 Kun Yang
Kun Yang Ting He
Ting He Wenbin Dong
Wenbin Dong- Department of Neonatology, Children’s Medical Center, The Affiliated Hospital of Southwest Medical University, Luzhou, China
Bronchopulmonary dysplasia is a prevalent respiratory disorder posing a significant threat to the quality of life in premature infants. Its pathogenesis is intricate, and therapeutic options are limited. Besides genetic coding, protein post-translational modification plays a pivotal role in regulating cellular function, contributing complexity and diversity to substrate proteins and influencing various cellular processes. Substantial evidence indicates that post-translational modifications of several substrate proteins are intricately related to the molecular mechanisms underlying bronchopulmonary dysplasia. These modifications facilitate the progression of bronchopulmonary dysplasia through a cascade of signal transduction events. This review outlines the relationships between substrate protein phosphorylation, acetylation, ubiquitination, SUMOylation, methylation, glycosylation, glycation, S-glutathionylation, S-nitrosylation and bronchopulmonary dysplasia. The aim is to provide novel insights into bronchopulmonary dysplasia's pathogenesis and potential therapeutic targets for clinical management.
Introduction
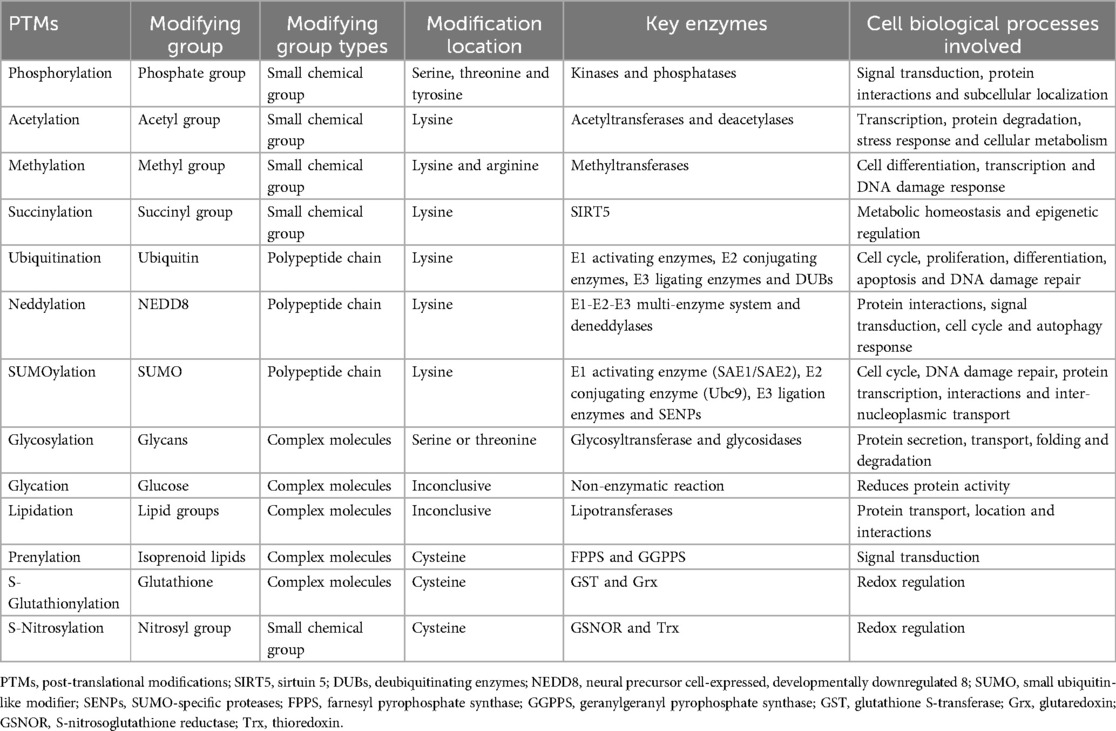
Bronchopulmonary dysplasia (BPD) represents a chronic lung ailment affecting premature infants, characterized by complex pathogenesis and clinical manifestations associated with elevated morbidity and mortality rates (1). Despite remarkable advancements in perinatal medicine, understanding the pathophysiology of BPD remains a formidable challenge. Therefore, investigating BPD's pathogenesis from multiple perspectives is of paramount importance. Post-translational modifications (PTMs) are distinct chemical alterations occurring at amino acid termini or side chains of proteins subsequent to translation. These modifications bestow proteins with high complexity and diverse biological functions. Over 200 PTMs have been identified, most of which are catalyzed by enzymes, with some being non-enzymatic reactions (2). Table 1 lists the characteristics of several common PTMs. A growing body of evidence indicates that PTMs play a significant role in the development of various diseases associated with prematurity, with BPD being of particular interest (6, 7). This review delves into the potential relationships between well-studied PTMs such as phosphorylation, acetylation, ubiquitination, and methylation, and the development of BPD. Beyond elucidating the fundamental principles of these PTMs, the focus lies in elucidating how these modifications influence specific substrate protein characteristics, thus impacting BPD. Understanding the molecular mechanisms of protein PTMs holds significance as it may unveil new avenues for therapeutic strategies in managing BPD.
Phosphorylation
Phosphorylation, one of the most common PTMs, plays a crucial role in various cellular processes. It influences substrate protein interactions, enzyme activity, subcellular localization, half-life and the turnover of target proteins through kinases (which add phosphate groups via phosphorylation) and phosphatases (which remove phosphate groups via dephosphorylation). Phosphorylation predominantly occurs at serine, threonine, and tyrosine residues on substrate proteins (8). In this section, we will discuss the significance of phosphorylation modifications on several substrate proteins, including members of the mitogen-activated protein kinases family, small mothers against decapentaplegic, dynamin-related protein 1, glycogen synthase kinase-3β, and nuclear factor-kappa B, particularly concerning their roles in BPD (Figure 1A).
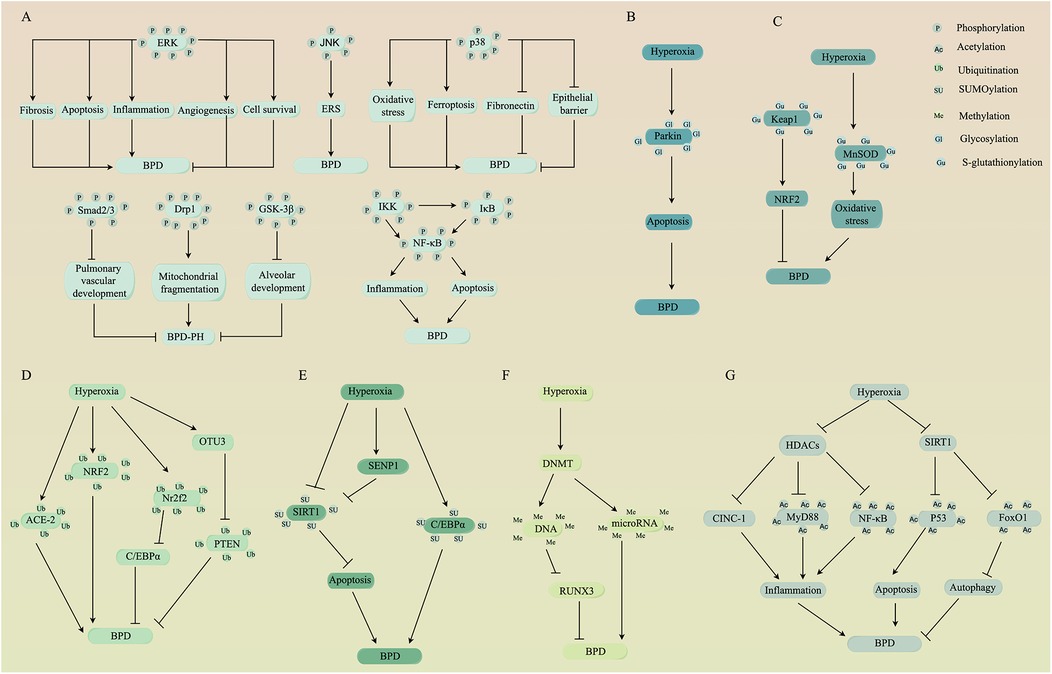
Figure 1. PTMs of several substrate proteins are involved in the pathogenesis of BPD. PTMs including (A) phosphorylation, (B) glycosylation, (C) S-glutathionylation, (D) ubiquitination, (E) SUMOylation, (F) methylation, and (G) acetylation alter the activity of substrate proteins, which can positively or negatively affect BPD through a series of signal transduction events. Schematic created by Figdraw (www.figdraw.com). PTMs, post-translational modifications; BPD, bronchopulmonary dysplasia; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; ERS, endoplasmic reticulum stress; Smad, small mothers against decapentaplegic; Drp1, dynamin-related protein 1; BPD-PH, BPD-pulmonary hypertension; GSK-3β, glycogen synthase kinase-3β; IκB, inhibitor of κB; IKK, inhibitor of κB kinase; NF-κB, nuclear factor-kappa B; Keap1, kelch-like ECH-associated protein 1; NRF2, nuclear factor erythroid 2-related factor 2; MnSOD, manganese superoxide dismutase; ACE-2, angiotensin-converting enzyme-2; Nr2f2, nuclear receptor subfamily 2, group F, member 2; C/EBPα, CCAAT/enhancer-binding protein alpha; OTUs, ovarian tumor proteases; PTEN, phosphatase and tensin homolog; SIRT1, sirtuin 1; SENPs, SUMO-specific proteases; DNMT, DNA methyltransferase; RUNX3, runt-related transcription factor 3; HDACs, histone deacetylases; CINC-1, cytokine-induced neutrophil chemoattractant-1; FoxO1, forkhead box O1.
Mitogen-activated protein kinases (MAPK)
MAPK, a pivotal family of proteins, includes extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 kinase. ERK is primarily associated with cell genesis, proliferation, and differentiation, p38 is responsible for inflammation, apoptosis, and autophagy, while JNK is involved in cellular stress responses, differentiation, proliferation, and apoptosis (9). A growing body of evidence suggests that phosphorylation modifications of MAPK family members are closely intertwined with BPD. Studies have revealed that ERK phosphorylation accelerates lung fibroblast proliferation, transdifferentiation, and migration. This acceleration is achieved by expediting the cell cycle progression from the G1 to S phase and enhancing the synthesis of α-SMA, contributing to hyperoxia-induced lung fibrosis in neonatal rats (10, 11). Additionally, ERK phosphorylation promotes mitochondrial apoptosis and inflammatory pathways, which are significant factors in BPD (12). Conversely, decreased levels of phosphorylated ERK have been found to offer protection against hyperoxia-induced lung injury in neonatal rats (13). These findings suggest that inhibiting ERK phosphorylation may be a potential protective strategy for BPD. However, conflicting studies propose that ERK phosphorylation could mitigate hyperoxia-induced lung injury in mice by promoting angiogenesis (14). Menon et al. (15) have indicated that phosphorylated ERK levels increase early during hyperoxia exposure, potentially as a compensatory mechanism to promote angiogenesis. Nevertheless, sustained hyperoxia reduces phosphorylated ERK, hindering cell cycle progression and, consequently, pulmonary angiogenesis. Moreover, phosphorylated ERK activates cell survival signaling to mitigate hyperoxia-induced damage to lung epithelial cells (16). These contradictory findings underscore the complexity of the role played by phosphorylated ERK in BPD pathogenesis. Thus, it is essential to further investigate whether additional factors influence the impact of phosphorylated ERK in BPD, beyond the considerations related to the study population and exposure factors.
The JNK signaling pathway is closely associated with endoplasmic reticulum stress (ERS), and plays a significant role in the pathogenesis of BPD. Specifically, the phosphorylation of inositol-requiring enzyme 1 alpha (IRE1α) and JNK leads to apoptosis in lung epithelial cells associated with ERS (17). This indicates that JNK signaling activation contributes to cell death and ERS in the context of BPD. Conversely, inhibiting the phosphorylated IRE1α/JNK pathway has been shown to attenuate lung tissue inflammation, oxidative stress and apoptosis in a rat model of hyperoxia-induced lung injury (18). Phosphorylated p38 promotes this transition, which is associated with the development of lung fibrosis. This transition involves the conversion of alveolar epithelial cells (AECs) into mesenchymal-like cells, contributing to tissue scarring (19). Phosphorylated p38 inhibits the expression of claudin-18, a protein that plays a role in maintaining the integrity of the alveolar epithelial barrier. This disruption can lead to increased permeability and tissue damage (20). Activation of p38 signaling can induce ferroptosis, a type of programmed cell death characterized by lipid peroxidation and iron-dependent cell damage (21). Phosphorylated p38 can activate oxidative stress pathways and promote apoptosis in AECs, contributing to tissue injury (22). Additionally, phosphorylated p38 can enhance the expression of IL-33, a cytokine that has the ability to degrade fibronectin via its receptor neutrophil extracellular traps. This process is relevant to the pathological progression of BPD (23). Conversely, down-regulation or inhibition of phosphorylated p38 signaling has shown promise in attenuating BPD in neonatal mice by reducing inflammation (24). This suggests that targeting the p38 pathway could be a therapeutic approach to mitigate the effects of BPD.
Small mothers against decapentaplegic (smad)
Smad proteins are crucial transcription factors that regulate gene expression. They are affected by various modifications, with phosphorylation being a key one. Phosphorylated Smad is a critical component of the transforming growth factor-β (TGF-β) pathway (25), which is closely related to BPD. Studies have identified that the TGF-β/phosphorylated Smad3 signaling pathway can trigger cell death and impair lung development, particularly in mice lacking the chaperone protein GRP78 in the endoplasmic reticulum (26). Elevated levels of phosphorylated Smad2/3 may also contribute to problems with pulmonary blood vessel development in BPD (27). Exposure to high levels of oxygen increases the phosphorylation of Smad2 and Smad3 in lung microvascular endothelial cells, leading to changes that promote endothelial-mesenchymal transition and, subsequently, pulmonary hypertension in mice (28). Therefore, targeting Smad phosphorylation shows promise as a therapeutic approach for BPD. In fact, inhibiting TGF-β-induced Smad2/3 phosphorylation has been shown to delay the development of BPD (29).
Dynamin-related protein 1 (Drp1)
Drp1 is a key regulator of mitochondrial dynamics and undergoes various PTMs, including phosphorylation, SUMOylation, S-palmitoylation, ubiquitination, S-nitrosylation, and O-GlcNAcylation (30). Phosphorylation of Drp1, specifically at serine 616, appears to be involved in the development of BPD. Increased phosphorylation at this site is associated with mitochondrial fragmentation in lung endothelial cells and the formation of BPD-related pulmonary hypertension (BPD-PH) in rats exposed to high oxygen levels (31). Interestingly, different phosphorylation sites on Drp1 respond differently to lung injury caused by hyperoxia. Reduced phosphorylation at serine 637, for instance, is related to mitochondrial dysfunction in a mouse model of acute lung injury (ALI) (32). In contrast, in another in vitro experiment, hyperoxia increased phosphorylation of Drp1 at the serine 616 site to cause increased mitochondrial fragmentation in lung endothelial cells (33).
Glycogen synthase kinase-3β (GSK-3β)
GSK-3β is a serine/threonine kinase that plays a role in various cellular processes, including cell proliferation, DNA repair, the cell cycle, and metabolic pathways, by phosphorylating target proteins (34). For instance, it can lead to sustained fibroblast activation by phosphorylating β-catenin at tyrosine 489 (6). This phosphorylated GSK-3β/β-catenin pathway can thicken alveolar septa and promote the differentiation of lung mesenchymal stromal cells (35). These are important events in the development of BPD. Rodent models further demonstrate that GSK-3β-mediated inflammation and oxidative stress play an important pathological role in hyperoxia lung injury (36). Interestingly, in a rat BPD model, increased GSK-3β phosphorylation led to lung inflammation and simplification of alveolar and pulmonary vascular structures (37). This suggests that, in addition to its role in modifying other proteins through phosphorylation, GSK-3β's own activity is influenced by phosphorylation. Specifically, increased phosphorylation at serine 9 promotes the nuclear translocation of β-catenin, which disrupts alveolar and vascular development and contributes to pulmonary hypertension associated with hyperoxia-induced lung injury in mice (38).
Nuclear factor-kappa B (Nf-κB)
NF-κB is a transcription factor that plays a crucial role in various cellular processes, including inflammation, cell survival, and differentiation. The phosphorylation modification of NF-κB is significant in regulating these processes (39). Elevated levels of NF-κB phosphorylation were associated with inflammation and apoptosis in a rat model of BPD (40). Conversely, inhibiting NF-κB phosphorylation reduced oxidative stress and inflammation induced by hyperoxia in AECs and alleviated lung injury caused by hyperoxia in neonatal mice (41, 42). It is important to consider the roles of two upstream factors, inhibitor of κB (IκB) and inhibitor of κB kinase (IKK), in activating NF-κB. IKK becomes activated through phosphorylation and subsequently inactivates IκB by phosphorylation, leading to the nuclear translocation of NF-κB (39). In fetal lung fibroblasts, hyperoxia induces the phosphorylation of IκBα at tyrosine 42, activating NF-κB and contributing to apoptosis and oxidative stress (43). Additionally, inhibiting phosphorylated IKK/NF-κB is associated with the protective effect of IL-3 deficiency against hyperoxia-induced lung injury in mice (44).
In fact, the phosphorylation modifications of substrate proteins involved in the pathogenesis of BPD may extend beyond the examples mentioned above. For instance, in a rat model of hyperoxia-induced BPD-PH, reduced phosphorylation of endothelial nitric oxide synthase (eNOS) at the serine 1,177 site may contribute to increased pulmonary vascular tone (45). Hyperoxia also inhibits the phosphorylation of GTP-cyclohydrolase-1 at the serine 51 site, impacting tetrahydrobiopterin synthesis and potentially playing a role in hyperoxia-induced lung injury in rats (46). Clinical studies have shown that low levels of phosphorylated vasodilator-stimulated phosphoprotein, which affects cell proliferation and migration, are observed in the lung tissue of infants who succumb to BPD (47). Protein kinase B (Akt) is involved in various biological activities, such as cell survival and proliferation. Its phosphorylated form mediates forkhead box O3 (FoxO3) and E26 oncogene homologue 1 signaling, which plays an important role in counteracting hyperoxia-induced apoptosis in AECs (48, 49). However, due to space limitations, we regret that we cannot provide a comprehensive summary of all phosphorylation modifications of substrate proteins related to BPD. This remains an avenue for future research. Nevertheless, gaining a comprehensive understanding of these phosphorylation modifications of substrate proteins is likely to enhance our comprehension of the pathogenesis of BPD. Additionally, finding a balance between phosphorylation and dephosphorylation of substrate proteins may represent a potential therapeutic strategy for BPD.
Acetylation
The process of acetylation modifications is controlled by two classes of enzymes: histone acetyltransferases, which are responsible for adding acetyl groups to lysine residues on target proteins, and histone deacetylases (HDACs), which catalyze the removal of acetyl groups (50). A growing number of studies have shown that acetylation modifications can influence the progression of BPD (Figure 1G). In a rat model of BPD, hyperoxia inhibits the activity of HDACs, leading to an increase in cytokine-induced neutrophil chemoattractant-1, which promotes inflammation and disrupts alveolar development (51). Moreover, a reduction in HDACs activity could attenuate the deacetylation of MyD88 and NF-κB, exacerbating sepsis-induced lung inflammation and impairing lung development in mice, leading to pathological changes similar to those observed in BPD (52). Conversely, enhanced HDACs activity alleviates lipopolysaccharide-induced alveolar developmental arrest in rats by inhibiting TGF-α (53). These compelling findings suggest that HDACs may serve as a promising therapeutic target for BPD. However, the role of HDACs in BPD appears to be controversial, as a previous animal study demonstrated that reduced HDACs activity could enhance the expression of acetylated histones H3 and H4, which supports recovery from hyperoxia-induced lung injury in neonatal rats (54). One possible explanation is that acetylation modifications of different target proteins may have different effects against BPD.
It is worth mentioning that the class III HDACs, which depend on NAD+ and are known as the sirtuins (SIRTs) family, may play an important protective role in BPD. Both human AECs and rat BPD models have shown that the activation of SIRT1 reduces hyperoxia-induced apoptosis by deacetylating p53 (55, 56). SIRT1 may also have the ability to reduce ERS associated with hyperoxia ALI in rats, but whether the exact mechanism involves the deacetylation of SIRT1 remains to be elucidated (57). Furthermore, in vitro and in vivo experiments have demonstrated that SIRTs can attenuate hyperoxia-induced AECs injury by deacetylating FoxO1 (58). This is supported by our recent study, which found that the attenuation of SIRT1's ability to deacetylate FoxO1 leads to hyperoxia-induced injury in umbilical vein endothelial cells by inhibiting autophagy (59).
Ubiquitination
Ubiquitination is a process in which ubiquitin molecules covalently bind to target proteins, leading to their degradation in a series of steps involving E1-activating enzymes, E2-binding enzymes, and E3-ligases. Deubiquitinating enzymes (DUBs) are responsible for removing ubiquitin from ubiquitinated proteins, allowing for the dynamic reversibility of ubiquitination. Common types of DUBs include ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases, ovarian tumor proteases (OTUs), and others (60). Experimental studies have shown that ubiquitination modifications of specific target proteins are associated with the pathophysiology of BPD (Figure 1D). For instance, exposure to hyperoxia increases the ubiquitination modification of angiotensin-converting enzyme-2, diminishing its protective effect on lung epithelial cells (61). Additionally, nuclear factor erythroid 2-related factor 2 (NRF2) plays a protective role in BPD by regulating the antioxidant response element, a process effective only when the ubiquitinated degradation of NRF2 by kelch-like ECH-associated protein 1 (Keap1) is attenuated (62). Another target protein such as nuclear receptor subfamily 2, group F, member 2, is inhibited through ubiquitination by S-phase kinase-associated protein 2, thereby inhibiting CCAAT/enhancer-binding protein alpha (C/EBPα), which is necessary for the protection of lung cells in mice with hyperoxia-induced lung injury (63). In addition to the modification of target proteins, ubiquitination has been linked to organelle activity. Increased total ubiquitinated proteins in AECs have been shown to promote ERS-mediated apoptosis under hyperoxia exposure (64).
On the other hand, DUBs have the potential to impact BPD, even though direct evidence is lacking. Studies have revealed that USP18 and USP25 attenuate oxidative stress and inflammatory responses induced by lipopolysaccharide in human pulmonary microvascular endothelial cells and lung epithelial cells by inhibiting toll-like receptor 4/NF-κB and tumor necrosis factor receptor-associated factor 6/MAPK signaling, respectively (65, 66). Furthermore, USP22 stabilizes SIRT1 activity through deubiquitination, thereby promoting SIRT1's ability to deacetylate p53 and inhibit apoptosis (67). These findings suggest that enhancing DUB activity may contribute to the recovery of BPD. However, in BPD mouse models, He et al. (68) found that OTU3 enhances the stability of phosphatase and tensin homolog protein through deubiquitination, exacerbating lung injury and inflammatory responses. This suggests that the relationship between DUBs and BPD is complex, with the effects of DUBs on BPD depending on the action of target proteins as well as the subclasses of DUBs.
SUMOylation
Similar to ubiquitination, SUMOylation is a process where the small ubiquitin-like modifier (SUMO) covalently attaches to lysine residues of substrate proteins in a cascade reaction catalyzed by three enzymes. In contrast, SUMO-specific proteases (SENPs) promote the maturation of the precursor SUMO and perform de-SUMOylation (69). An important consideration is that reduced SUMOylation of SIRT1 may be involved in the pathogenesis of BPD (7). There is evidence that hyperoxia upregulates SENP1 to increase the nucleoplasmic shuttling of SIRT1 and inhibits its deacetylase activity, thus promoting apoptosis in AECs (70). Conversely, the combination of budesonide and Poractant Alfa injection reduces the hyperoxia-induced increase in SENP1 in peripheral blood mononuclear cells of preterm infants, which mitigates the nucleoplasmic shuttling of SIRT1 and ultimately prevents BPD (71). These findings suggest that enhancing the SUMOylation of SIRT1 is beneficial for BPD. In addition to SIRT1, SUMOylation of other target proteins may also contribute to the pathogenesis of BPD. For example, SENP3 induces de-SUMOylation of hypoxia-inducible factor-1 alpha (HIF-1α), activating JNK and promoting M1 macrophage polarization and tissue factor-mediated coagulation cascade, leading to lipopolysaccharide-induced ALI in mice (72, 73). Moreover, fibrosis-inducing E3 ligase 1 promotes TGF-β signaling through the ubiquitinated degradation of SUMO-E3 ligase protein inhibitor of activated signal transducer and activator of transcription 4, thereby contributing to bleomycin-induced lung fibrosis in mice (74). However, it is worth noting that not all SUMOylation of target proteins is beneficial for BPD (Figure 1E). In hyperoxia-induced neonatal rat lung injury, as reported by Zhu et al. (75), the SUMOylation of C/EBPα has a negative effect on the differentiation and secretion of AECs.
Methylation
DNA methylation, histone modifications and microRNA expression, along with the crosstalk between them, play significant roles in lung development at various stages by regulating phenotypic programming (76). As a result, epigenetic changes are pivotal in BPD development (77). Among the common complications of prematurity, BPD is one of the most likely to experience abnormal DNA methylation (78). Differential DNA methylation has been implicated in hyperoxia-exposed rat lung tissues (79), as well as during alveolar septation formation in mice and humans (80). Notably, in rats with hyperoxia-induced lung injury, DNA methyltransferase (DNMT) 3b-mediated DNA methylation and enhancer of zeste homolog 2-mediated tri-methylation of lysine 27 on histone H3 decrease runt-related transcription factor 3 proteins associated with lung epithelial and vascular development, possibly contributing to the pathogenesis of BPD (81). Furthermore, hyperoxia enhances DNA hypermethylation of TGF-β pathway-related genes and immune system-related genes, particularly the PI3K-AKT pathway, affecting alveolar development and later abnormal responses to respiratory infections in mice (82, 83). Additionally, DNMT-mediated methylation of certain microRNA promoters is associated with BPD severity (84). These findings suggest that targeting epigenetics may be a potential strategy to alleviate BPD (Figure 1F). Indeed, it has been shown that vitamin D promotes the methylation status of vitamin D-responsive elements in the interferon-γ-promoter region, thereby inhibiting interferon-γ expression and attenuating lipopolysaccharide-induced BPD in rats (85). In BPD mouse models, the application of DNA methylase inhibitors potentially reduces phosphorylated Smad2/3 and increases surface-active protein C levels, thereby improving alveolarization (86). Furthermore, DNA methylation inhibitors reverse hyperoxia-induced aberrant methylation of the cell cycle inhibitor p16 gene, thereby attenuating pulmonary fibrosis in neonatal rats (87).
Glycosylation
Glycosylation refers to the process of attaching glycans to proteins and lipids, which is carried out by glycosyltransferases. Conversely, glycosidases are responsible for breaking down glycosidic bonds, leading to de-glycosylation (88). Glycosylation plays a crucial role in various biological processes, including the secretion and translocation of proteins. It also ensures the proper folding of proteins and shields them from degradation by proteases (89). There is evidence indicating that exposure to high levels of oxygen, known as hyperoxia, can temporarily disrupt the glycosylation balance in mouse lung microvascular endothelial cells. This disruption may impact cellular interactions and signal transduction (90). Furthermore, a protein called endoplasmic reticulum protein 57, associated with glycosylation modifications, has been found to contribute to hyperoxia-induced apoptosis in human endothelial cells by inhibiting GRP78 and activating caspase-3 (91). Clinical studies have shown that preterm infants, especially those born at less than 28 weeks of gestational age, exhibit increased glycosylation of IgG Fc in their peripheral blood. This increased glycosylation may play a role in the development of inflammatory diseases such as BPD (92).
O-GlcNAcylation is a specific type of glycosylation involving the binding of O-linked β-N-acetylglucosamine (O-GlcNAc) to serine or threonine residues of substrate proteins. O-GlcNAc transferase and O-GlcNAcase are enzymes responsible for adding and removing O-GlcNAcylation, respectively (93). This process is implicated in various essential cellular activities, including gene expression, signal transduction, cell cycle regulation, nutrient sensing, protein homeostasis, cellular stress response and neuronal function (94). O-GlcNAcylation is particularly closely related to oxidative stress (95). For instance, reduced O-GlcNAcylation of lung tissue proteins has been suggested as an essential response to injury in sepsis-induced ALI models in mice (96). Conversely, O-GlcNAcylation of specific target proteins, like SIRT1 at the serine 549 site, can enhance its deacetylation activity (97), promoting recovery in BPD by reducing apoptosis (56). However, in cases of hyperoxia-induced injury in AECs, increased O-GlcNAcylation of the mitophagy regulator Parkin can disrupt mitochondrial homeostasis and lead to apoptosis (98) (Figure 1B). This indicates that O-GlcNAcylation can have both positive and negative effects in BPD, depending on the specific substrate proteins, and requires further investigation to understand its precise mechanisms of action.
Glycation
In contrast to glycosylation, glycation (also known as the Maillard Reaction) is a non-enzymatic reaction where free sugars covalently bind to proteins. This reaction produces advanced glycation end products (AGEs), which are important stabilizing substances (99). The receptor for AGEs (RAGE) is abundant in AECs, where it plays a positive role in lung development. However, under pathological conditions, RAGE interacts with its ligands, including AGEs, leading to inflammation and oxidative stress that can induce lung diseases (100). RAGE makes the developing lung more susceptible to the harmful effects of hyperoxia (101). Studies involving RAGE-deficient mice have shown delayed hyperoxia-induced mortality and ALI (102). RAGE also disrupts the alveolar-capillary barrier (103) and promotes inflammatory signals, such as high mobility group box 1 (104) and NF-κB (105), which are significant mechanisms for RAGE's involvement in BPD. Clinical studies have identified serum RAGE as a promising biomarker for predicting BPD, suggesting that targeting RAGE may be a new approach to treating lung diseases (106).
S-glutathionylation
S-Glutathionylation is a process where mixed disulfides are formed between glutathione and specific cysteine residues on target proteins (5). It plays a crucial role in regulating cellular redox signaling. For instance, in lipopolysaccharide-induced ALI in mice, S-glutathionylation of macrophage fatty acid-binding protein 5 at cysteine 127 is enhanced. This modification activates peroxisome proliferator-activated receptor (PPAR), facilitating the cells to resist oxidative stress (107). Significant changes in S-glutathionylation of proteins are observed in mouse models of hyperoxia-induced lung injury, underlining its critical role in BPD (108). S-glutathionylation of the substrate protein Keap1 is necessary for activating the antioxidant NRF2, which may protect experimental neonatal rats against BPD (109) (Figure 1C). However, S-glutathionylation of proteins can also have adverse effects under certain conditions. For example, S-glutathionylation of certain apoptotic proteins in lung epithelial cells has potential pathophysiological significance in pulmonary fibrosis (110). Additionally, hyperoxia-induced S-glutathionylation of manganese superoxide dismutase increases mitochondrial oxidative stress in macrophages, leading to increased mortality in mice with ventilator-associated pneumonia (111). These findings suggest that the process by which S-glutathionylation of proteins affects BPD may involve a complex regulatory network, and more evidence is needed to elucidate the specific mechanisms.
Glutaredoxin (Grx), primarily responsible for deglutathionylation, also plays a role in cellular redox signaling. Grx1 gene deficiency has been found to stabilize HIF-1α and inhibit NF-κB, promoting angiogenesis and reducing apoptosis to alleviate hyperoxia-induced lung injury in mice (112). However, another study suggests that Grx deficiency may increase the expression of TGF-β in respiratory epithelial cells, potentially raising the risk of developing fibrotic lung disease (113). The differing effects of Grx may depend on oxidative inducers and the specific substrate proteins involved.
S-nitrosylation
S-nitrosylation is a reaction where the nitrosyl group of nitric oxide (NO) covalently binds to the thiol group of cysteine, forming S-nitrosothiol. This process promotes pulmonary vasodilation but can lead to alveolitis and constriction of pulmonary artery smooth muscle in the presence of high inhaled NO doses and oxidative stress. Therefore, maintaining a dynamic balance of S-nitrosylation is beneficial for alveolar integrity and NO-mediated vasodilation (114). S-nitrosoglutathione (GSNO) is a critical donor of NO that promotes S-nitrosylation, while S-nitrosoglutathione reductase (GSNOR) is responsible for de-S-nitrosylation by acting on GSNO (5). Previous studies have shown that GSNO contributes to airway relaxation in BPD mice, whereas GSNOR is associated with increased airway hyperresponsiveness related to BPD (115, 116). Knockout of the alcohol dehydrogenase-5 gene encoding GSNOR has been shown to ameliorate BPD and BPD-PH (117). Clinical studies have revealed high expression of GSNOR in the airways and pulmonary vasculature of BPD patients (117), suggesting its significance in BPD pathogenesis and its potential as a therapeutic target for BPD-PH.
Thioredoxin (Trx) is another important regulator of S-nitrosylation, and exposure to hyperoxia can impact Trx function. The inhibition of Trx can interfere with heat shock protein 90-mediated oxidative responses, negatively affecting cell survival signaling pathways associated with BPD (118). Conversely, promoting Trx expression can prevent hyperoxia-induced loss of uncoupling protein 2 by activating the mitogen-activated protein kinase kinase 4/p38/PPAR-gamma coactivator 1 alpha pathway, thus reducing superoxide anion production (119). Trx also suppresses hyperoxia-induced inflammatory responses (120). These effects of Trx have been shown to be beneficial in mouse models of BPD. Furthermore, Trx enhances the ability of mesenchymal stromal cells (MSCs) to resist hyperoxia injury by inhibiting apoptosis-regulating kinase-1/p38-mediated apoptosis signaling. This improvement in MSC efficacy holds promise for treating BPD (121).
Conclusion
BPD is indeed a significant and challenging complication in preterm infants, and gaining a deeper understanding of the molecular mechanisms underlying its pathogenesis is crucial. PTMs influence almost all cellular biological processes by altering the function of substrate proteins in response to environmental changes. Investigating the relationship between PTMs and BPD is a promising avenue of research, as it can lead to early detection of BPD and the development of innovative therapeutic strategies. In this article, we have discussed the significance of several common PTMs, including phosphorylation, acetylation, ubiquitination, SUMOylation, methylation, glycosylation, glycation, S-glutathionylation, and S-nitrosylation, in the context of BPD (Figure 1). However, several important questions remain unanswered. Firstly, many substrate proteins can undergo multiple PTMs, and understanding how crosstalk between different PTMs influences BPD requires further investigation. Secondly, the same PTMs can act on different substrate proteins, resulting in diverse effects on BPD. Deciphering the roles of both PTMs and substrate proteins in influencing BPD is a complex task. Finally, establishing a comprehensive network of PTMs related to BPD and harnessing PTMs to develop novel diagnostic markers and therapeutic targets for BPD is an intriguing prospect that holds the potential to transform our approach to managing this condition.
Author contributions
KY: Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Visualization, Writing – original draft, Writing – review & editing. TH: Conceptualization, Data curation, Formal Analysis, Project administration, Supervision, Writing – original draft. XS: Formal Analysis, Investigation, Methodology, Software, Validation, Writing – original draft. WD: Data curation, Formal Analysis, Funding acquisition, Investigation, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This article was supported by the National Natural Science Foundation of China (82371710), Sichuan Science and Technology Department Major Science and Technology Special Project (2022YFS0062) and Luzhou Science and Technology Programme Project (2022-JYJ-122).
Acknowledgments
The authors would like to express their gratitude to EditSprings (https://www.editsprings.com/) for the expert linguistic services provided.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BPD, bronchopulmonary dysplasia; PTMs, post-translational modifications; MAPK, mitogen-activated protein kinases; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; ERS, endoplasmic reticulum stress; IRE1α, inositol-requiring enzyme 1 alpha; AECs, alveolar epithelial cells; Smad, small mothers against decapentaplegic; TGF-β, transforming growth factor-β; GRP78, glucose-regulated protein 78; Drp1, dynamin-related protein 1; BPD-PH, BPD-pulmonary hypertension; ALI, acute lung injury; GSK-3β, glycogen synthase kinase-3β; NF-κB, nuclear factor-kappa B; IκB, inhibitor of κB; IKK, inhibitor of κB kinase; eNOS, endothelial nitric oxide synthase; Akt, protein kinase B; FoxO3, forkhead box O3; HDACs, histone deacetylases; SIRTs, sirtuins; DUBs, deubiquitinating enzymes; USPs, ubiquitin-specific proteases; OTUs, ovarian tumor proteases; NRF2, nuclear factor erythroid 2-related factor 2; Keap1, kelch-like ECH-associated protein 1; C/EBPα, CCAAT/enhancer-binding protein alpha; SUMO, small ubiquitin-like modifier; SENPs, SUMO-specific proteases; HIF-1α, hypoxia-inducible factor-1 alpha; DNMT, DNA methyltransferase; O-GlcNAc, O-linked β-N-acetylglucosamine; AGEs, advanced glycation end products; RAGE, receptor for AGEs; Grx, glutaredoxin; NO, nitric oxide; GSNO, S-nitrosoglutathione; GSNOR, S-nitrosoglutathione reductase; Trx, thioredoxin; MSCs, mesenchymal stromal cells.
References
1. Shukla VV, Ambalavanan N. Recent advances in bronchopulmonary dysplasia. Indian J Pediatr. (2021) 88:690–5. doi: 10.1007/s12098-021-03766-w
2. Pan S, Chen R. Pathological implication of protein post-translational modifications in cancer. Mol Aspects Med. (2022) 86:101097. doi: 10.1016/j.mam.2022.101097
3. Gao J, Shao K, Chen X, Li Z, Liu Z, Yu Z, et al. The involvement of post-translational modifications in cardiovascular pathologies: focus on SUMOylation, neddylation, succinylation, and prenylation. J Mol Cell Cardiol. (2020) 138:49–58. doi: 10.1016/j.yjmcc.2019.11.146
4. Salas-Lloret D, González-Prieto R. Insights in post-translational modifications: ubiquitin and SUMO. Int J Mol Sci. (2022) 23(6):3281. doi: 10.3390/ijms23063281
5. Kalinina E, Novichkova M. Glutathione in protein redox modulation through S-glutathionylation and S-nitrosylation. Molecules. (2021) 26(2):435. doi: 10.3390/molecules26020435
6. Sucre JM, Vijayaraj P, Aros CJ, Wilkinson D, Paul M, Dunn B, et al. Posttranslational modification of β-catenin is associated with pathogenic fibroblastic changes in bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L186–l195. doi: 10.1152/ajplung.00477.2016
7. Tan F, Dong W, Lei X, Liu X, Li Q, Kang L, et al. Attenuated SUMOylation of sirtuin 1 in premature neonates with bronchopulmonary dysplasia. Mol Med Rep. (2018) 17(1):1283–8. doi: 10.3892/mmr.2017.8012
8. Hepworth EMW, Hinton SD. Pseudophosphatases as regulators of MAPK signaling. Int J Mol Sci. (2021) 22(22):12595. doi: 10.3390/ijms222212595
9. Wei J, Liu R, Hu X, Liang T, Zhou Z, Huang Z. MAPK signaling pathway-targeted marine compounds in cancer therapy. J Cancer Res Clin Oncol. (2021) 147:3–22. doi: 10.1007/s00432-020-03460-y
10. Hu Y, Fu J, Liu X, Xue X. ERK1/2 signaling pathway activated by EGF promotes proliferation, transdifferentiation, and migration of cultured primary newborn rat lung fibroblasts. Biomed Res Int. (2020) 2020:7176169. doi: 10.1155/2020/7176169
11. Hu Y, Fu J, Xue X. Association of the proliferation of lung fibroblasts with the ERK1/2 signaling pathway in neonatal rats with hyperoxia-induced lung fibrosis. Exp Ther Med. (2019) 17(1):701–8. doi: 10.3892/etm.2018.6999
12. Hu J, Wu Z, Wang H, Geng H, Huo J, Zhu X, et al. Vitamin D ameliorates apoptosis and inflammation by targeting the mitochondrial and MEK1/2-ERK1/2 pathways in hyperoxia-induced bronchopulmonary dysplasia. J Inflamm Res. (2022) 15:4891–906. doi: 10.2147/JIR.S371906
13. Liang Z, Zhang X, Liu Y, Wu Q, You C. SEMA3A protects against hyperoxia-induced lung injury in a bronchopulmonary dysplasia model of newborn rat by inhibiting ERK pathway. Allergol Immunopathol (Madr). (2021) 49:8–15. doi: 10.15586/aei.v49i6.478
14. Kim YE, Park WS, Ahn SY, Sung DK, Sung SI, Kim JH, et al. WKYMVm hexapeptide, a strong formyl peptide receptor 2 agonist, attenuates hyperoxia-induced lung injuries in newborn mice. Sci Rep. (2019) 9:6815. doi: 10.1038/s41598-019-43321-4
15. Menon RT, Shrestha AK, Barrios R, Shivanna B. Hyperoxia disrupts extracellular signal-regulated kinases 1/2-induced angiogenesis in the developing lungs. Int J Mol Sci. (2018) 19(5):1525. doi: 10.3390/ijms19051525
16. Xu D, Guthrie JR, Mabry S, Sack TM, Truog WE. Mitochondrial aldehyde dehydrogenase attenuates hyperoxia-induced cell death through activation of ERK/MAPK and PI3K-akt pathways in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. (2006) 291:L966–975. doi: 10.1152/ajplung.00045.2006
17. Tong X, Li M, Liu N, Huang W, Xue X, Fu J. Hyperoxia induces endoplasmic reticulum stress-associated apoptosis via the IRE1α; pathway in rats with bronchopulmonary dysplasia. Mol Med Rep. (2021) 23(1):33. doi: 10.3892/mmr.2020.11671
18. Zhang S, Li X, Yuan T, Guo X, Jin C, Jin Z, et al. Glutamine inhibits inflammation, oxidative stress, and apoptosis and ameliorates hyperoxic lung injury. J Physiol Biochem. (2023) 79:613–23. doi: 10.1007/s13105-023-00961-5
19. Zhao S, Luo G, Wu H, Zhang L. Placental growth factor gene silencing mitigates the epithelial-to-mesenchymal transition via the p38 MAPK pathway in rats with hyperoxia-induced lung injury. Mol Med Rep. (2019) 20(6):4867–74. doi: 10.3892/mmr.2019.10785
20. Shen CH, Lin JY, Lu CY, Yang SS, Peng CK, Huang KL. SPAK-p38 MAPK signal pathway modulates claudin-18 and barrier function of alveolar epithelium after hyperoxic exposure. BMC Pulm Med. (2021) 21:58. doi: 10.1186/s12890-021-01408-7
21. Guo B, Zuo Z, Di X, Huang Y, Gong G, Xu B, et al. Salidroside attenuates HALI via IL-17A-mediated ferroptosis of alveolar epithelial cells by regulating Act1-TRAF6-p38 MAPK pathway. Cell Commun Signal. (2022) 20:183. doi: 10.1186/s12964-022-00994-1
22. Wang X, Lv S, Sun J, Zhang M, Zhang L, Sun Y, et al. Caffeine reduces oxidative stress to protect against hyperoxia-induced lung injury via the adenosine A2A receptor/cAMP/PKA/src/ERK1/2/p38MAPK pathway. Redox Rep. (2022) 27:270–8. doi: 10.1080/13510002.2022.2143114
23. Jin R, Xu J, Gao Q, Mao X, Yin J, Lu K, et al. IL-33-induced neutrophil extracellular traps degrade fibronectin in a murine model of bronchopulmonary dysplasia. Cell Death Discov. (2020) 6:33. doi: 10.1038/s41420-020-0267-2
24. Zhang Z, Sun C, Wang J, Jiang W, Xin Q, Luan Y. Timing of erythropoietin modified mesenchymal stromal cell transplantation for the treatment of experimental bronchopulmonary dysplasia. J Cell Mol Med. (2018) 22:5759–63. doi: 10.1111/jcmm.13843
25. Tzavlaki K, Moustakas A. TGF-β signaling. Biomolecules. (2020) 10(3):487. doi: 10.3390/biom10030487
26. Flodby P, Li C, Liu Y, Wang H, Marconett CN, Laird-Offringa IA, et al. The 78-kD glucose-regulated protein regulates endoplasmic Reticulum homeostasis and distal epithelial cell survival during lung development. Am J Respir Cell Mol Biol. (2016) 55:135–49. doi: 10.1165/rcmb.2015-0327OC
27. Lee Y, Lee J, Nam SK, Hoon Jun Y. S-endoglin expression is induced in hyperoxia and contributes to altered pulmonary angiogenesis in bronchopulmonary dysplasia development. Sci Rep. (2020) 10:3043. doi: 10.1038/s41598-020-59928-x
28. Gong J, Feng Z, Peterson AL, Carr JF, Vang A, Braza J, et al. Endothelial to mesenchymal transition during neonatal hyperoxia-induced pulmonary hypertension. J Pathol. (2020) 252:411–22. doi: 10.1002/path.5534
29. Kunzmann S, Ottensmeier B, Speer CP, Fehrholz M. Effect of progesterone on smad signaling and TGF-β/smad-regulated genes in lung epithelial cells. PLoS One. (2018) 13:e0200661. doi: 10.1371/journal.pone.0200661
30. Jin JY, Wei XX, Zhi XL, Wang XH, Meng D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin. (2021) 42:655–64. doi: 10.1038/s41401-020-00518-y
31. Dai Y, Yu B, Ai D, Yuan L, Wang X, Huo R, et al. Mitochondrial fission-mediated lung development in newborn rats with hyperoxia-induced bronchopulmonary dysplasia with pulmonary hypertension. Front Pediatr. (2021) 8:619853. doi: 10.3389/fped.2020.619853
32. Soundararajan R, Hernández-Cuervo H, Stearns TM, Griswold AJ, Patil SS, Fukumoto J, et al. A-kinase anchor protein 1 deficiency causes mitochondrial dysfunction in mouse model of hyperoxia induced acute lung injury. Front Pharmacol. (2022) 13:980723. doi: 10.3389/fphar.2022.980723
33. Ma C, Beyer AM, Durand M, Clough AV, Zhu D, Norwood Toro L, et al. Hyperoxia causes mitochondrial fragmentation in pulmonary endothelial cells by increasing expression of pro-fission proteins. Arterioscler Thromb Vasc Biol. (2018) 38:622–35. doi: 10.1161/ATVBAHA.117.310605
34. Lin J, Song T, Li C, Mao W. GSK-3β in DNA repair, apoptosis, and resistance of chemotherapy, radiotherapy of cancer. Biochim Biophys Acta Mol Cell Res. (2020) 1867:118659. doi: 10.1016/j.bbamcr.2020.118659
35. Popova AP, Bentley JK, Anyanwu AC, Richardson MN, Linn MJ, Lei J, et al. Glycogen synthase kinase-3β/β-catenin signaling regulates neonatal lung mesenchymal stromal cell myofibroblastic differentiation. Am J Physiol Lung Cell Mol Physiol. (2012) 303:L439–448. doi: 10.1152/ajplung.00408.2011
36. Li J, Shi J, Li P, Guo X, Wang T, Liu A. Genipin attenuates hyperoxia-induced lung injury and pulmonary hypertension via targeting glycogen synthase kinase-3 β in neonatal rats. Nutrition. (2019) 57:237–44. doi: 10.1016/j.nut.2018.05.017
37. Hummler SC, Rong M, Chen S, Hehre D, Alapati D, Wu S. Targeting glycogen synthase kinase-3β to prevent hyperoxia-induced lung injury in neonatal rats. Am J Respir Cell Mol Biol. (2013) 48:578–88. doi: 10.1165/rcmb.2012-0383OC
38. Chen S, Rong M, Platteau A, Hehre D, Smith H, Ruiz P, et al. CTGF disrupts alveolarization and induces pulmonary hypertension in neonatal mice: implication in the pathogenesis of severe bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. (2011) 300:L330–340. doi: 10.1152/ajplung.00270.2010
39. Motolani A, Martin M, Sun M, Lu T. Phosphorylation of the regulators, a complex facet of NF-κB signaling in cancer. Biomolecules. (2020) 11(1):15. doi: 10.3390/biom11010015
40. Liu D, Wang Y, Li L, Zhao H, Li L, Liu Y, et al. Celecoxib protects hyperoxia-induced lung injury via NF-κB and AQP1. Front Pediatr. (2019) 7:228. doi: 10.3389/fped.2019.00228
41. Li XG, Song X, Wang JY, Sun CH, Li ZQ, Meng LL, et al. Fibroblast growth factor 18 alleviates hyperoxia-induced lung injury in mice by adjusting oxidative stress and inflammation. Eur Rev Med Pharmacol Sci. (2021) 25(3):1485–94. doi: 10.26355/eurrev20210224856
42. Xie B, Li S, Bai W, Li Z, Lou F. Artesunate alleviates hyperoxia-induced lung injury in neonatal mice by inhibiting NLRP3 inflammasome activation. Evid Based Complement Alternat Med. (2023) 2023:7603943. doi: 10.1155/2023/7603943
43. Wright CJ, Zhuang T, La P, Yang G, Dennery PA. Hyperoxia-induced NF-kappaB activation occurs via a maturationally sensitive atypical pathway. Am J Physiol Lung Cell Mol Physiol. (2009) 296:L296–306. doi: 10.1152/ajplung.90499.2008
44. Huang Z, Zhang W, Yang J, Sun F, Zhou H. Interleukin-3 plays a vital role in hyperoxic acute lung injury in mice via mediating inflammation. BMC Pulm Med. (2018) 18:164. doi: 10.1186/s12890-018-0725-2
45. Dumas de la Roque E, Smeralda G, Quignard JF, Freund-Michel V, Courtois A, Marthan R, et al. Altered vasoreactivity in neonatal rats with pulmonary hypertension associated with bronchopulmonary dysplasia: implication of both eNOS phosphorylation and calcium signaling. PLoS One. (2017) 12:e0173044. doi: 10.1371/journal.pone.0173044
46. Jing X, Huang YW, Jarzembowski J, Shi Y, Konduri GG, Teng RJ. Caffeine ameliorates hyperoxia-induced lung injury by protecting GCH1 function in neonatal rat pups. Pediatr Res. (2017) 82:483–9. doi: 10.1038/pr.2017.89
47. Ali M, Heyob K, Tipple TE, Pryhuber GS, Rogers LK. Alterations in VASP phosphorylation and profilin1 and cofilin1 expression in hyperoxic lung injury and BPD. Respir Res. (2018) 19:229. doi: 10.1186/s12931-018-0938-1
48. Wu D, Liang M, Dang H, Fang F, Xu F, Liu C. Hydrogen protects against hyperoxia-induced apoptosis in type II alveolar epithelial cells via activation of PI3K/akt/Foxo3a signaling pathway. Biochem Biophys Res Commun. (2018) 495:1620–7. doi: 10.1016/j.bbrc.2017.11.193
49. He F, Wang QF, Li L, Yu C, Liu CZ, Wei WC, et al. Melatonin protects against hyperoxia-induced apoptosis in alveolar epithelial type II cells by activating the MT2/PI3K/AKT/ETS1 signaling pathway. Lung. (2023) 201:225–34. doi: 10.1007/s00408-023-00610-0
50. Ding P, Ma Z, Liu D, Pan M, Li H, Feng Y, et al. Lysine acetylation/deacetylation modification of immune-related molecules in cancer immunotherapy. Front Immunol. (2022) 13:865975. doi: 10.3389/fimmu.2022.865975
51. Zhu L, Li H, Tang J, Zhu J, Zhang Y. Hyperoxia arrests alveolar development through suppression of histone deacetylases in neonatal rats. Pediatr Pulmonol. (2012) 47:264–74. doi: 10.1002/ppul.21540
52. Menden H, Xia S, Mabry SM, Noel-MacDonnell J, Rajasingh J, Ye SQ, et al. Histone deacetylase 6 regulates endothelial MyD88-dependent canonical TLR signaling, lung inflammation, and alveolar remodeling in the developing lung. Am J Physiol Lung Cell Mol Physiol. (2019) 317:L332–l346. doi: 10.1152/ajplung.00247.2018
53. Ni W, Lin N, He H, Zhu J, Zhang Y. Lipopolysaccharide induces up-regulation of TGF-α through HDAC2 in a rat model of bronchopulmonary dysplasia. PLoS One. (2014) 9:e91083. doi: 10.1371/journal.pone.0091083
54. Cetinkaya M, Cansev M, Cekmez F, Tayman C, Canpolat FE, Kafa IM, et al. Protective effects of valproic acid, a histone deacetylase inhibitor, against hyperoxic lung injury in a neonatal rat model. PLoS One. (2015) 10:e0126028. doi: 10.1371/journal.pone.0126028
55. Zhu X, Wang F, Lei X, Dong W. Resveratrol alleviates alveolar epithelial cell injury induced by hyperoxia by reducing apoptosis and mitochondrial dysfunction. Exp Biol Med (Maywood). (2021) 246:596–606. doi: 10.1177/1535370220975106
56. Zhu X, Lei X, Wang J, Dong W. Protective effects of resveratrol on hyperoxia-induced lung injury in neonatal rats by alleviating apoptosis and ROS production. J Matern Fetal Neonatal Med. (2020) 33:4150–8. doi: 10.1080/14767058.2019.1597846
57. Sun Q, Han W, Hu H, Fan D, Li Y, Zhang Y, et al. Hydrogen alleviates hyperoxic acute lung injury related endoplasmic reticulum stress in rats through upregulation of SIRT1. Free Radic Res. (2017) 51:622–32. doi: 10.1080/10715762.2017.1351027
58. Zang L, Chi J, Bi S, Tao Y, Wang R, Li L. SIRT3 improves alveolar epithelial cell damage caused by bronchopulmonary dysplasia through deacetylation of FOXO1. Allergol Immunopathol (Madr). (2023) 51:191–204. doi: 10.15586/aei.v51i2.710
59. Zhu X, He S, Zhang R, Kang L, Lei X, Dong W. Protective effect and mechanism of autophagy in endothelial cell injury induced by hyperoxia. Am J Perinatol. (2024) 41(S 01):e2365–75. doi: 10.1055/s-0043-1771258
60. Sun T, Liu Z, Yang Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol Cancer. (2020) 19:146. doi: 10.1186/s12943-020-01262-x
61. Mohamed T, Abdul-Hafez A, Uhal BD. Regulation of ACE-2 enzyme by hyperoxia in lung epithelial cells by post-translational modification. J Lung Pulm Respir Res. (2021) 8:47–52.34825051
62. Liu Q, Gao Y, Ci X. Role of Nrf2 and its activators in respiratory diseases. Oxid Med Cell Longev. (2019) 2019:7090534. doi: 10.1155/2019/7090534
63. Wu Y, Zhang Z, Li J, Zhong H, Yuan R, Deng Z, et al. Mechanism of adipose-derived mesenchymal stem cell-derived extracellular vesicles carrying miR-21-5p in hyperoxia-induced lung injury. Stem Cell Rev Rep. (2022) 18:1007–24. doi: 10.1007/s12015-021-10311-x
64. Zhu Y, Ju H, Lu H, Tang W, Lu J, Wang Q. The function role of ubiquitin proteasome pathway in the ER stress-induced AECII apoptosis during hyperoxia exposure. BMC Pulm Med. (2021) 21:379. doi: 10.1186/s12890-021-01751-9
65. Jiang Z, Shen J, Ding J, Yuan Y, Gao L, Yang Z, et al. USP18 mitigates lipopolysaccharide-induced oxidative stress and inflammation in human pulmonary microvascular endothelial cells through the TLR4/NF-κB/ROS signaling. Toxicol in Vitro. (2021) 75:105181. doi: 10.1016/j.tiv.2021.105181
66. Xu Z, Peng Q, Wang Y, Chen X, Zhang J. Ubiquitin specific peptidase 25 alleviates acute lung injury and suppresses the inflammatory response in lung epithelial cells. Gen Physiol Biophys. (2022) 41:569–77. doi: 10.4149/gpb_2022038
67. Lin Z, Yang H, Kong Q, Li J, Lee SM, Gao B, et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol Cell. (2012) 46:484–94. doi: 10.1016/j.molcel.2012.03.024
68. He X, Kuang J, Wang Y, Lan G, Shi X. Bone marrow stromal cell-secreted extracellular vesicles containing miR-34c-5p alleviate lung injury and inflammation in bronchopulmonary dysplasia through promotion of PTEN degradation by targeting OTUD3. Immunol Invest. (2023) 52(6):681–702. doi: 10.1080/08820139.2023.2217854
69. Sheng Z, Zhu J, Deng YN, Gao S, Liang S. SUMOylation modification-mediated cell death. Open Biol. (2021) 11:210050. doi: 10.1098/rsob.210050
70. Dong W, Zhu X, Liu X, Zhao X, Lei X, Kang L, et al. Role of the SENP1-SIRT1 pathway in hyperoxia-induced alveolar epithelial cell injury. Free Radic Biol Med. (2021) 173:142–50. doi: 10.1016/j.freeradbiomed.2021.07.027
71. Du FL, Dong WB, Zhang C, Li QP, Kang L, Lei XP, et al. Budesonide and poractant alfa prevent bronchopulmonary dysplasia via triggering SIRT1 signaling pathway. Eur Rev Med Pharmacol Sci. (2019) 23(24):11032–42. doi: 10.26355/eurrev20191219811
72. He S, Fan C, Ji Y, Su Q, Zhao F, Xie C, et al. SENP3 facilitates M1 macrophage polarization via the HIF-1α/PKM2 axis in lipopolysaccharide-induced acute lung injury. Innate Immun. (2023) 29:25–34. doi: 10.1177/17534259231166212
73. Chen X, Lao Y, Yi J, Yang J, He S, Chen Y. SENP3 in monocytes/macrophages up-regulates tissue factor and mediates lipopolysaccharide-induced acute lung injury by enhancing JNK phosphorylation. J Cell Mol Med. (2020) 24:5454–62. doi: 10.1111/jcmm.15199
74. Lear T, McKelvey AC, Rajbhandari S, Dunn SR, Coon TA, Connelly W, et al. Ubiquitin E3 ligase FIEL1 regulates fibrotic lung injury through SUMO-E3 ligase PIAS4. J Exp Med. (2016) 213:1029–46. doi: 10.1084/jem.20151229
75. Zhu Y, Chen X, Mi L, Wang Q, Zhu H, Ju H, et al. Sumoylation of CCAAT-enhancer-binding protein α inhibits lung differentiation in bronchopulmonary dysplasia model rats. J Cell Mol Med. (2020) 24:7067–71. doi: 10.1111/jcmm.15310
76. Tong Y, Zhang S, Riddle S, Zhang L, Song R, Yue D. Intrauterine hypoxia and epigenetic programming in lung development and disease. Biomedicines. (2021) 9(8):944. doi: 10.3390/biomedicines9080944
77. Wang X, Cho HY, Campbell MR, Panduri V, Coviello S, Caballero MT, et al. Epigenome-wide association study of bronchopulmonary dysplasia in preterm infants: results from the discovery-BPD program. Clin Epigenetics. (2022) 14:57. doi: 10.1186/s13148-022-01272-0
78. Everson TM, O’Shea TM, Burt A, Hermetz K, Carter BS, Helderman J, et al. Serious neonatal morbidities are associated with differences in DNA methylation among very preterm infants. Clin Epigenetics. (2020) 12:151. doi: 10.1186/s13148-020-00942-1
79. Chen CM, Liu YC, Chen YJ, Chou HC. Genome-wide analysis of DNA methylation in hyperoxia-exposed newborn rat lung. Lung. (2017) 195:661–9. doi: 10.1007/s00408-017-0036-z
80. Cuna A, Halloran B, Faye-Petersen O, Kelly D, Crossman DK, Cui X, et al. Alterations in gene expression and DNA methylation during murine and human lung alveolar septation. Am J Respir Cell Mol Biol. (2015) 53:60–73. doi: 10.1165/rcmb.2014-0160OC
81. Zhu Y, Fu J, Yang H, Pan Y, Yao L, Xue X. Hyperoxia-induced methylation decreases RUNX3 in a newborn rat model of bronchopulmonary dysplasia. Respir Res. (2015) 16:75. doi: 10.1186/s12931-015-0239-x
82. Bik-Multanowski M, Revhaug C, Grabowska A, Dobosz A, Madetko-Talowska A, Zasada M, et al. Hyperoxia induces epigenetic changes in newborn mice lungs. Free Radic Biol Med. (2018) 121:51–6. doi: 10.1016/j.freeradbiomed.2018.04.566
83. Revhaug C, Bik-Multanowski M, Zasada M, Rognlien AGW, Günther CC, Ksiązek T, et al. Immune system regulation affected by a murine experimental model of bronchopulmonary dysplasia: genomic and epigenetic findings. Neonatology. (2019) 116:269–77. doi: 10.1159/000501461
84. Robbins ME, Dakhlallah D, Marsh CB, Rogers LK, Tipple TE. Of mice and men: correlations between microRNA-17∼92 cluster expression and promoter methylation in severe bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. (2016) 311:L981–l984. doi: 10.1152/ajplung.00390.2016
85. Liu C, Chen Z, Li W, Huang L, Zhang Y. Vitamin D enhances alveolar development in antenatal lipopolysaccharide-treated rats through the suppression of interferon-γ production. Front Immunol. (2018) 8:1923. doi: 10.3389/fimmu.2017.01923
86. Heyob KM, Khuhro Z, Khan AQ, Brown D, Tipple TE, Rogers LK. Effects of DNA methylase inhibitors in a murine model of severe BPD. Respir Physiol Neurobiol. (2023) 313:104060. doi: 10.1016/j.resp.2023.104060
87. Zhao SM, Wu HM, Cao ML, Han D. 5-aza-2'-deoxycytidine, a DNA methylation inhibitor, attenuates hyperoxia-induced lung fibrosis via re-expression of P16 in neonatal rats. Pediatr Res. (2018) 83:723–30. doi: 10.1038/pr.2017.291
88. Zhu X, Al-Danakh A, Zhang L, Sun X, Jian Y, Wu H, et al. Glycosylation in renal cell carcinoma: mechanisms and clinical implications. Cells. (2022) 11(16):2598. doi: 10.3390/cells11162598
89. Masbuchin AN, Rohman MS, Liu PY. Role of glycosylation in vascular calcification. Int J Mol Sci. (2021) 22(18):9829. doi: 10.3390/ijms22189829
90. Tiboldi A, Führer J, Schaubmayr W, Hunyadi-Gulyas E, Zach ML, Hochreiter B, et al. Oxygen-dependent changes in the N-glycome of murine pulmonary endothelial cells. Antioxidants (Basel). (2021) 10(12):1947. doi: 10.3390/antiox10121947
91. Xu D, Perez RE, Rezaiekhaligh MH, Bourdi M, Truog WE. Knockdown of ERp57 increases BiP/GRP78 induction and protects against hyperoxia and tunicamycin-induced apoptosis. Am J Physiol Lung Cell Mol Physiol. (2009) 297:L44–51. doi: 10.1152/ajplung.90626.2008
92. Twisselmann N, Bartsch YC, Pagel J, Wieg C, Hartz A, Ehlers M, et al. Igg fc glycosylation patterns of preterm infants differ with gestational age. Front Immunol. (2019) 9:3166. doi: 10.3389/fimmu.2018.03166
93. Chang YH, Weng CL, Lin KI. O-GlcNAcylation and its role in the immune system. J Biomed Sci. (2020) 27:57. doi: 10.1186/s12929-020-00648-9
94. Lee BE, Suh PG, Kim JI. O-GlcNAcylation in health and neurodegenerative diseases. Exp Mol Med. (2021) 53:1674–82. doi: 10.1038/s12276-021-00709-5
95. Chen PH, Chi JT, Boyce M. Functional crosstalk among oxidative stress and O-GlcNAc signaling pathways. Glycobiology. (2018) 28:556–64. doi: 10.1093/glycob/cwy027
96. Hwang JS, Kim KH, Park J, Kim SM, Cho H, Lee Y, et al. Glucosamine improves survival in a mouse model of sepsis and attenuates sepsis-induced lung injury and inflammation. J Biol Chem. (2019) 294:608–22. doi: 10.1074/jbc.RA118.004638
97. Han C, Gu Y, Shan H, Mi W, Sun J, Shi M, et al. O-GlcNAcylation of SIRT1 enhances its deacetylase activity and promotes cytoprotection under stress. Nat Commun. (2017) 8:1491. doi: 10.1038/s41467-017-01654-6
98. Xuefei Y, Dongyan L, Tianming L, Hejuan Z, Jianhua F. O-linked N-acetylglucosamine affects mitochondrial homeostasis by regulating parkin-dependent mitophagy in hyperoxia-injured alveolar type II cells injury. Respir Res. (2023) 24:16. doi: 10.1186/s12931-022-02287-0
99. Jansen FAC, Fogliano V, Rubert J, Hoppenbrouwers T. Dietary advanced glycation End products interacting with the intestinal epithelium: what do we really know? Mol Metab. (2023) 73:101734. doi: 10.1016/j.molmet.2023.101734
100. Khaket TP, Kang SC, Mukherjee TK. The potential of receptor for advanced glycation End products (RAGE) as a therapeutic target for lung associated diseases. Curr Drug Targets. (2019) 20:679–89. doi: 10.2174/1389450120666181120102159
101. Kindermann A, Baier J, Simm A, Haase R, Bartling B. Receptor for advanced glycation end-products modulates lung development and lung sensitivity to hyperoxic injury in newborn mice. Pflugers Arch. (2019) 471:983–94. doi: 10.1007/s00424-019-02267-2
102. Reynolds PR, Schmitt RE, Kasteler SD, Sturrock A, Sanders K, Bierhaus A, et al. Receptors for advanced glycation end-products targeting protect against hyperoxia-induced lung injury in mice. Am J Respir Cell Mol Biol. (2010) 42:545–51. doi: 10.1165/rcmb.2008-0265OC
103. Ota C, Ishizawa K, Yamada M, Tando Y, He M, Takahashi T, et al. Receptor for advanced glycation end products expressed on alveolar epithelial cells is the main target for hyperoxia-induced lung injury. Respir Investig. (2016) 54:98–108. doi: 10.1016/j.resinv.2015.08.009
104. Qiao J, Chen L, Huang X, Guo F. Effects of nebulized N–acetylcystein on the expression of HMGB1 and RAGE in rats with hyperoxia–induced lung injury. J Cell Physiol. (2019) 234:10547–53. doi: 10.1002/jcp.27724
105. Tian Z, Li Y, Ji P, Zhao S, Cheng H. Mesenchymal stem cells protects hyperoxia-induced lung injury in newborn rats via inhibiting receptor for advanced glycation end-products/nuclear factor κB signaling. Exp Biol Med (Maywood). (2013) 238:242–7. doi: 10.1177/1535370212473706
106. Go H, Ohto H, Nollet KE, Sato K, Miyazaki K, Maeda H, et al. Biomarker potential of the soluble receptor for advanced glycation End products to predict bronchopulmonary dysplasia in premature newborns. Front Pediatr. (2021) 9:649526. doi: 10.3389/fped.2021.649526
107. Guo Y, Liu Y, Zhao S, Xu W, Li Y, Zhao P, et al. Oxidative stress-induced FABP5 S-glutathionylation protects against acute lung injury by suppressing inflammation in macrophages. Nat Commun. (2021) 12:7094. doi: 10.1038/s41467-021-27428-9
108. Zhang T, Day NJ, Gaffrey M, Weitz KK, Attah K, Mimche PN, et al. Regulation of hyperoxia-induced neonatal lung injury via post-translational cysteine redox modifications. Redox Biol. (2022) 55:102405. doi: 10.1016/j.redox.2022.102405
109. Teng RJ, Jing X, Martin DP, Hogg N, Haefke A, Konduri GG, et al. N-acetyl-lysyltyrosylcysteine amide, a novel systems pharmacology agent, reduces bronchopulmonary dysplasia in hyperoxic neonatal rat pups. Free Radic Biol Med. (2021) 166:73–89. doi: 10.1016/j.freeradbiomed.2021.02.006
110. Corteselli E, Aboushousha R, Janssen-Heininger Y. S-Glutathionylation-controlled apoptosis of lung epithelial cells; potential implications for lung fibrosis. Antioxidants (Basel). (2022) 11(9):1789. doi: 10.3390/antiox11091789
111. Gauthier AG, Lin M, Zefi S, Kulkarni A, Thakur GA, Ashby CR Jr, et al. GAT107-mediated Α7 nicotinic acetylcholine receptor signaling attenuates inflammatory lung injury and mortality in a mouse model of ventilator-associated pneumonia by alleviating macrophage mitochondrial oxidative stress via reducing MnSOD-S-glutathionylation. Redox Biol. (2023) 60:102614. doi: 10.1016/j.redox.2023.102614
112. Liu X, Li K, Zhang F, Zhang Y, Deng C, Guo C. Ablation of glutaredoxin 1 promotes pulmonary angiogenesis and alveolar formation in hyperoxia-injured lungs by modifying HIF-1α stability and inhibiting the NF-κB pathway. Biochem Biophys Res Commun. (2020) 525:528–35. doi: 10.1016/j.bbrc.2020.02.129
113. Chia SB, Nolin JD, Aboushousha R, Erikson C, Irvin CG, Poynter ME, et al. Glutaredoxin deficiency promotes activation of the transforming growth factor beta pathway in airway epithelial cells, in association with fibrotic airway remodeling. Redox Biol. (2020) 37:101720. doi: 10.1016/j.redox.2020.101720
114. Bhatia V, Elnagary L, Dakshinamurti S. Tracing the path of inhaled nitric oxide: biological consequences of protein nitrosylation. Pediatr Pulmonol. (2021) 56:525–38. doi: 10.1002/ppul.25201
115. Einisman HJ, Gaston B, Wijers C, Smith LA, Lewis TH, Lewis SJ, et al. Tracheomalacia in bronchopulmonary dysplasia: trachealis hyper-relaxant responses to S-nitrosoglutathione in a hyperoxic murine model. Pediatr Pulmonol. (2019) 54:1989–96. doi: 10.1002/ppul.24513
116. Raffay TM, Dylag AM, Di Fiore JM, Smith LA, Einisman HJ, Li Y, et al. S-nitrosoglutathione attenuates airway hyperresponsiveness in murine bronchopulmonary dysplasia. Mol Pharmacol. (2016) 90:418–26. doi: 10.1124/mol.116.104125
117. Raffay TM, Bonilla-Fernandez K, Jafri A, Sopi RB, Smith LA, Cui F, et al. Bronchopulmonary dysplasia and pulmonary hypertension. The role of smooth muscle adh5. Am J Respir Cell Mol Biol. (2021) 65:70–80. doi: 10.1165/rcmb.2020-0289OC
118. Floen MJ, Forred BJ, Bloom EJ, Vitiello PF. Thioredoxin-1 redox signaling regulates cell survival in response to hyperoxia. Free Radic Biol Med. (2014) 75:167–77. doi: 10.1016/j.freeradbiomed.2014.07.023
119. Raghavan S, Kundumani-Sridharan V, Kumar S, White CW, Das KC. Thioredoxin prevents loss of UCP2 in hyperoxia via MKK4-p38 MAPK-PGC1α signaling and limits oxygen toxicity. Am J Respir Cell Mol Biol. (2022) 66:323–36. doi: 10.1165/rcmb.2021-0219OC
120. Nagano N, Tanaka K, Ozawa J, Watanabe T, Miyake F, Matsumura S, et al. Attenuation of hyperoxic lung injury in newborn thioredoxin-1-overexpressing mice through the suppression of proinflammatory cytokine mRNA expression. Biomedicines. (2020) 8(3):66. doi: 10.3390/biomedicines8030066
Keywords: post-translational modification, bronchopulmonary dysplasia, hyperoxia, lung injury, substrate protein
Citation: Yang K, He T, Sun X and Dong W (2025) Post-translational modifications and bronchopulmonary dysplasia. Front. Pediatr. 12:1426030. doi: 10.3389/fped.2024.1426030
Received: 30 April 2024; Accepted: 13 December 2024;
Published: 3 January 2025.
Edited by:
Javier Rodriguez Fanjul, Pediatric Service, Hospital Germans Trias i Pujol, SpainReviewed by:
Vladimir Pohanka, Slovak Medical University, SlovakiaGary C. Mouradian, Medical College of Wisconsin, United States
Copyright: © 2025 Yang, He, Sun and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenbin Dong, ZG9uZ3dlbmJpbjIwMDBAMTYzLmNvbQ==