
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 04 July 2024
Sec. Pediatric Immunology
Volume 12 - 2024 | https://doi.org/10.3389/fped.2024.1415020
This article is part of the Research TopicReviews in Pediatric Primary ImmunodeficienciesView all 11 articles
 Tatiana P. Volodashchik1
Tatiana P. Volodashchik1 Ekaterina A. Polyakova1
Ekaterina A. Polyakova1 Taisia M. Mikhaleuskaya1Inga S. Sakovich1Aleksandra N. Kupchinskaya1Aliaxandr Ch. Dubrouski2
Taisia M. Mikhaleuskaya1Inga S. Sakovich1Aleksandra N. Kupchinskaya1Aliaxandr Ch. Dubrouski2 Mikhail V. Belevtsev1
Mikhail V. Belevtsev1 Joseph F. Dasso3,4
Joseph F. Dasso3,4 Dzmitry S. Varabyou5
Dzmitry S. Varabyou5 Luigi D. Notarangelo6
Luigi D. Notarangelo6 Jolan E. Walter3,4
Jolan E. Walter3,4 Svetlana O. Sharapova1*
Svetlana O. Sharapova1*
Background and aims: There is an increased risk of lymphomas in inborn errors of immunity (IEI); however, germline genetic testing is rarely used in oncological patients, even in those with early onset of cancer. Our study focuses on a child with a recombination-activating gene 1 (RAG1) deficiency who was identified through a screening program for Slavic founder genetic variants among patients who died with malignancy at an early age in Belarus.
Results: We identified one homozygous founder RAG1 variant out of 24 available DNA samples from 71 patients who developed lymphoma aged <3 years from the Belarusian cancer registry between 1986 and 2023. Our patient had an episode of pneumonia at 3 months of age and was hospitalized for respiratory distress, candida-positive lung disease, and lymphadenopathy at 14 months of age. The diagnosis of Epstein–Barr virus (EBV)-positive diffuse large B-cell lymphoma (DLBCL) was established. The patient had a normal lymphocyte count that decreased over time. One month after chemotherapy initiation, the patient died due to sepsis and multiple organ failure without a genetic diagnosis. In a retrospective analysis, T-cell receptor excision circles (TRECs) and kappa-deleting recombination excision circles (KRECs) were undetectable in peripheral blood.
Conclusions: A targeted screening program designed to detect a Slavic founder variant in the RAG1 gene among children revealed a 14-month-old Belarusian male infant with low TREC levels who died of EBV-driven DLBCL and complications of chemotherapy including infections. This case highlights how patients with IEI and recurrent infections may develop serious non-infectious complications, such as fatal malignancy. It also emphasizes the importance of early identification, such as newborn screening for severe combined immune deficiency. Earlier diagnosis of RAG deficiency could have prompted hematopoietic stem cell transplant well before the DLBCL occurrence. This likely would impact the onset and/or management strategies for the cancer.
Patients with inborn errors of immunity (IEI) have a much higher risk of developing cancer than the general population, in the range of 4%–25% (1, 2). There is some variability regarding the type of malignancy and its association with specific IEI. In patients with IEI, malignancies may occur at any age, including childhood, but on average they tend to manifest earlier than in the general population. In addition, there is a narrower age range with higher incidence of hematological malignancies than in the general population (3). Increased susceptibility to malignant neoplasms in IEI patients is mediated by a combination of intrinsic and extrinsic factors. Genetic disorders associated with impaired cell differentiation or apoptosis, cytoskeleton function, lymphocyte co-signaling, metabolism, cytotoxicity, and disorders leading to increased genotoxicity, such as chromosome instability, defective telomere maintenance and DNA repair, along with extrinsic causes, such as transforming viral infections and chronic tissue inflammation, contribute to the malignancy development (4, 5).
The most common IEI associated with increased cancer predisposition include DNA repair defects and/or IEI with susceptibility to oncogenic herpes group virus infection. Ataxia telangiectasia, Nijmegen breakage syndrome, and Bloom syndrome are the most frequently encountered DNA repair defects. Beside malignancy predisposition, these patients also have distinctive syndromic features that may expedite the diagnosis. In patients with combined immunodeficiencies (CIDs) with Epstein–Barr virus (EBV) susceptibility, infection may be one of the main triggers for the development of malignancy. These disorders include autoimmune lymphoproliferative syndromes, X-linked lymphoproliferative syndrome type I and regulatory T-cell disorders, and malignancy, mostly lymphoma (6–10).
Lymphoma is the most common tumor type in patients with IEI, with the risk of lymphoid malignancies being overall 8–10-fold higher (11). Non-Hodgkin lymphomas [diffuse large B-cell lymphoma (DLBCL), marginal zone, and Burkitt lymphomas] account for the vast majority of lymphomas observed in patients with IEI (12), whereas leukemia, Hodgkin's lymphoma, and solid tumors are less common in this group of patients (13).
We previously described 18 Slavic patients who were homozygous for the deleterious RAG1 p.K86VfsX33 (c.256_257delAA) variant. This study is the largest report in the literature of RAG variants within a geographically restricted population (14).
Here we report the case of a 14-month-old male infant affected by EBV-positive DLBCL in whom homozygosity for the same RAG1 Slavic founder variant was identified postmortem after targeted Sanger sequencing of 24 available DNA samples out of 71 patients who developed lymphoma aged <3 years from the Belarusian cancer registry between 1986 and 2023. In addition, we performed a literature search of malignancy occurrence in RAG deficiency.
We analyzed the data of the Belarusian cancer registry and found 71 patients (48 boys, 23 girls) who had been diagnosed with lymphoma before the age of 3 years in the period between 1986 and 2023. There were 48 non-Hodgkin's lymphomas, 9 Hodgkin's lymphomas, and 14 unspecified lymphomas (diagnosed before 1995). The age at clinical diagnosis was in the range of 1 month to 2 years 10 months (median 2 years 2 months). Of these patients, 38% were reported to be alive in 2024. Genetic material for DNA extraction was available in only 24 patients who comprised the study cohort. The cohort for sequencing included 17 boys and 7 girls, of whom 62.5% were alive at the time of the study (median age 11 years). The age at clinical manifestation of lymphoma in this group of patients ranged from 7 months to 2 years 10 months (median 2 years).
Homozygosity for the founder Slavic RAG1 p.K86VfsX33 variant was found in 1 of 24 samples of available DNA.
Previous studies in the same cohort of 24 patients had identified one homozygous patient for the founder Slavic UNC13D variant [p.Arg782SerfsTer12] and two patients had disease-associated deleterious variants [p.Pro465ArgfsTer82 and p.His321Tyr] in the FOXN1 gene. In total, 4 of the 24 patients were diagnosed with IEI.
A male infant was born full-term from non-consanguineous parents who originated from Brest (Western Belarus), after a second pregnancy and second delivery. The patient’s family history was unrevealing. He had received hepatitis B virus (HBV) (first dose) and bacille Calmette-Guérin (BCG) vaccine without adverse effects.
From the age of 3 months, the patient presented with a history of recurrent upper and lower (pneumonias) respiratory tract infections. At the age of 1 year 1 month, the patient was hospitalized because of fever, cough, dyspnea, lymphadenopathy, weakness, and refusal to eat. At physical examination, muscular hypotrophy of the second degree (weight 7.5 kg and height 75 cm), flabby skin, cyanosis of the nasolabial triangle, and respiratory distress were found. Chest radiograph showed bilateral polysegmental pneumonia (Figure 1B). A microbiological analysis of sputum found Candida.
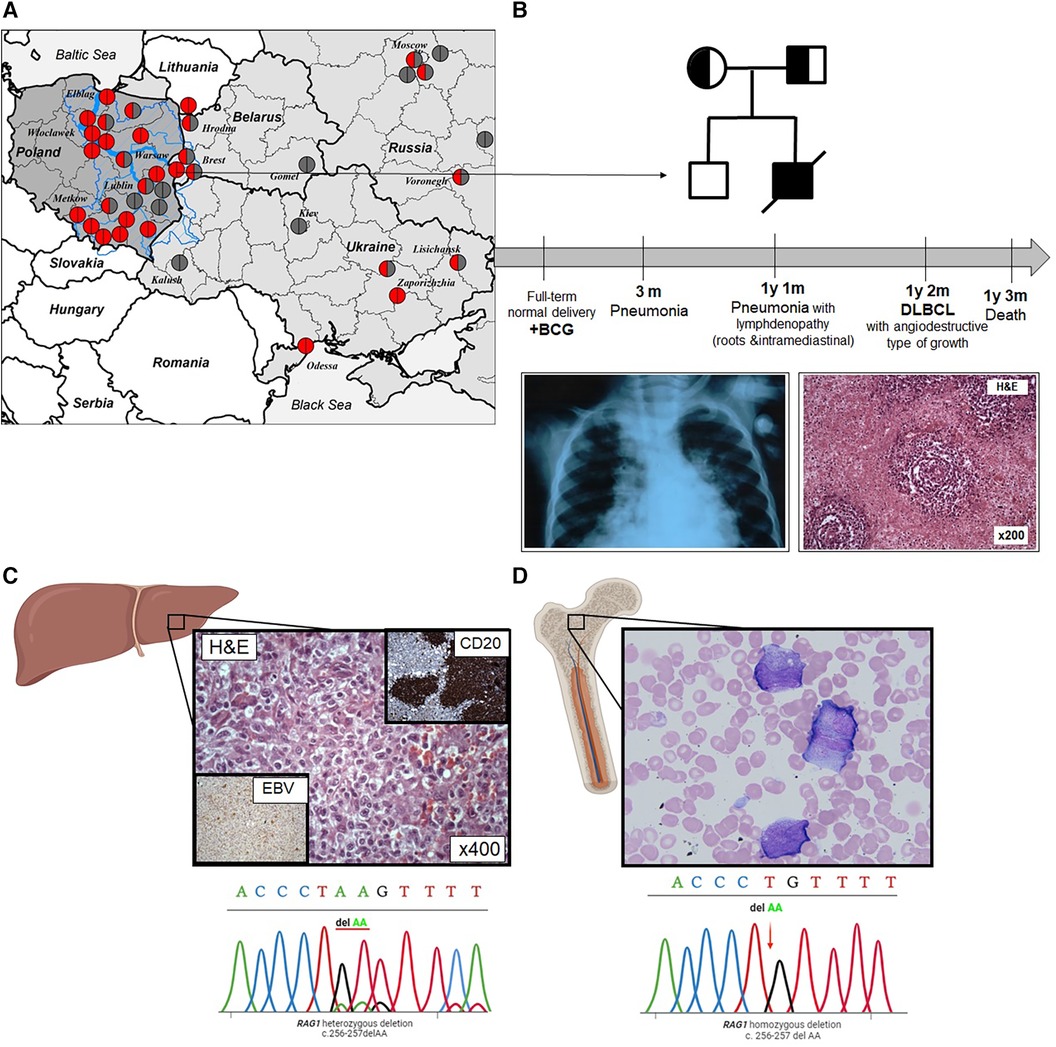
Figure 1 (A) Distribution map of Western and Eastern Slavic families with the RAG1 p.K86VfsX33 variant in Slavic countries; the Vistula river basin is overlaid on the map of Poland. The blue line is the Vistula River basin and its largest tributaries (Bug, Narev, San, Wieprz, Pilica, and Muchowiec), the geographic area coincides with the region of the largest concentration of families where patients with p.K86VfsX33 homozygous variants were born. The birthplace of the patients was indicated by the location of the circles; homozygous p.K86VfsTer33 variant is represented by red circles; heterozygous p.K86VfsTer33 variant is half red/half gray; and other variants are gray. (B) Pedigree of the proband; the mother and father are carriers; deceased (line through). Timeline summarizing main turning point of patient's clinical history. Chest radiograph at the age of 3 months: pneumonia; liver biopsy at the age of 1 year 2 months: lymphoma with angiodestructive type of growth. (C) Liver biopsy: solid shut of lymphoma cells, some of them resemble Reed–Sternberg cells and electropherograms of Sanger sequencing of RAG1 p.K86VfsX33 variant from lymphoma tissue. (D) Bone marrow smear: dissociation of neutrophil maturation cells and electropherograms of Sanger sequencing of RAG1 p.K86VfsX33 variant in the bone marrow.
An ultrasound examination showed hepato-(+3 cm)-spleno-(+3 cm)-megaly and lymphadenopathy. A lymph node biopsy was suggestive of DLBCL. Angiocentric lymphoid infiltrates of large, atypical cells and Hodgkin-like cells were accompanied by foci of coagulative necrosis. Large cells showed LCA, CD20, and patchy CD30 positivity. Immunostaining for EBV latent membrane protein was positive (Figure 1C). The lymphoma involved multiple lymph nodes (cervical, retroperitoneal, mediastinal, and abdominal) and multiple organs (liver, lungs, large and small intestines). Bone marrow investigation revealed dissociation of neutrophil maturation, hypochromia, and microanisocytosis of erythrocytes (Figure 1D).
Treatment was started according to the NHL-BFM-95 protocol with a dose reduction of one-third.
Laboratory investigation at the age of 1 year 1 month revealed progressive leukopenia (from 3,100 to 200 cells/µl), an imbalance in the ratio of stab and segmented neutrophils, and decreased hemoglobin (96 g/L). Lymphopenia (400 cells/µl), decreased T cells (300 cells/µl), and elevated activated T cells (41%) were detected. The percentage of B cells was elevated (33%), but the absolute number was slightly decreased (100 cells/µl). T-cell receptor excision circles (TRECs) and kappa-deleting recombination excision circles (KRECs) were undetectable (0 copies/106 leukocytes for both) and DNA was isolated from archived patient peripheral blood samples obtained at the time of hospitalization (1 year 3 months).
During the course of the lymphoma treatment, the patient's condition worsened. He developed sepsis (Pseudomonas aeruginosa and Enterococcus faecium), fibrinous-purulent peritonitis, and intestinal perforation. Resection of the ileocecal angle was performed. During surgery, biopsies of the liver, ileum, cecum, and ascending colon were taken. Extensive necrosis in the center and multiple nodular proliferations of diffuse large B-cell lymphoma were found in the tissues of the liver and intestines.
The patient died at the age of 1 year 3 months due to septic shock and multiple organ failure, 1 month after starting chemotherapy.
At 18 years after his death, targeted Sanger sequencing revealed a homozygous deletion in RAG1 gene (NM_000448.3) c.256_257del (p.Lys86ValfsTer33).
The recombination-activating gene 1 (RAG1) and 2 (RAG2) encode lymphoid-specific proteins that are expressed during the early stages of T-cell and B-cell development and initiate the process of V(D)J recombination by introducing DNA double-strand breaks (DSBs). The process of V(D)J recombination generates diverse T-cell and B-cell receptors capable of recognizing millions of possible antigens (15). Genotype–phenotype correlation is strong, as null variants of RAG1 and RAG2 genes result in the T-B-severe combined immune deficiency (SCID) phenotype, whereas hypomorphic RAG variants have been associated with distinct clinical entities, including Omenn syndrome (OS) and combined immunodeficiency with granuloma and/or autoimmunity (CID/G-AI) with herpesvirus infections and lymphoproliferation (16).
However, in RAG1/RAG2-deficient patients, as well as in overall SCID patients, hematopoietic neoplasms are extremely rare. This is presumably explained by the high frequency of life-threatening infections that require hematopoietic stem cell transplantation (HSCT) early in life. Currently, there are only six published cases of malignancy in five patients with RAG1 deficiency (Table 1) (17–21).
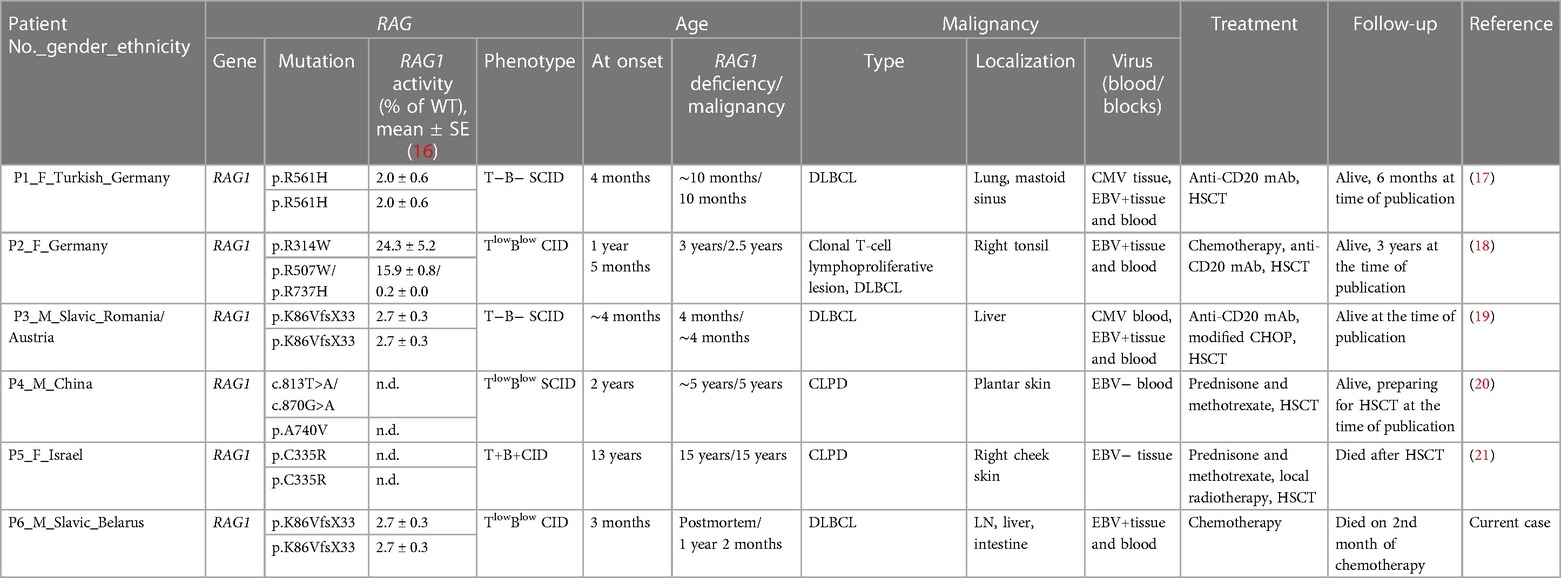
Table 1 Clinical data of patients with RAG deficiency and malignancies.
These patients had only two types of neoplasms: DLBCL (three cases) and cutaneous lymphoproliferative disease (CLPD) (three cases). CLPD manifested at a later age (2.5, 5, and 15 years) in patients with milder phenotype, which can be described as combined immunodeficiency or “leaky” SCID (20, 21). In contrast, DLBCL manifested earlier: in two cases, the diagnosis was established in the first year of life and occurred even in patients with typical SCID phenotype (17, 19). In only one patient with “leaky” SCID phenotype was DLBCL diagnosed at the age of 5 years (18). In the current case, DLBCL was established at the age of 1 year 2 months. Even though the patient had a history of severe infectious episodes and lymphopenia, a diagnosis of SCID was contemplated, but was not formally established during the patient's lifetime. EBV was detected in peripheral blood and tumor tissue in all four patients with DLBCL, including the current case. Rituximab therapy was initiated to rapidly control the EBV load. Patients with CLPD were treated with a combination of prednisone, methotrexate, and local radiation therapy (patient 5). All patients underwent HSCT except patient 4 who was being prepared for transplantation at the time of publication. Patient 5 did not recover owing to severe infection after HSCT.
Numerous lymphoma sequencing studies show that somatic mutations are often found in IEI genes. The most recurrently mutated genes in Hodgkin’s lymphoma and DLBCL include TNFAIP3, SOCS1, ITPKB, B2M, KMT2D, ATM, TP53, ACTB, and IRF4 (22–24).
Many studies showed that RAG endonucleases are involved in the pathogenesis of lymphomas and leukemias. Dysregulation of RAG expression may cause chromosomal translocations, which are a hallmark of hematopoietic neoplasms (25). RAG endonucleases are able to bind to nonamer-like sequences (cryptic nonamer) and cleave at adjacent mismatches resulting from activation-induced cytidine deaminase (AID)-mediated deamination of methylated CpG sites (26), thereby generating genomic instability. Whole genome sequencing of cutaneous T-cell lymphomas (CTCLs) revealed that RAG binding sites flanked a significant number of deletion breakpoints (27). Since CTCLs occur in older adults, this suggests that dysregulated re-expression of the RAG genes may occur in mature CD4+ T cells, leading to tumorigenesis. Complex genomic rearrangements such as chromothripsis, in which DSBs are widespread, have also been found in CTCLs (27).
Somatic mutations in the RAG1 and RAG2 genes are rare in lymphogenesis and leukemogenesis. According to data from the catalogue of somatic mutations in cancer (COSMIC), out of 8,381 samples of hematopoietic and lymphoid tissue studied, only 24 (0.29%) had point mutations in the RAG1 gene [https://cancer.sanger.ac.uk/cosmic, accessed May 2024]. These mutations have been found in acute myeloid leukemia, acute lymphoblastic B-cell leukemia, adult T-cell lymphoma/leukemia, plasma cell myeloma, diffuse large B-cell lymphoma, chronic lymphocytic leukemia-small lymphocytic lymphoma, mycosis fungoides-Sezary syndrome, and other cancers. A similar pattern is observed for the RAG2 gene: RAG2 somatic mutations have been reported in patients with Burkitt lymphoma, mycosis fungoides-Sezary syndrome, acute lymphoblastic B-cell leukemia, DLBCL, chronic lymphocytic leukemia, small lymphocytic lymphoma, and so on. Somatic point mutations in the RAG2 gene were detected in 18 (0.21%) of 8,393 hematopoietic and lymphoid tissue samples studied.
Approximately 15% of human cancers worldwide are caused by oncoviruses. Human oncogenic viruses include EBV, HBV, hepatitis C virus (HCV), high-risk human papillomaviruses (HPVs), human T-cell lymphotropic virus-1 (HTLV-1), Kaposi sarcoma-associated herpesvirus [KSHV; also known as human herpesvirus 8 (HHV-8), and Merkel cell polyomavirus (MCPyV)] (28). However, oncovirus infection alone is not sufficient to cause cancer. Within the context of multistep carcinogenesis, viral infection provides only a subset of the required oncogenic hits (29).
To successfully evade the immune response, oncoviruses have evolved powerful anti-apoptotic and proliferative programs. There are several basic oncogenic mechanisms. First, viruses encode proteins that are able to subvert, in a dominant manner, host-signaling mechanisms that regulate cell growth and survival. Second, recognition of viral genomes or replicative intermediates by the host leads to induction of the DNA damage response (DDR), which many oncoviruses need for their replication. Third, chronic inflammatory responses to persistent viral infection cause the formation of reactive oxygen species (ROS) that promote the acquisition of mutations (29).
Sheng et al. demonstrate that in a group of 329 patients with DLBCL, only 2.4% were EBV positive (23). According to our data, all patients with RAG1 deficiency had EBV-associated DLBCL. Evidence of the association between EBV positivity and mutation burden is mixed. Some studies have shown a strikingly lower number of somatic mutations in EBV-positive Hodgkin’s lymphomas (30).
A targeted screening program for searching the Slavic founder variant in RAG1 gene among Belarusian children who developed lymphoma aged <3 years revealed a 14-month-old Belarusian boy with low TREC levels who died of EBV-driven DLBCL and complications of chemotherapy including infections. This case highlights how patients with IEI and recurrent infections may develop serious non-infectious complications, such as fatal malignancy. It also stresses the importance of early identification, such as newborn screening for SCID, especially in the regions with domination of founder mutations. In addition, our case underlines the importance of resequencing the genomic DNA in patients with malignancy and somatic mutations in IEI genes.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by this study has been approved by the ethics committee of the Belarusian Research Center for Pediatric Oncology, Hematology and Immunology, Minsk, Belarus. Biological material was obtained on informed consent in accordance with the Declaration of Helsinki (IRB0012/2022). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
TV: Investigation, Writing – original draft, Writing – review & editing. EP: Investigation, Writing – original draft, Writing – review & editing. TM: Investigation, Writing – original draft, Writing – review & editing. IS: Writing – original draft, Writing – review & editing. AK: Writing – original draft, Writing – review & editing. AD: Investigation, Writing – original draft, Writing – review & editing. MB: Resources, Writing – original draft, Writing – review & editing. JD: Writing – original draft, Writing – review & editing. DV: Visualization, Writing – original draft, Writing – review & editing. LN: Writing – original draft, Writing – review & editing. JW: Writing – original draft, Writing – review & editing. SS: Conceptualization, Supervision, Visualization, Writing – original draft, Writing – review & editing.
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
LN is supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA (grant AI001222). JW is supported by National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA (grant # R01AI153830), Jeffrey Modell Foundation, and the Robert A. Good Endowment at the University of South Florida.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mueller BU, Pizzo PA. Cancer in children with primary or secondary immunodeficiencies. J Pediatr. (1995) 126(1):1–10. doi: 10.1016/S0022-3476(95)70491-4
2. Shapiro RS. Malignancies in the setting of primary immunodeficiency: implications for hematologists/oncologists. Am J Hematol. (2011) 86(1):48–55. doi: 10.1002/ajh.21903
3. Mastio J, Saeed MB, Wurzer H, Krecke M, Westerberg LS, Thomas C. Higher incidence of B cell malignancies in primary immunodeficiencies: a combination of intrinsic genomic instability and exocytosis defects at the immunological synapse. Front Immunol. (2020) 11:581119. doi: 10.3389/fimmu.2020.581119
4. Hauck F, Voss R, Urban C, Seidel MG. Intrinsic and extrinsic causes of malignancies in primary immunodeficiency disorders. J Allergy Clin Immunol. (2018) 141(1):59–68.e4. doi: 10.1016/j.jaci.2017.06.009
5. Kebudi R, Kiykim A, Sahin MK. Primary immunodeficiency and cancer in children; a review of the literature. Curr Pediatr Rev. (2019) 15(4):245–50. doi: 10.2174/1573396315666190917154058
6. Sharapova SO, Pashchenko OE, Bondarenko AV, Vakhlyarskaya SS, Prokofjeva T, Fedorova AS, et al. Geographical distribution, incidence, malignancies, and outcome of 136 Eastern Slavic patients with Nijmegen breakage syndrome and NBN founder variant c. 657_661del5. Front Immunol. (2021) 11:602482. doi: 10.3389/fimmu.2020.602482
7. Sharapova SO, Valochnik AV, Guryanova IE, Sakovich IS, Aleinikova OV. Novel biallelic ATM mutations coexist with a mosaic form of triple X syndrome in an 11-year-old girl at remission after T cell acute leukemia. Immunogenetics. (2018) 70(9):613–7. doi: 10.1007/s00251-018-1056-4
8. Sharapova SO, Fedorova AS, Pashchenko OE, Vahliarskaya SS, Guryanova IE, Migas AA, et al. Novel mutations in SH2D1A gene in X-linked lymphoproliferative syndrome, diagnosed after B-cell non-Hodgkin lymphoma. J Pediatr Hematol Oncol. (2017) 39(4):e203–6. doi: 10.1097/MPH.0000000000000815
9. Sharapova SO, Golovataya EI, Shepelevich EV, Mareika YE, Guryanova IE, Stegantseva MV, et al. Nijmegen breakage syndrome in two half sibs with peripheral T-cell lymphoma and cortical T-cell acute lymphoid leukemia. Cent Eur J Immunol. (2020) 45(4):507–10. doi: 10.5114/ceji.2020.103387
10. Barmettler S, Sharapova SO, Milota T, Greif PA, Magg T, Hauck F. Genomics driving diagnosis and treatment of inborn errors of immunity with cancer predisposition genomics driving diagnosis and treatment of inborn errors of immunity with cancer predisposition. J Allergy Clin Immunol Pract. (2022) 10(7):1725–36.e2. doi: 10.1016/j.jaip.2022.03.012
11. Mayor PC, Eng KH, Singel KL, Abrams SI, Odunsi K, Moysich KB, et al. Cancer in primary immunodeficiency diseases: cancer incidence in the United States immune deficiency network registry. J Allergy Clin Immunol. (2018) 141(3):1028–35. doi: 10.1016/j.jaci.2017.05.024
12. Riaz IB, Faridi W, Patnaik MM, Abraham RS. A systematic review on predisposition to lymphoid (B and T cell) neoplasias in patients with primary immunodeficiencies and immune dysregulatory disorders (inborn errors of immunity). Front Immunol. (2019) 10:777. doi: 10.3389/fimmu.2019.00777
13. Tavakol M, Delavari S, Salami F, Ansari S, Rasouli SE, Chavoshzadeh Z, et al. Diversity of malignancies in patients with different types of inborn errors of immunity. Allergy Asthma Clin Immunol. (2022) 18(1):106. doi: 10.1186/s13223-022-00747-2
14. Sharapova SO, Skomska-Pawliszak M, Rodina YA, Wolska-Kuśnierz B, Dabrowska-Leonik N, Mikołuć B, et al. The clinical and genetic spectrum of 82 patients with RAG deficiency including a c.256_257delAA founder variant in slavic countries. Front Immunol. (2020) 11:900. doi: 10.3389/fimmu.2020.00900
15. Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. (2016) 16(4):234–46. doi: 10.1038/nri.2016.28
16. Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol. (2014) 133(4):1099–108. doi: 10.1016/j.jaci.2013.10.007
17. Ehl S, Schwarz K, Enders A, Duffner U, Pannicke U, Kühr J, et al. A variant of SCID with specific immune responses and predominance of γδ T cells. J Clin Invest. (2005) 115(11):3140–8. doi: 10.1172/JCI25221
18. Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. New Engl J Med. (2008) 358(19):2030–8. doi: 10.1056/NEJMoa073966
19. Maas C, Lüftinger R, Krois W, Matthes-Martin S, Bayer G, Boztug K, et al. EBV-positive B-cell lymphoma manifestation of the liver in an infant with RAG1 severe combined immunodeficiency disease. Pediatr Blood Cancer. (2018) 65(9):e27258. doi: 10.1002/pbc.27258
20. Xu CC, Chen ZM, Xiong JS, Gan L, Zhang Y, Chen H, et al. Novel compound heterozygous mutations in RAG1 in a patient with cutaneous lymphoproliferative disease. Acta Derm Venereol. (2019) 99(1):105–6. doi: 10.2340/00015555-3042
21. Avitan-Hersh E, Stepensky P, Zaidman I, Nevet MJ, Hanna S, Bergman R. Primary cutaneous clonal CD8+T-cell lymphoproliferative disorder associated with immunodeficiency due to RAG1 mutation. Am J Dermatopathol. (2020) 42(1):e11–5. doi: 10.1097/DAD.0000000000001492
22. Gomez F, Fisk B, McMichael JF, Mosior M, Foltz JA, Skidmore ZL, et al. Ultra-deep sequencing reveals the mutational landscape of classical Hodgkin lymphoma. Cancer Res Commun. (2023) 3(11):2312–30. doi: 10.1158/2767-9764.CRC-23-0140
23. Sheng L, Fu D, Cao Y, Huo Y, Wang S, Shen R, et al. Integrated genomic and transcriptomic analyses of diffuse large B-cell lymphoma with multiple abnormal immunologic markers. Front Oncol. (2022) 12:790720. doi: 10.3389/fonc.2022.790720
24. Spina V, Bruscaggin A, Cuccaro A, Martini M, Di Trani M, Forestieri G, et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood. (2018) 131(22):2413–25. doi: 10.1182/blood-2017-11-812073
25. Desai SS, Whadgar S, Raghavan SC, Choudhary B. MiRAGDB: a knowledgebase of RAG regulators. Front Immunol. (2022) 13:863110. doi: 10.3389/fimmu.2022.863110
26. Paranjape AM, Desai SS, Nishana M, Roy U, Nilavar NM, Mondal A, et al. Nonamer dependent RAG cleavage at CpGs can explain mechanism of chromosomal translocations associated to lymphoid cancers. PLoS Genet. (2022) 18(10):e1010421. doi: 10.1371/journal.pgen.1010421
27. Damsky WE, Choi J. Genetics of cutaneous T cell lymphoma: from bench to bedside. Curr Treat Options Oncol. (2016) 17(7):33. doi: 10.1007/s11864-016-0410-8
28. Morales-Sánchez A, Fuentes-Pananá EM. Human viruses and cancer. Viruses. (2014) 6(10):4047–79. doi: 10.3390/v6104047
29. Mesri EA, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe. (2014) 15(3):266–82. doi: 10.1016/j.chom.2014.02.011
Keywords: RAG deficiency, lymphoma, malignancy in SCID, infant, case report
Citation: Volodashchik TP, Polyakova EA, Mikhaleuskaya TM, Sakovich IS, Kupchinskaya AN, Dubrouski Aliaxandr Ch., Belevtsev MV, Dasso JF, Varabyou DS, Notarangelo LD, Walter JE and Sharapova SO (2024) Infant with diffuse large B-cell lymphoma identified postmortem with homozygous founder Slavic RAG1 variant: a case report and literature review. Front. Pediatr. 12:1415020. doi: 10.3389/fped.2024.1415020
Received: 9 April 2024; Accepted: 13 June 2024;
Published: 4 July 2024.
Edited by:
Catharina Schuetz, University Hospital Carl Gustav Carus, GermanyReviewed by:
Andrew R. Gennery, Newcastle University, United Kingdom© 2024 Volodashchik, Polyakova, Mikhaleuskaya, Sakovich, Kupchinskaya, Dubrouski, Belevtsev, Dasso, Varabyou, Notarangelo, Walter and Sharapova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Svetlana O. Sharapova, c2hhcmFwb3Zhc3ZAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.