz, Cimasoni, Kremer Hovinga, Coppo and Ansari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lara Chavaz, bGFyYS5jaGF2YXpAaGN1Z2UuY2g=
†ORCID:
Lara Chavaz
orcid.org/0000-0002-8253-465X
Laurent Cimasoni
orcid.org/0000-0003-2586-932X
Johanna A. Kremer Hovinga
orcid.org/0000-0002-1300-7135
Paul Coppo
orcid.org/0000-0002-4618-2095
Marc Ansari
orcid.org/0000-0002-9649-6498
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.