
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 09 September 2024
Sec. Neonatology
Volume 12 - 2024 | https://doi.org/10.3389/fped.2024.1441891
This article is part of the Research TopicThe Effects of Emerging and Commonly Used Medications on the Developing BrainView all 5 articles
 Eric Witherspoon1
Eric Witherspoon1 Nicholas Zuczek1
Nicholas Zuczek1 Gabrielle Williams1
Gabrielle Williams1 Briana Bernstein2
Briana Bernstein2 Anjik Ghosh1Marko Culjat1
Anjik Ghosh1Marko Culjat1 Suhasini Kaushal1
Suhasini Kaushal1 Patrick A. Forcelli1,2,3*
Patrick A. Forcelli1,2,3*
Introduction: Exposure to a range of anti-seizure medications (ASMs) during early brain development adversely impacts neurodevelopmental outcomes in both animal models and in clinical studies. Many ASMs, including phenobarbital, phenytoin, valproate (VPA), and benzodiazepines, are associated with acute neurotoxicity (cell death), impaired synaptic development, and long-term behavioral changes following gestational or neonatal exposure in animals. This is mirrored in clinical studies which show lasting neurodevelopmental deficits following early-life or gestational exposure to these drugs. Brivaracetam (BRV) and perampanel (PER) are two newer generation anti-seizure medications and are of interest based on their mechanisms of action (SV2A modulator, AMPA antagonist, respectively), as other drugs with these mechanisms of action do not trigger acute neurotoxicity. Both BRV and PER show anti-seizure efficacy in developing animals, but potential neurotoxicity of these drugs is unexplored.
Methods: To address this gap, we treated postnatal day (P)7 Sprague-Dawley rats with BRV (20, 40, 80 mg/kg) and PER (0.1, 0.9, 2.7 mg/kg), and assessed the induction of cell death across a range of vulnerable brain regions 24 h after exposure. Cell death was assessed using pathogreen staining.
Results: In each of the regions examined (dorsal striatum, nucleus accumbens, motor cortex, cingulate cortex, lateral thalamus, septum, hippocampus), VPA, which served as a positive control, significantly increased cell death as measured by the numer of pathogreen positive cells. By contrast, neither BRV nor PER increased the number of pathogreen positive cells in any region examined.
Discussion: Our results suggest that BRV and PER may have a positive safety profile–at least with respect to acute induction of cell death - and therefore may offer a safer option for the treatment of early life seizures.
The treatment of seizures during critical periods of brain development poses a unique set of challenges—both the treatment of neonatal seizures, and the treatment of pregnant women with epilepsy requires balancing seizure control with the potential for adverse effects of pharmacotherapy on the developing brain.
Many classic and first-line anti-seizure medications (ASMs) are associated with both acute and long-term toxicities (1, 2). One notably acute toxicity shared by many ASMs is the acute induction of cell death in postnatal day (P)7 rats. Valproate, phenobarbital, phenytoin, diazepam, clonazepam, lamotrigine, vigabatrin—as well as ethanol and anesthetic agents, all induce acute neurotoxicity after a single exposure at this age. The induction of neuronal apoptosis by these drugs occurs during a narrow postnatal window of brain development in rodents, with a peak around P7 (3–5). This timepoint, which corresponds to the peak of the brain growth spurt (6), models a period spanning from the third trimester through early infancy in humans.
Many of these drugs are also associated with impaired synaptic development, and long-term behavioral disruption (7–10). These findings have been mirrored in clinical studies, which show long-term alterations in brain volume (11), and neurodevelopmental delays following in utero or post-natal treatment (12, 13).
Fortunately, therapeutic agents like levetiracetam (LEV) have grown in use due to its favorable clinical safety and efficacy profile. Moreover, LEV is one of only a few ASMs that does not trigger cell death in the developing brain, suggesting that it may have a more benign safety profile (14). While many ASMs have been examined in preclinical assays for developmental neurotoxicity following acute drug exposure, many remain unexplored. Two drugs of considerable interest are brivaracetam and perampanel.
Brivaracetam, like levetiracetam, is an antagonist at the synaptic-vesicle 2A (SV2A) site (15). Moreover, brivaracetam has shown preclinical and clinical efficacy against neonatal seizures (16–18) making it a promising drug candidate. Perampanel is likewise of interest; it is a selective antagonist of α-amino−3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, and displays efficacy over a range of postnatal ages in rodents (16). It is currently approved for pediatric patients with focal onset seizures older than 4 years of age (19). While N-methyl-D-aspartate (NMDA) receptor antagonists are potent inducers of apoptosis in neonatal animals, other AMPA receptor antagonists do not induce cell death (4).
In the present study we sought to determine if acute exposure to brivaracetam or perampanel would induce cell death in the P7 rat brain. Whether perampanel and brivaracetam would share the neurotoxic profile described for many other ASMs (see above), or rather would match the safer profile seen for other drugs in their respective classes remains unknown. To address this gap, we treated P7 rat pups with brivaracetam or perampanel, and measured induction of cell death 24 h after treatment.
Timed-pregnant Sprague-Dawley rats were acquired from Harlan/Envigo (Frederick, MD) on gestational day 15 and housed in the Department of Comparative Medicine (DCM) at Georgetown University. Animals were kept in a temperature-controlled environment at 21°C under a 12-hour light cycle (Lights on: 6:00–18:00 h). All procedures were performed in accordance with protocol (#2016-1306), which was approved by the Georgetown University Animal Care and Use Committee. The day of birth was defined as postnatal day (P) 0. On postnatal day (P) 7, pups were treated with drugs and euthanized 24 h later at postnatal day (P) 8. P7 rats were chosen as this age and model corresponds to a period spanning the late third trimester through early infancy in humans (6). Moreover, P7 also represents the developmental age where there is maximum proapoptotic effects induced by ASMs, anesthetics, and ethanol (3, 4, 20–22), and is the time that has been examined for essentially all of the ASMs that have been evaluated for pro-apoptotic effects. This allows for a direct comparison between our present findings and prior studies. All brain tissue was processed at the same time and treatments were counterbalanced across sex and within litters.
Vehicle solution was prepared using the following combination: methylparaben (0.2 mg/ml), citric acid (0.18 mg/ml), sodium citrate dihydrate (0.58 mg/ml), sodium carboxymethylcellulose (1 mg/ml), sucralose (8 mg/ml), sorbitol (48 mg/ml), and glycerin (30.4 mg/ml). Vehicle stock solution was then diluted with deionized water in a 1:4 ratio to maintain the osmolarity at <1,000 mOsm/dl.
Valproate was used as a positive control for the cell death assay (see below) as it is a well-established neurotoxic agent in the developing brain (3, 23). Valproate was diluted in vehicle solution at a concentration of 40 mg/ml to deliver a dose of 400 mg/kg intraperitoneally (ip), which is consistent with prior reports (3, 23, 24).
Brivaracetam was obtained as a stock solution from Union Chimique Belge (UCB; Brussels, Belgium) and diluted in vehicle to an initial concentration of 8 mg/ml. 4 and 2 mg/ml solutions of brivaracetam were then prepared using a 1:2 serial dilution to deliver final doses of 80, 40, and 20 mg/kg (ip) of drug.
Perampanel was obtain from Eisai (Woodcliff Lake, New Jersey) and diluted from 0.5 mg/mL stock solution into vehicle to make an initial concentration of .27 mg/ml drug. A 1:9 and 1:3 serial dilution was then prepared to make a final concentration of 0.09 and 0.01 mg/ml of perampanel, respectively. Final doses of perampanel were delivered at of 2.7, 0.9, and 0.1 mg/kg (ip) bodyweight. All drugs were administered intraperitoneally (ip) at a volume of 0.01 ml/g bodyweight and fell within the anticonvulsant dose range in neonatal rodents that we have previously reported (16).
The dose range we used for both brivaracetam and perampanel included a dose that was higher than the therapeutically relevant range by allometric scaling. We used the allometric scaling approach described in (25), which converts a rat dose (in mg/kg) to a human dose (in mg/kg) by dividing the animal dose by 6.2. For brivaracetam, our highest dose was 80 mg/kg. The human equivalent dose (HED) for this treatment was 12.9 mg/kg. Note that the label recommends 6 mg/kg/day in pediatric patients weighing less than 11 kg (26). Thus, our highest dose was twice the recommended maximum dose. For perampanel, our highest dose was 2.7 mg/kg. The HED for this treatment was 0.435 mg/kg. The label recommends a starting dose of 2 mg/day with a maximum of 12 mg/day (27). For an adult, the scaled dose would result in a dose of 26 mg/day. There are no established dosing guidelines for patients under the age of 12.
Thus, the high end of the dose range we selected for each drug represents a extreme dose and was chosen as the most robust test of induction of toxicity.
Twenty-four hours (24 h) following drug treatment, pups were anesthetized with an overdose of euthasol (40 mg/kg) and perfused with phosphate buffered saline followed by 4% paraformaldehyde. Brains were extracted and fixed in paraformaldehyde for 24 h before being transferred to a 30% sucrose solution for three days. The 24 h survival time is consistent with prior studies using late-stage markers of cell death such as fluorojade, TUNEL, and silver staining to detect drug-induced cell death in neonatal rats (3, 23, 28–30).
Following cryoprotection in sucrose, brains were sliced coronally at 35 microns thick sections using a Leica CM1800 cryostat.
Pathogreen histofluorescent dye (Biotin, catalog #: 80027) was applied to detect degenerating neuronal cell bodies. The procedure consisted of several steps involving submerging slides in decreasing concentrations of ethanol (80% and 70%) followed by submersion of slides in diH20 for 2 min. Slides were then placed in 0.06%KmO4 for roughly 10mins followed by an additional submersion in diH20 for 2 min, where the diH20 step was done twice. Pathogreen was made according to the manufacturer's protocol (PI-80027) at a 1:1,000 mix and applied to slides containing tissue samples for roughly 10mins. Slides were again washed with diH20 and air-dried prior to imaging.
Photomicrographs were taken at 10× magnification from 3 consecutive brain regions using a Nikon 80i light microscope. The neonatal rat brain atlas (31) was used as a guide to define the following brain regions of interest (ROI): striatum, septum, cingulate cortex, nucleus accumbens, motor cortex, lateral thalamus, and hippocampus. These regions were selected based on prior studies from our group and others (3, 32) demonstrating vulnerability to ASM induced cell death.
QuPath v0.3.2 software was used to automate counting of pathogreen-positive cells and the total number of positive cells was averaged across the three consecutive brain regions imaged. Analysis was also performed using 2 observers who were blind to treatment groups.
GraphPad Prism 9 (GraphPad Software; La Jolla, Ca) was used for statistical analysis of histology data. To assess if the data were normally distributed across groups, we used the Shapiro Wilk normality test. The number of pathogreen positive cells were analyzed using the Analysis of Variance (ANOVA) followed by Holm-Sidak multiple comparisons test. P-values <0.05 were considered statistically significant.
To evaluate the safety profile of brivaracetam and perampanel with respect to induction of cell death in the neonatal rodent brain, we examined cortical, subcortical, and limbic regions.
Vehicle treated rats displayed a low baseline level of pathogreen-positive cells in basal ganglia sub-regions (i.e., striatum, nucleus accumbens) 24 h after pretreatment (Figure 1). As expected, when pups were treated with valproate (400 mg/kg; positive control), the number of pathogreen positive cells within the striatum was significantly elevated (Figure 1B, P = 0.0001, Holm-Sidak; 4.7-fold increase).
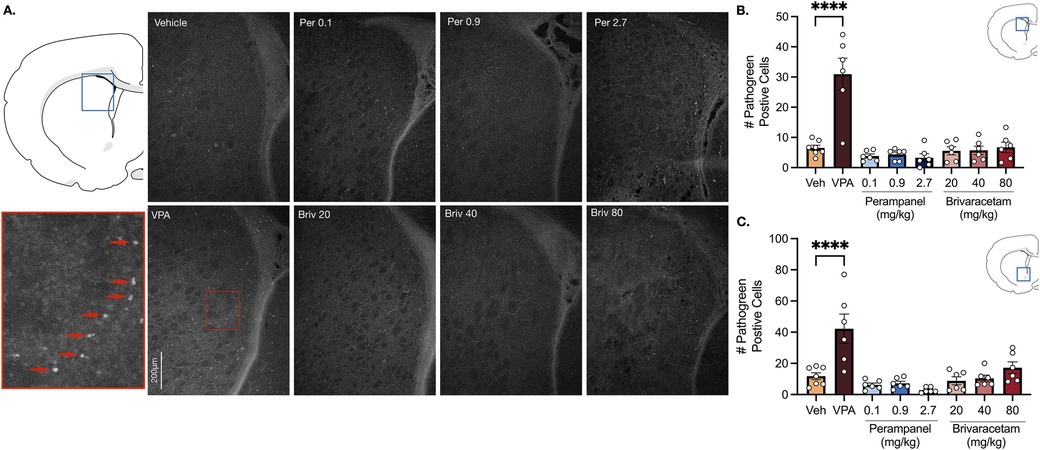
Figure 1. Exposure to valproate (VPA) but not brivaracetam (BRIV) or perampanel (PER) induces cell death in basal ganglia regions. (A) Photomicrographs displaying pathogreen-positive cell bodies in the striatum of postnatal day 8 rats. Blue box = representative region of interest of captured photomicrograph; red box = magnified striatal photomicrograph with positive-pathogreen cell bodies indicated by red arrows (B) mean (± SEM) number of pathogreen-positive cells. Valproate-treated rodents displayed higher levels of neurodegeneration compared to vehicle-treated groups (p < 0.0001, Holm-Sidak). Scale bar = 200 μm. (Veh n = 7; VPA n = 6; PER 0.1 mg/kg n = 6; PER 0.9 mg/kg n = 6; PER 2.7 mg/kg n = 6; BRIV 20 mg/kg n = 6; BRIV 40 mg/kg n = 6; BRIV 80 mg/kg n = 6) (C) Mean (± SEM) number of pathogreen-positive cells in nucleus accumbens. VPA-treated animals had significantly higher levels of degenerating neurons compared to vehicle-treated groups (p < 0.0001, Holm-Sidak). Scale bar = 200 μm. (Veh n = 7; VPA n = 6; PER 0.1 mg/kg n = 6; PER 0.9 mg/kg n = 6; PER 2.7 mg/kg n = 6; BRIV 20 mg/kg n = 6; BRIV 40 mg/kg n = 6; BRIV 80 mg/kg n = 6).
Analysis of variance (ANOVA) revealed a main of effect of drug (F7,41 = 18.05, P = 0.0001), which was driven by the valproate condition. No significant difference was observed after exposure to perampanel (0.1, 0.9, 2.7 mg/kg) or brivaracetam (20, 40, 80 mg/kg) compared to vehicle treated animals (Figure 1B, Ps > 0.86, Holm-Sidak).
Similar to the striatum, we found low levels of pathogreen-positive cells in the nucleus accumbens after vehicle treatment (Figure 1C). When postnatal-day (P) 7 pups were treated with valproate (400 mg/kg), we observed a significant (3.6-fold) increase compared to control groups (Figure 1C, P = 0.0001, Holm-Sidak). While analysis of variance (ANOVA) revealed a main effect of drug (F7,41 = 10.23, P = 0.0001), again driven by the valproate condition, neither perampanel nor brivaracetam increased the number of pathogen positive cells relative to vehicle controls, at any dose tested (Figure 1C, Ps > 0.47, Holm-Sidak).
In the motor cortex, vehicle treated animals displayed low levels of neuronal cell death (Figure 2B). Valproate induced a 5.2-fold increase in pathogreen positive cells (Figure 2B, F7,41 = 6.94, P = 0.0001; P = 0.0001, Holm-Sidak), an effect that was not observed after treatment with either perampanel or brivaracetam (Figure 2B, Ps > 0.99; Holm-Sidak corrected). We found a similar pattern in the cingulate cortex (Figure 2C). Exposure following valproate pretreatment revealed a 4.3-fold increase in cell death (P = 0.0081, Holm-Sidak), while no significant difference was observed after exposure to perampanel or brivaracetam (Figure 2C, Ps > 0.98, F7,41 = 4.56, Holm-Sidak).
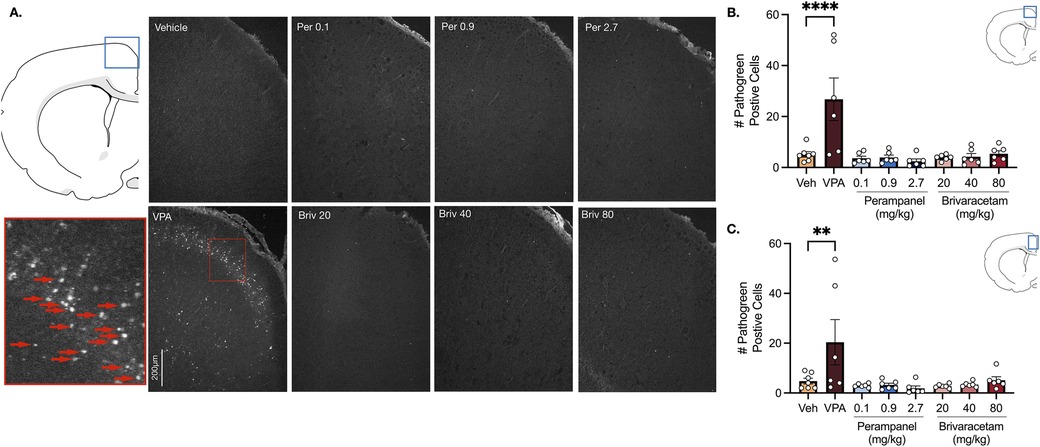
Figure 2. Exposure to valproate (VPA) but not brivaracetam (BRIV) or perampanel (PER) induces cell death in cortical regions. (A) Photomicrographs displaying pathogreen-positive cell bodies in the motor cortex of postnatal day 8 rats. Blue box = representative region of interest of captured photomicrograph; red box = magnified photomicrograph with positive-pathogreen cell bodies indicated by red arrows (B) mean (± SEM) number of pathogreen-positive cells in motor cortex. VPA-treated animals displayed significantly higher levels of pathogreen positive compared to vehicle-treated groups (p < 0.0001, Holm-Sidak). Scale bar = 200 μm. (Veh n = 7; VPA n = 6; PER 0.1 mg/kg n = 6; PER 0.9 mg/kg n = 6; PER 2.7 mg/kg n = 6; BRIV 20 mg/kg n = 6; BRIV 40 mg/kg n = 6; BRIV 80 mg/kg n = 6). (C) Mean (± SEM) number of pathogreen-positive cells in the cingulate cortex. VPA-treated animals had significantly higher levels of pathogreen-positive compared to vehicle-treated groups (p = 0.0081, Holm-Sidak). Scale bar = 200 μm. (Veh n = 7; VPA n = 6; PER 0.1 mg/kg n = 6; PER 0.9 mg/kg n = 6; PER 2.7 mg/kg n = 6; BRIV 20 mg/kg n = 6; BRIV 40 mg/kg n = 6; BRIV 80 mg/kg n = 6).
Analysis of variance (ANOVA) revealed a main effect of drug treatment in the lateral thalamus (Figure 3B, F7,41 = 20.69, P < 0.0001), septum (Figure 3C, F7,41 = 10.70 P < 0.0001) and hippocampus (Figure 3D, F7,41 = 6.12, P < 0.0001). Holm-Sidak multiple comparisons test confirmed that valproate (400 mg/kg) significantly increased cell death in all limbic (i.e., hippocampus, septum) and subcortical (i.e., lateral thalamus) regions compared to vehicle-treated controls (Figure 3, Ps for the Holm Sidak here). Neither perampanel (0.1, 0.9, 2.7 mg/kg) or brivaracetam (20, 40, 80 mg/kg) significantly increased cell death in the lateral thalamus (Figure 3B, Ps > 0.99, Holm-Sidak), septum (Figure 3C, Ps > 0.57, Holm-Sidak) or hippocampus (Figure 3D, Ps > 0.95, Holm-Sidak) compared to vehicle treated controls.
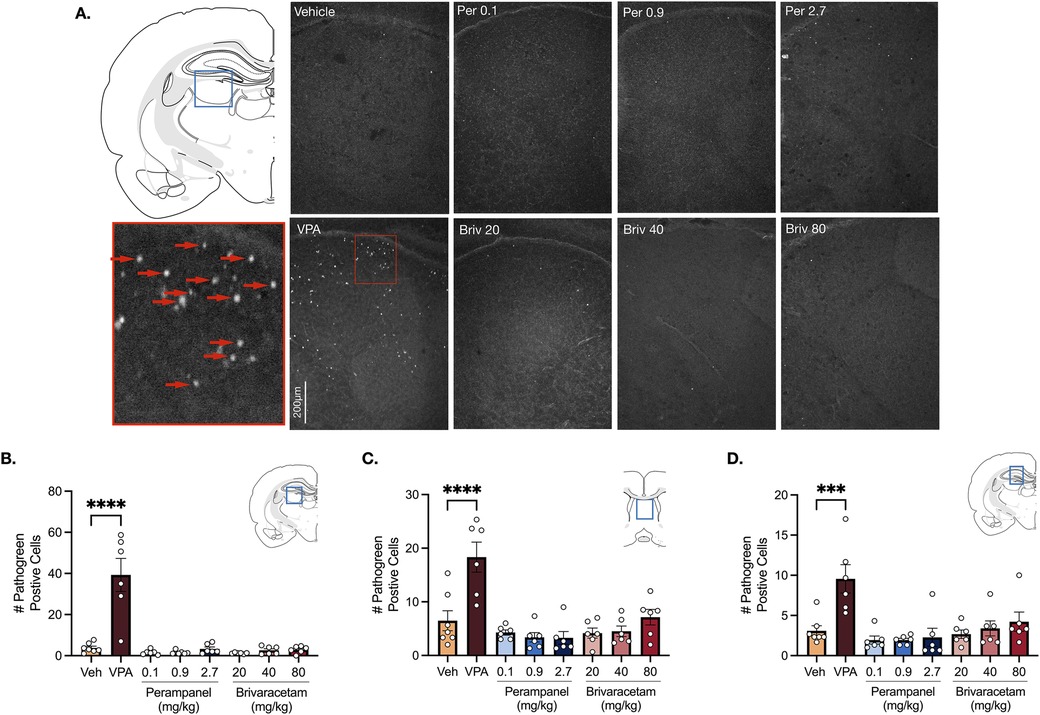
Figure 3. Exposure to valproate (VPA) but not brivaracetam (BRIV) or perampanel (PER) induces cell death in limbic and subcortical regions. (A) Photomicrographs displaying pathogreen-positive cell bodies in the thalamus of postnatal day 8 rats. Blue box = representative region of interest of captured photomicrograph; red box = magnified photomicrograph with positive-pathogreen cell bodies indicated by red arrows. (B) Mean (± SEM) number of pathogreen-positive cells in the lateral thalamus. VPA-treated animals had significantly higher levels of pathogreen-positive cells compared to vehicle-treated groups (p < 0.0001, Holm-Sidak). Scale bar = 200 μm. (Veh n = 7; VPA n = 6; PER 0.1 mg/kg n = 6; PER 0.9 mg/kg n = 6; PER 2.7 mg/kg n = 6; BRIV 20 mg/kg n = 6; BRIV 40 mg/kg n = 6; BRIV 80 mg/kg n = 6). (C) Mean (± SEM) number of pathogreen-positive cells in the septum. VPA-treated pups had significantly higher levels of pathogreen positive cells compared to vehicle-treated groups (p < 0.0001, Holm-Sidak). Scale bar = 200 μm. (Veh n = 7; VPA n = 6; PER 0.1 mg/kg n = 6; PER 0.9 mg/kg n = 6; PER 2.7 mg/kg n = 6; BRIV 20 mg/kg n = 6; BRIV 40 mg/kg n = 6; BRIV 80 mg/kg n = 6). (D) Mean (± SEM) number of pathogreen-positive cells in hippocampus. VPA-treated pups had significantly higher levels of pathogreen positive cells compared to vehicle-treated groups (p = 0.0001, Holm-Sidak). Scale bar = 200 μm. (Veh n = 7; VPA n = 6; PER 0.1 mg/kg n = 6; PER 0.9 mg/kg n = 6; PER 2.7 mg/kg n = 6; BRIV 20 mg/kg n = 6; BRIV 40 mg/kg n = 6; BRIV 80 mg/kg n = 6).
Here, we report a lack of significant cell death induced by brivaracetam and perampanel in neonatal (P7) rodents. Consistent with prior reports, we found that valproate, a prototypical antiseizure drug, induced robust cell death in limbic, basal ganglia, cortical, and subcortical regions. By contrast, brivaracetam and perampanel did not elicit the same effect at doses that are effective at suppressing seizures in early stages of postnatal development (16).
Brivaracetam, like levetiracetam also selectively binds to synaptic vesicle 2A (SV2A) sites, resulting in a reduction in excitatory neurotransmitter release during enhanced neuronal excitability (33). Our group as well as others have extensively reported that levetiracetam does not trigger cell death in neonatal rodents (14, 23, 34).
Similarly, while it is well established that drugs that target glutamatergic neurotransmission (i.e., particularly NMDA receptors) display efficacy against neonatal seizures (35), the impact of NMDA receptor antagonist on neuronal apoptosis in neonatal rodents is unfavorable (30, 36). For example, MK801—a common NMDA antagonist—protects against kainic-acid induced seizures, but also induces significant apoptosis in cortical and subcortical regions in the immature brain (30, 37). By contrast, other AMPA receptor antagonists do not induce cell death in neonatal rodents (38, 39).
In some respects, our data while encouraging, are not surprising. While mechanism or drug-class specific safety/toxicity profiles would expedite neurotoxicity assessment, unfortunately, drug mechanism of action is not a perfect predictor of neurotoxic potential. For example, voltage gated sodium channel blockers display wide-ranging profiles with respect to cell death in neonatal animals. While phenytoin triggers cell death at therapeutic doses, lamotrigine and carbamazepine only triggers cell death at supratherapeutic doses, or, in the case of lamotrigine, when given as part of a polytherapeutic cocktail.
Brivaracetam has been only sparsely investigated in preclinical models of neonatal seizures. However, pharmacokinetic studies show that brivaracetam has a greater lipophilicity and is roughly 20 times more potent than its analog levetiracetam (15, 40). In the clinical setting, brivaracetam displays efficacy coupled with a favorable side effect profile at nearly 10 times lower than the concentration of levetiracetam (41, 42).
In the present study, we examined only one marker of cell death, pathogreen histoflourescence. This approach, which is similar to fluorojade staining (43) marks degenerating neurons. We and others have previously used a wide range of methods for detecting cell death following neonatal anti-seizure medication exposure, including TUNEL staining, Amino Cupric Silver staining, morphology by electron microscopy, activated caspse-3 immunohistochemistry, and fluorojade staining. The profile across all of these approaches has been uniformly consistent, suggesting that these findings are not specific to a particular method of cell death detection.
One obvious limitation of the present study is that while there is a lack of acute cell death following brivaracetam and perampanel exposure, it has yet to be determined what impact chronic exposure to these ASMs has on neurotoxicity. Whether either BRIV or PER would display cell death with even higher doses, or as part of a polytherapy cocktail remains to be explored. Moreover, cell death likely only represents the “tip of the iceberg” in terms of developmental neurotoxicity. We have previously found that drug doses that are below the threshold for induction of cell death still can impair synaptic development (8, 36). Moreover, many ASMs produce long-term effects on behavior following early life exposure in rodent models (9, 36, 44). Whether or not brivaracetam or perampanel would show a safe profile with respect to synaptic or behavioral functions remains unknown. However, at least with respect to the induction of cell death, which is observed across a wide range of ASMs, brivaracetam and perampanel have a safer profile. These drugs merit further exploration for the treatment of neonatal seizures and seizures in pregnant women with epilepsy, including further evaluation of long-term outcomes following drug exposure.
Here we find that perampanel and brivaracetam do not increase cell death in neonatal rats, even at doses that extend into the supratherapeutic range. This finding is strikingly different from many other ASMs, but mirrors the established safety profile of AMPA antagonists and SV2A modulators in the developing brain. These data suggest promise for these drugs for use during critical periods of brain development, and open the door to studies examining longer term-exposure as well as behavioral outcomes. If these drugs continue to display a favorable toxicity profile, clinical examination of them for use in neonates and pregnancy may be warranted.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was approved by Georgetown University Animal Care and Use committee. The study was conducted in accordance with the local legislation and institutional requirements.
EW: Data curation, Formal Analysis, Project administration, Supervision, Writing – review & editing, Investigation, Methodology, Visualization, Writing – original draft. NZ: Investigation, Writing – review & editing. GW: Investigation, Writing – review & editing. BB: Investigation, Writing – review & editing. AG: Investigation, Writing – review & editing. MC: Investigation, Writing – review & editing, Conceptualization. SK: Conceptualization, Writing – review & editing. PF: Conceptualization, Writing – review & editing, Data curation, Formal Analysis, Funding acquisition, Project administration, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grant R01HD091994 from the National Institutes of Health (NIH)/Eunice Kennedy Schriver National Institute of Child Health and Human Development (NICHD) to PF. EW was supported by grant T32GM142520 from the National Institutes of Health (NIH)/National Institute of General Medical Sciences (NIGMS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Berg AT, Rychlik K. The course of childhood-onset epilepsy over the first two decades: a prospective, longitudinal study. Epilepsia. (2015) 56(1):40–8. doi: 10.1111/epi.12862
2. Ikonomidou C, Turski L. Antiepileptic drugs and brain development. Epilepsy Res. (2010) 88(1):11–22. doi: 10.1016/j.eplepsyres.2009.09.019
3. Bittigau P, Sifringer M, Genz K, Reith E, Pospischil D, Govindarajalu S, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci U S A. (2002) 99(23):15089–94. doi: 10.1073/pnas.222550499
4. Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. (1999) 283(5398):70–4. doi: 10.1126/science.283.5398.70
5. Soriano SG, Liu Q, Li J, Liu J-R, Han XH, Kanter JL, et al. Ketamine activates cell cycle signaling and apoptosis in the neonatal rat brain. Anesthesiology. (2010) 112(5):1155–63. doi: 10.1097/ALN.0b013e3181d3e0c2
6. Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. (1979) 3(1):79–83. doi: 10.1016/0378-3782(79)90022-7
7. Al-Muhtasib N, Sepulveda-Rodriguez A, Vicini S, Forcelli PA. Neonatal phenobarbital exposure disrupts GABAergic synaptic maturation in rat CA1 neurons. Epilepsia. (2018) 59(2):333–44. doi: 10.1111/epi.13990
8. Forcelli PA, Janssen MJ, Vicini S, Gale K. Neonatal exposure to antiepileptic drugs disrupts striatal synaptic development. Ann Neurol. (2012) 72(3):363–72. doi: 10.1002/ana.23600
9. Forcelli PA, Kozlowski R, Snyder C, Kondratyev A, Gale K. Effects of neonatal antiepileptic drug exposure on cognitive, emotional, and motor function in adult rats. J Pharmacol Exp Ther. (2012) 340(3):558–66. doi: 10.1124/jpet.111.188862
10. Gutherz SB, Kulick CV, Soper C, Kondratyev A, Gale K, Forcelli PA. Brief postnatal exposure to phenobarbital impairs passive avoidance learning and sensorimotor gating in rats. Epilepsy Behav. (2014) 37:265–9. doi: 10.1016/j.yebeh.2014.07.010
11. Ikonomidou C, Scheer I, Wilhelm T, Juengling FD, Titze K, Stöver B, et al. Brain morphology alterations in the basal ganglia and the hypothalamus following prenatal exposure to antiepileptic drugs. Eur J Paediatr Neurol. (2007) 11(5):297–301. doi: 10.1016/j.ejpn.2007.02.006
12. Baker GA, Bromley RL, Briggs M, Cheyne CP, Cohen MJ, García-Fiñana M, et al. IQ at 6 years after in utero exposure to antiepileptic drugs: a controlled cohort study. Neurology. (2015) 84(4):382–90. doi: 10.1212/WNL.0000000000001182
13. Meador KJ, Baker GA, Browning N, Cohen MJ, Bromley RL, Clayton-Smith J, et al. Fetal antiepileptic drug exposure and cognitive outcomes at age 6 years (NEAD study): a prospective observational study. Lancet Neurol. (2013) 12(3):244–52. doi: 10.1016/S1474-4422(12)70323-X
14. Kaushal S, Tamer Z, Opoku F, Forcelli PA. Anticonvulsant drug induced cell death in the developing white matter of the rodent brain. Epilepsia. (2016) 57(5):727–34. doi: 10.1111/epi.13365
15. Nicolas J-M, Hannestad J, Holden D, Kervyn S, Nabulsi N, Tytgat D, et al. Brivaracetam, a selective high-affinity synaptic vesicle protein 2A (SV2A) ligand with preclinical evidence of high brain permeability and fast onset of action. Epilepsia. (2016) 57(2):201–9. doi: 10.1111/epi.13267
16. Culjat M, Huizenga MN, Forcelli PA. Age-dependent anticonvulsant actions of perampanel and brivaracetam in the methyl-6,7-dimethoxy-4-ethyl-beta-carboline-3-carboxylate (DMCM) model of seizures in developing rats. Pharmacol Rep. (2021) 73(1):296–302. doi: 10.1007/s43440-020-00189-w
17. McGuire S, Silva G, Lal D, Khurana DS, Legido A, Hasbani D, et al. Safety and efficacy of brivaracetam in pediatric refractory epilepsy: a single-center clinical experience. J Child Neurol. (2020) 35(2):102–5. doi: 10.1177/0883073819879276
18. Nissenkorn A, Tzadok M, Bar-Yosef O, Ben-Zeev B. Treatment with brivaracetam in children—the experience of a pediatric epilepsy center. Epilepsy Behav. (2019) 101:106541. doi: 10.1016/j.yebeh.2019.106541
19. FYCOMPA® Newly Approved By U.S. FDA As Treatment For Partial-Onset Seizures In Pediatric Patients With Epilepsy | News Release: 2018. (n.d.). Eisai Co., Ltd. Available online at: https://www.eisai.com/news/2018/news201879.html (Accessed December 7, 2023).
20. Bittigau P, Sifringer M, Ikonomidou C. Antiepileptic drugs and apoptosis in the developing brain. Ann N Y Acad Sci. (2003) 993(1):103–14. doi: 10.1111/j.1749-6632.2003.tb07517.x
21. Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. (2000) 287(5455):1056–60. doi: 10.1126/science.287.5455.1056
22. Olney JW, Tenkova T, Dikranian K, Qin Y-Q, Labruyere J, Ikonomidou C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res. (2002) 133(2):115–26. doi: 10.1016/s0165-3806(02)00279-1
23. Kim J, Kondratyev A, Gale K. Antiepileptic drug-induced neuronal cell death in the immature brain: effects of carbamazepine, topiramate, and levetiracetam as monotherapy versus polytherapy. J Pharmacol Exp Ther. (2007) 323(1):165–73. doi: 10.1124/jpet.107.126250
24. Kabakus N, Ay I, Aysun S, Söylemezoglu F, Ozcan A, Celasun B. Protective effects of valproic acid against hypoxic-ischemic brain injury in neonatal rats. J Child Neurol. (2005) 20(7):582–7. doi: 10.1177/08830738050200070801
25. Center for Drug Evaluation and Research (CDER). Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Silver Spring, MD: U.S. Department of Health and Human Services Food and Drug Administration (2005). Available online at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estimating-maximum-safe-starting-dose-initial-clinical-trials-therapeutics-adult-healthy-volunteers (Accessed August 5, 2023).
26. Briviact (brivaracetam) Label—Accessdata.fda.gov. (n.d.). Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/205836s009,205837s007,205838s006lbl.pdf (Accessed July 22, 2024).
27. FYCOMPA (perampanel) Label—Accessdata.fda.gov. (n.d.). Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/202834s011lbl.pdf (Accessed July 22, 2024).
28. Brown L, Gutherz S, Kulick C, Soper C, Kondratyev A, Forcelli PA. Profile of retigabine-induced neuronal apoptosis in the developing rat brain. Epilepsia. (2016) 57(4):660–70. doi: 10.1111/epi.13335
29. Ghosh A, Quinlan S, Forcelli PA. Anti-seizure medication-induced developmental cell death in neonatal rats is unaltered by history of hypoxia. Epilepsy Res. (2024) 201:107318. doi: 10.1016/j.eplepsyres.2024.107318
30. Katz I, Kim J, Gale K, Kondratyev A. Effects of lamotrigine alone and in combination with MK-801, phenobarbital, or phenytoin on cell death in the neonatal rat brain. J Pharmacol Exp Ther. (2007) 322(2):494–500. doi: 10.1124/jpet.107.123133
31. Ramachandra R, Subramanian T. Atlas of the Neonatal Rat Brain. Boca Raton, FL: CRC Press (2011). p. 1–164.
32. Forcelli PA, Kim J, Kondratyev A, Gale K. Pattern of antiepileptic drug-induced cell death in limbic regions of the neonatal rat brain. Epilepsia. (2011) 52(12):e207–11. doi: 10.1111/j.1528-1167.2011.03297.x
33. Steinhoff BJ, Staack AM. Levetiracetam and brivaracetam: a review of evidence from clinical trials and clinical experience. Ther Adv Neurol Disord. (2019) 12:1756286419873518. doi: 10.1177/1756286419873518
34. Komur M, Okuyaz C, Celik Y, Resitoglu B, Polat A, Balci S, et al. Neuroprotective effect of levetiracetam on hypoxic ischemic brain injury in neonatal rats. Childs Nerv Syst. (2014) 30(6):1001–9. doi: 10.1007/s00381-014-2375-x
35. Mares P, Mikulecká A. Different effects of two N-methyl-D-aspartate receptor antagonists on seizures, spontaneous behavior, and motor performance in immature rats. Epilepsy Behav. (2009) 14(1):32–9. doi: 10.1016/j.yebeh.2008.08.013
36. Stefovska VG, Uckermann O, Czuczwar M, Smitka M, Czuczwar P, Kis J, et al. Sedative and anticonvulsant drugs suppress postnatal neurogenesis. Ann Neurol. (2008) 64(4):434–45. doi: 10.1002/ana.21463
37. Stafstrom CE, Tandon P, Hori A, Liu Z, Mikati MA, Holmes GL. Acute effects of MK801 on kainic acid-induced seizures in neonatal rats. Epilepsy Res. (1997) 26(2):335–44. doi: 10.1016/s0920-1211(96)00904-7
38. Aujla PK, Fetell MR, Jensen FE. Talampanel suppresses the acute and chronic effects of seizures in a rodent neonatal seizure model. Epilepsia. (2009) 50(4):694–701. doi: 10.1111/j.1528-1167.2008.01947.x
39. Koh S, Tibayan FD, Simpson JN, Jensen FE. NBQX or topiramate treatment after perinatal hypoxia-induced seizures prevents later increases in seizure-induced neuronal injury. Epilepsia. (2004) 45(6):569–75. doi: 10.1111/j.0013-9580.2004.69103.x
40. Gillard M, Fuks B, Leclercq K, Matagne A. Binding characteristics of brivaracetam, a selective, high affinity SV2A ligand in rat, mouse and human brain: relationship to anti-convulsant properties. Eur J Pharmacol. (2011) 664(1):36–44. doi: 10.1016/j.ejphar.2011.04.064
41. Klitgaard H, Matagne A, Nicolas J-M, Gillard M, Lamberty Y, De Ryck M, et al. Brivaracetam: rationale for discovery and preclinical profile of a selective SV2A ligand for epilepsy treatment. Epilepsia. (2016) 57(4):538–48. doi: 10.1111/epi.13340
42. Yates SL, Fakhoury T, Liang W, Eckhardt K, Borghs S, D’Souza J. An open-label, prospective, exploratory study of patients with epilepsy switching from levetiracetam to brivaracetam. Epilepsy Behav. (2015) 52(Pt A):165–8. doi: 10.1016/j.yebeh.2015.09.005
43. Neuronal Probes. (n.d.). Retrieved April 1, 2024. Available online at: https://biotium.com/wp-content/uploads/2018/08/Neuronal-Probes.pdf (Accessed April 1, 2024).
Keywords: developmental neurotoxicity (DNT), neonatal seizures, pregnancy, anti-seizure medication, apoptosis, programmed cell death
Citation: Witherspoon E, Zuczek N, Williams G, Bernstein B, Ghosh A, Culjat M, Kaushal S and Forcelli PA (2024) A single exposure to brivaracetam or perampanel does not cause cell death in neonatal rats. Front. Pediatr. 12:1441891. doi: 10.3389/fped.2024.1441891
Received: 31 May 2024; Accepted: 13 August 2024;
Published: 9 September 2024.
Edited by:
Jennifer Burnsed, University of Virginia, United StatesReviewed by:
Raul Chavez-Valdez, Johns Hopkins Medicine, United StatesCopyright: © 2024 Witherspoon, Zuczek, Williams, Bernstein, Ghosh, Culjat, Kaushal and Forcelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick A. Forcelli, cGFmMjJAZ2VvcmdldG93bi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.