 Jianling Cai1,†
Jianling Cai1,† Dongqing Zhang
Dongqing Zhang- 1Department of Pediatrics, The First Affiliated Hospital of Shantou University Medical College, Shantou, Guangdong, China
- 2Department of Laboratory Medicine, The First Affiliated Hospital of Shantou University Medical College, Shantou, Guangdong, China
- 3Department of Clinical Medicine, Shantou University Medical College, Shantou, Guangdong, China
- 4Department of Pharmacology, Shantou University Medical College, Shantou, Guangdong, China
Background: Congenital sideroblastic anemia (CSA) constitutes a group of inherited erythropoietic disorders. Some affect mainly or exclusively erythroid cells; other syndromic forms occur within multisystem disorders with extensive nonhematopoietic manifestations. In this study, we have performed clinical and molecular investigations on a 10-year-old boy suspected of having CSA.
Methods: Routine blood examination, peripheral blood and bone marrow smears, and serum iron tests were performed. Gene mutation analysis was conducted using whole-exome sequencing (WES) and the results were confirmed using Sanger sequencing. Furthermore, the functional impact of the identified variant was assessed/predicted with bioinformatics methods.
Results: The patient presented with severe microcytic anemia (hemoglobin, 50 g/L), iron overload and ring sideroblasts in the bone marrow. Moreover, WES revealed the presence of a hemizygous missense variant in ALAS2 (c.1102C > T), changing an encoded arginine to tryptophan (p. Arg368Trp). This variant was verified via Sanger sequencing, and neither of the parents carried this variant, which was suspected to be a de novo variant. Using in silico analysis with four different software programs, the variant was predicted to be harmful. PyMol and LigPlot software showed that the p. Arg368Trp variant may result in changes in hydrogen bonds. The patient was treated with vitamin B6 combined with deferasirox. After 6 months, the hemoglobin increased to 99 g/L and the serum ferritin decreased significantly.
Conclusion: We report a novel pathogenic variant in the ALAS2 gene (c.1102C > T:p. Arg368Trp), which caused CSA in a 10-year-old boy. Mutational analysis is important in patients with CSA when family history data are unavailable. Anemia due to the ALAS2 Arg368Trp variant responds to pyridoxine supplements.
1 Introduction
Sideroblastic anemias (SAs) are a heterogeneous group of disorders characterized by the presence of abnormal erythroid precursors with perinuclear mitochondrial iron deposition in the bone marrow. Although their prevalence has not been defined, they are uncommon and occur in congenital and acquired forms (1–3). In general, there are two clinical manifestations of congenital sideroblastic anemia (CSA). One form affects mainly or exclusively erythroid cells, which is non-syndromic with only presenting hematopoietic disorders. Another form has associated syndromic features, which involves multiple system disorders. Nevertheless, many of CSA have mutations in genes involved in heme biosynthesis, iron-sulfur cluster biosynthesis, and mitochondrial protein synthesis. To date, over 10 pathogenic genes associated with CSA have been reported, including ALAS2, SLC25A38, GLRX5, HSPA9, HSCB, ABCB7, PUS1, YARS2, LARS2, TRNT1, MT-ATP6, NDUFB11, and SLC19A2 (4–7). X-linked sideroblastic anemia (XLSA), which is the most common form of non-syndromic CSA. It was caused by variants in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene (1). Here, we report the case of a patient presenting with characteristics of CSA in whom we identified a novel pathogenic variant in the ALAS2 gene.
2 Materials and methods
2.1 Patient
A 10-year-old boy was admitted to another hospital with a 3-year history of chronic anemia. Due to born in the area, Guangdong province in PRC, with high incidence of thalassemia, this patient was conducted with the thalassemia genetic testing. The result was normal, with no large deletion/duplications in the alpha-globin gene cluster or pathogenic variants in the beta-globin gene. Bone marrow examination showed erythroid-predominant trilineage hematopoiesis. Although his condition improved after several red blood cell transfusions, the diagnosis was unclear. However, when symptoms worsened after 3 months, he visited our hospital for further evaluation.
2.2 Routine examination
Samples were collected from the patient and his parents. Blood tests were performed using an LH 780 automated hematology analyzer (Beckman Coulter, CA, USA). Iron metabolism tests, including serum iron and ferritin, were performed using a 7,600 Fully Automated Biochemical Analyzer (Hitachi, Ltd., Tokyo, Japan). Bone marrow smears were obtained, and Perls’ staining was performed.
2.3 Whole-exome sequencing
Genomic DNA was analyzed using whole-exome sequencing (WES). To this end, 2 ml of peripheral blood was collected from the patient and his parents. Genomic DNA was isolated from leukocytes using an QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Exomes were captured using a TruSight One Sequencing Panel (Illumina Inc., USA), and sequencing was conducted using an Illumina HiSeq (Illumina Inc., USA). The results were aligned with the University of California, Santa Cruz Human Genome Assembly 19 (UCSC.hg19; http://genome.ucsc.edu/) reference sequence. Variants were filtered using several common variant databases, including the 1,000 genome database, dbSNP database, ClinVar database, Polymorphism Phenotyping v2 software, and Sorting Intolerant from Tolerant algorithm. The pathogenicity of the filtered candidate variants was determined according to the ACMG (American College of Medical Genetics and Genomics) guidelines (8). The ACMG classifications of variations include pathogenic, likely pathogenic, uncertain significance, likely benign, and benign. Based on the patient's clinical history, variants in candidate genes linked to CSA were prioritized.
2.4 Validation of the candidate variant by sanger sequencing
The variant that most likely contributed to the disease was verified by Sanger sequencing. Briefly, the selected ALAS2 gene region, including the Arg368Trp variant, was amplified by Polymerase chain reactions (PCR) and sequenced by Sanger sequencing. PCR amplification was performed using an ABI system. The PCR products were loaded onto an ABI3500XL. The results were analyzed using Variant Reporter v 1.1 software and Sequencing Analysis 5.2 software. Primer pairs were designed using the PrimerQuest Tool (http://sg.idtdna.com/Primerquest/Home/Index). The primers used to confirm the ALAS2 Arg368Trp variant were as follows:
Forward primer: 5′AGCTGGGGAAGGGTTATGAT3′
Reverse primer: 5′GGGAGGAGGCAGAAAAGAAT3′
2.5 Mutation analysis for ALAS2
Bioinformatics software was used to analyze and predict the effect of the missense variant (p. Arg368Trp) on ALAS2 functioning. For this purpose, the PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.bii.a-star.edu.sg, FATHMM (http://fathmm.biocompute.org.uk), and Mutation Assessor (http://mutationassessor.org) bioinformatics programs were selected. To study the effect of c.1102C > T on the three-dimensional (3D) crystal structure of the protein, computational modeling was performed for normal and mutant ALAS2 molecules using the online software program Swiss-model (https://swissmodel.expasy.org/). The template protein structure was obtained from the Protein Data Bank (https://www.rcsb.org/pdb/home/home.do). PyMol and LigPlot software were employed to perform a comparative analysis, make necessary modifications, and visualize the molecular structures of both the wild-type and variant forms of the ALAS2 protein.
3 Results
3.1 Clinical features
Clinical examination was normal except for severe anemia and splenomegaly. Abdominal ultrasonographic findings revealed splenomegaly (spleen length: 148 mm, thickness: 51 mm, subcostal: 38 mm). The complete blood count was as follows: white blood cells (WBC), 4.2 × 109/L (normal value, 4.00–10.00 × 109/l); red blood cells (RBC), 2.51 × 1012/l (normal value, 4.20–5.70 × 1012/L); Hemoglobin (Hb), 50 g/L (normal value, 118–156 g/L); mean corpuscular volume (MCV), 65.3 fL (normal value, 77.0–92.0 fL); mean corpuscular hemoglobin (MCH), 19.9 pg (normal value, 25.0–34.0 pg); mean corpuscular hemoglobin concentration (MCHC), 305 g/L (normal value, 310–355 g/L); red cell distribution width (RDW), 32.0% (normal value, 11.5–14.5%); reticulocyte (RET), 2.53% (normal value, 0.50%–2.50%); platelet (PLT), 170 × 109/L (normal value, 167–453 × 109/L). Tests for iron levels indicated iron overload, with a serum iron level of 40.33 µmol/L (normal value, 9.00–22.00 µmol/L) and a ferritin level of 2,267.0 µg/L (normal value, 6.1–252.5 µg/L). Red cell morphology showed marked microcytosis, a few red cell fragmentations, and anisopoikilocytosis. Bone marrow aspiration revealed erythroid hyperplasia with no dysplastic features. Perls’ staining revealed increased iron stores with the presence of ring sideroblasts in 36% of the developing erythrocytes (Figure 1A).
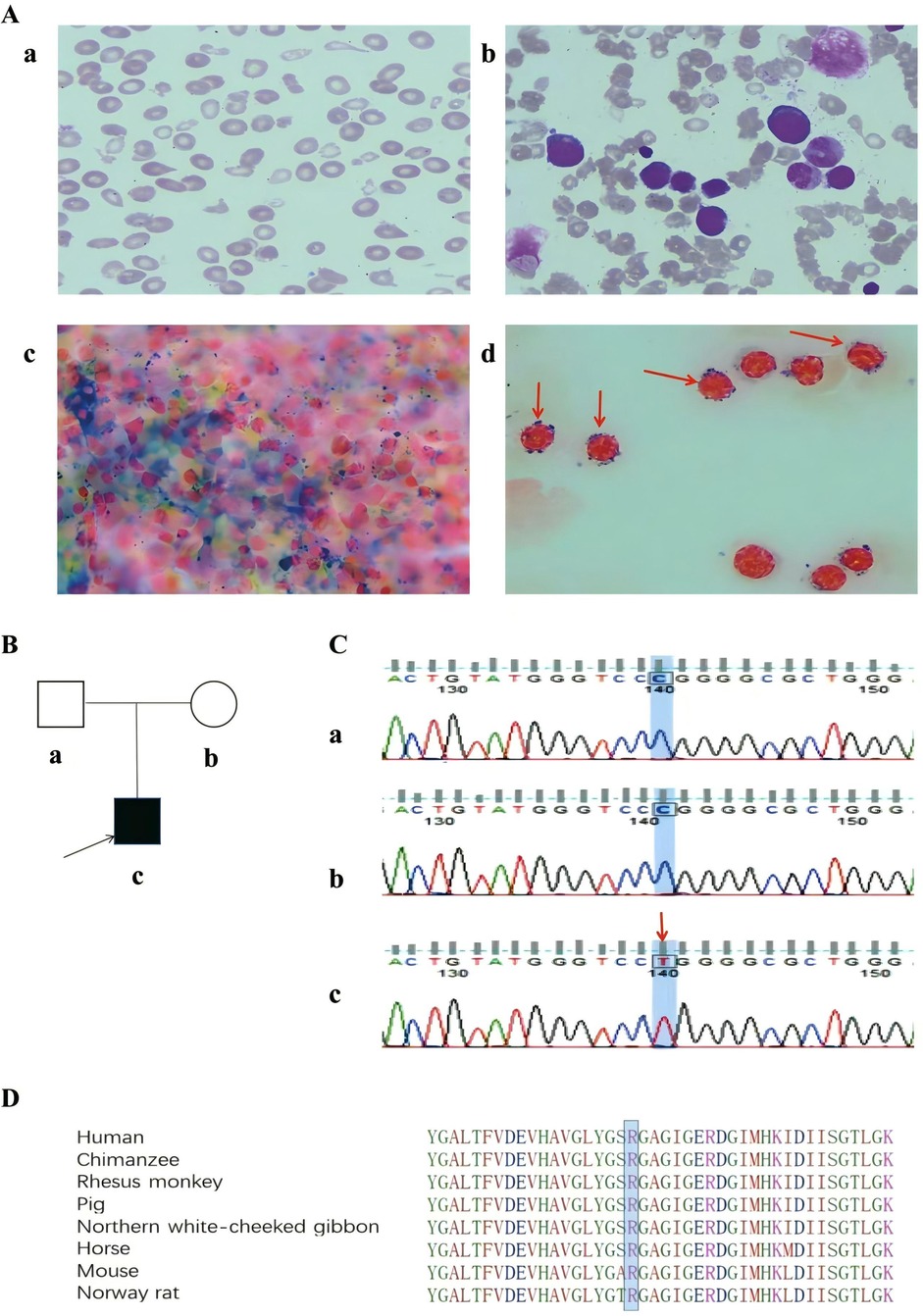
Figure 1. (A) Peripheral blood and bone marrow morphology of the patient. (a) Peripheral blood smear showing anisopoikilocytosis, microcytosis, and hypochromia (Wright-Giemsa stain, ×1000). (b) Bone marrow smear showing erythroid hyperplasia without dysplasia (Wright-Giemsa stain, ×1000). (c, d) Perls' staining showing increased iron stores with the presence of ring sideroblasts (Perls' stain, ×1000); the red arrow indicates ring sideroblasts. (B) Pedigree of the family. (a) Patient's father, (b) Patient's mother, (c) Patient. (C) Genetic analysis of the family: A novel variant in exon 8 (c.1102C>T) of the ALAS2 gene in the patient. The red arrow indicates point variant. (a) Patient's father, (b) Patient's mother, (c) Patient; (D) Bioinformatics conservative analysis among humans, mice, Norway rats, chimpanzees, rhesus monkeys, horses, pigs, and northern white-cheeked gibbons at the variant position.
3.2 DNA analysis
WES was performed on the proband, and 13G of raw data was obtained. The proportions of base quality values ≥30 (Q30) were 93.38%. The average sequencing depth for the target region was 136X, with more than 99% of the target sequence reaching up to 20X. A missense variant (c.1102C > T: p. Arg368Trp) in exon 8 of the ALAS2 gene was identified. Sanger sequencing confirmed that the variant was hemizygous in the proband but absent from his parents (Figures 1B,C). According to the ACMG guidelines, the variant was classified as variant of uncertain significance (PM6 + PM2_supporting + PP4). Based on the amino acid sequence of ALAS2 protein in the PUBMED website, we selected eight species, including humans, mice, Norway rats, chimpanzees, rhesus monkeys, horses, pigs, and northern white-cheeked gibbons, for the homology analysis of ALAS2 protein sequences. We found that ALAS2 is highly conserved across these species at the variant position (Figure 1D).
3.3 Prediction and analysis of online protein structure
Polyphen-2, SIFT, FATHMM, and MutationAssessor were used to predict the pathogenicity of the ALAS2 variant (c.1102C > T), as shown in Table 1. PyMol and LigPlot were used to predict changes in hydrogen bonds when tryptophan was substituted with arginine at position 368 of ALAS2 protein. Arg368 is involved in the formation of hydrogen bonds with glutamic acid (Glu) residues at positions 245 and 374. PyMol analyses indicated that the Arg368Trp variant had no effect on hydrogen bonds with Glu374, but it disrupted the hydrogen bonds with Glu245. LigPlot analyses indicated that the variant disrupted hydrogen bonds with both Glu245 and Glu374 (Figure 2).
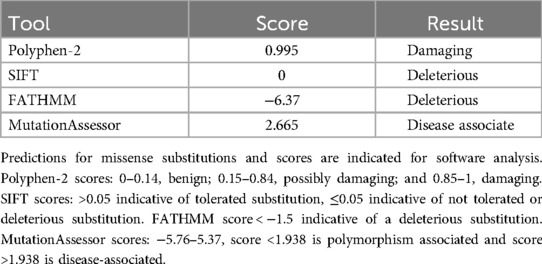
Table 1. In silico analysis of ALAS2 variant (c.1102C>T).
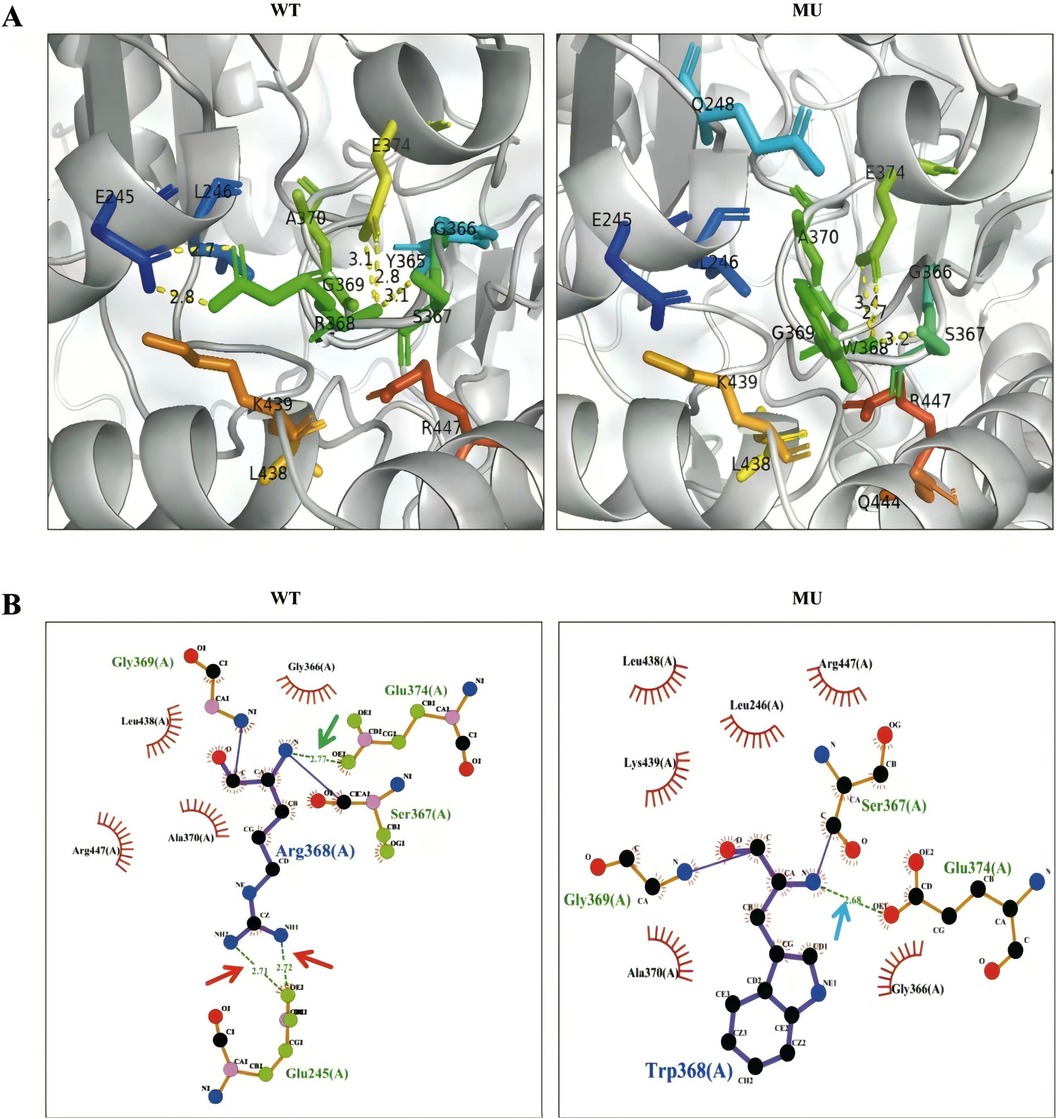
Figure 2. Structural analysis between wild-type (WT) and mutant-type ALAS2 protein. (A) PyMol software showed that the Arg368Trp variant disrupted hydrogen bonds with Glu245. The WT ALAS2 protein R368 forms two hydrogen bonds with E245 and two hydrogen bonds with E374, whereas the ALAS2 mutant protein W368 forms only two hydrogen bonds with E374. (B) LigPlot software showed that the Arg368Trp variant disrupted hydrogen bonds with both Glu245 and Glu374. The WT ALAS2 protein Arg368 forms two hydrogen bonds with Glu245 (red arrow) and one hydrogen bond with Glu374 (green arrow), whereas the ALAS2 mutant protein Trp368 forms only one hydrogen bond with Gly366 (blue arrow).
3.4 Case follow-up
The patient was treated with vitamin B6 (60 mg/day) and deferasirox (250 mg/day). He responded well, with a gradual increase in Hb and a gradual decrease in ferritin. After 6 months, his Hb level increased to 99 g/L and ferritin level decreased to 924.3 µg/L. However, the microscopic hypochromic features of erythrocytes remained, which showed little improvement (Table 2). Abdominal ultrasonographic findings showed a considerable reduction in the size of the spleen (spleen length: 137 mm, thickness: 42 mm, subcostal: 12 mm).
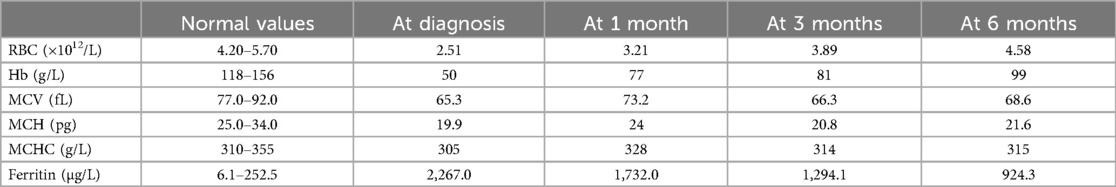
Table 2. Hematological findings before and after treatment with vitamin B6.
4 Discussion
Ring sideroblasts in the bone marrow are the hallmark of sideroblastic anemias. The child presented with microcytic anemia, high serum ferritin levels, and notably ring sideroblasts in the bone marrow. The possibility of CSA was considered based on young age at disease onset, spleen enlargement, and lack of a history of exposure to drugs or toxins. However, the parents of the child were healthy. Currently, only molecular testing can be performed to determine the cause. Analysis using WES revealed a hemizygous missense variant in exon 8 of the ALAS2 gene (c.1102C > T:p. Arg368Trp), which has not been previously reported. Because the variant was classified as of unknown significance according to ACMG, the functional impact of the variant was further analyzed using bioinformatics methods. Multiple sequence alignment of ALAS2 from several species revealed that Arg368 was located within a highly conserved region. The p. Arg368Trp variant was predicted to be harmful by four separate bioinformatic software programs. In addition, the molecular visualization system revealed the changes in hydrogen bonds. Hydrogen bonds are essential interatomic bonds to protein molecules because they maintain the shape of proteins. Generally, more hydrogen bonds make the protein more stable (9). When tryptophan is substituted for arginine at position 368, the hydrogen bonds are reduced, potentially leading to structural and functional alterations in the protein. Consequently, such a substitution is considered damaging. We have not formally validated the Arg368Trp variant as pathogenic with a functional assay. However, based on our in silico analysis, combined with the significant therapeutic improvement of the patient after vitamin B6 supplementation, which is typical of XLSA, we concluded that the Arg368Trp variant is likely pathogenic.
Because ALAS2 is located on the X chromosome, CSA caused by the mutation of the ALAS2 exhibits an X-linked inheritance pattern (10). Most males carrying the XLSA allele will express the disease, and many females carrying the allele will do as well (11, 12). In our case, although the child carried a pathogenic variant of the ALAS2 gene, sequencing data showed that the parents did not carry this variant; thus, the variant is likely be de novo. An et al. reported that 3 of 9 patients with XLSA had de novo mutations, suggesting that the disease has a somewhat high de novo mutation frequency (13). To date, approximately 119 pathogenic variants in the ALAS2 have been reported in patients with XLSA, and the mutations are mainly attributed to the hypermutability of CpG dinucleotides, which also contributes to the relative frequency of de novo mutations in this disease (2, 14).
ALAS2 is the first enzyme in the heme biosynthesis pathway in erythroid cells. It uses pyridoxal phosphate (PLP) as a coenzyme to catalyze the synthesis of glycine and succinyl-coA into delta aminolevulinic acid (ALA) (15). ALAS2 consists of 587 amino acids, comprising an N-terminal region (aa 1–142), a conserved catalytic core (aa 143–544), and a C-terminal extension (aa 545–587) (16). Researchers reported that most of the pathogenic variants are located in exon 5 and exon 9, the latter containing the PLP-binding binding site, lysine 391. Generally, patients with variants close to the PLP-binding site respond favourably to pyridoxine therapy, whereas those with variants distant from the site are refractory to treatment (17–19). In our patient, the Arg368Trp variant being located in the catalytic domain and near the PLP-binding site along with the weakened hydrogen bonds would explain the favorable response to pyridoxine supplements.
Blood transfusion is currently the primary treatment in patients with pyridoxine-resistant severe CSA. However, long-term transfusion is associated with the risk of transfusion-related complications and iron overload, which can lead to organ dysfunction (20, 21). Hematopoietic stem cell transplantation is the only curative option, but there are challenges, such as graft-versus-host disease (GVHD) (22). Luspatercept is a specific activin receptor fusion protein that acts as a ligand trap to neutralize negative regulators of late-stage erythropoiesis and is mainly used to manage and treat anemia in myelodysplastic syndrome, primary myelofibrosis, and beta-thalassemia (23, 24). Recently, luspatercept has been used to treat CSA. Van et al. reported that in a transfusion-dependent 51-year-old man with XLSA, the use of luspatercept led to a 30% increase in Hb levels and a decreased need for transfusion, suggesting its efficacy in the treatment of CSA. However, more clinical trials are needed to validate this approach (25).
In summary, we report a novel pathogenic variant in the ALAS2 gene (c.1102C > T: p. Arg368Trp), causing XLSA in a 10-year-old boy and expanding the spectrum of ALAS2 variants. The patient responded well to pyridoxine supplementation. Rapid and accurate diagnosis led to effective treatment and a favorable clinical course, thereby reducing long-term complications.
Data availability statement
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (https://bigd.big.ac.cn/). After publication of study findings, the data will be available for others to request. The research team will provide accession number of the database once the data are approved to be shared with others. The proposal with description of study objectives will be needed for evaluation of the reasonability to request for the data. The corresponding authors and the Ethical Committee of the First Affiliated Hospital of Shantou University Medical College will make a decision based on these materials.
Ethics statement
The studies involving humans were approved by the First Affiliated Hospital of Shantou University Medical College. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
JC: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. TL: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. YH: Data curation, Writing – original draft. HC: Data curation, Writing – original draft. MY: Data curation, Writing – original draft. DZ: Conceptualization, Investigation, Methodology, Writing – original draft, Writing – review & editing. ZH: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bottomley SS, Fleming MD. Sideroblastic anemia: diagnosis and management. Hematol Oncol Clin North Am. (2014) 28:653–70, v. doi: 10.1016/j.hoc.2014.04.008
2. Ducamp S, Fleming MD. The molecular genetics of sideroblastic anemia. Blood. (2019) 133:59–69. doi: 10.1182/blood-2018-08-815951
3. Rodriguez-Sevilla JJ, Calvo X, Arenillas L. Causes and pathophysiology of acquired sideroblastic anemia. Genes (Basel). (2022) 13:1562. doi: 10.3390/genes13091562
4. Long Z, Li H, Du Y, Han B. Congenital sideroblastic anemia: advances in gene mutations and pathophysiology. Gene. (2018) 668:182–9. doi: 10.1016/j.gene.2018.05.074
5. Heeney MM, Berhe S, Campagna DR, Oved JH, Kurre P, Shaw PJ, et al. SLC25A38 Congenital sideroblastic anemia: phenotypes and genotypes of 31 individuals from 24 families, including 11 novel mutations, and a review of the literature. Hum Mutat. (2021) 42:1367–83. doi: 10.1002/humu.24267
6. Colin E, Courtois G, Brouzes C, Pulman J, Rabant M, Rötig A, et al. Biallelic mutations in the SARS2 gene presenting as congenital sideroblastic anemia. Haematologica. (2021) 106:3202–5. doi: 10.3324/haematol.2021.279138
7. Xiong S, Jia Y, Li S, Huang P, Xiong J, Mao D, et al. The first case report of X-linked sideroblastic anemia with ataxia of Chinese origin and literature review. Front Pediatr. (2021) 9:692459. doi: 10.3389/fped.2021.692459
8. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
9. Mi H, Muruganujan A, Thomas PD. PANTHER In 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. (2013) 41:D377–86. doi: 10.1093/nar/gks1118
10. Cotter PD, Willard HF, Gorski JL, Bishop DF. Assignment of human erythroid delta-aminolevulinate synthase (ALAS2) to a distal subregion of band Xp11.21 by PCR analysis of somatic cell hybrids containing X; autosome translocations. Genomics. (1992) 13:211–2. doi: 10.1016/0888-7543(92)90223-f
11. Qiu Y, Cai H, Cui L, Liu YX, Wang YN, Li J, et al. Identification of a novel heterozygous ALAS2 mutation in a young Chinese female with X-linked sideroblastic anemia. Ann Hematol. (2020) 99:371–3. doi: 10.1007/s00277-019-03894-6
12. Nzelu D, Shangaris P, Story L, Smith F, Piyasena C, Alamelu J, et al. X-linked sideroblastic anaemia in a female fetus: a case report and a literature review. BMC Med Genomics. (2021) 14:296. doi: 10.1186/s12920-021-01146-z
13. An WB, An WB, Liu C, Wan Y, Guo Y, Wang SC, et al. [Clinical features and gene mutation spectrum in children with sideroblastic anemia]. Zhongguo Dang Dai Er Ke Za Zhi. (2019) 21:1016–21. doi: 10.7499/j.issn.1008-8830.2019.10.012
14. HGMD® gene result. (2023). Available online at: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ALAS2 (cited May 10).
15. Cotter PD, Rucknagel DL, Bishop DF. X-linked sideroblastic anemia: identification of the mutation in the erythroid-specific delta-aminolevulinate synthase gene (ALAS2) in the original family described by cooley. Blood. (1994) 84:3915–24.7949148
16. Bailey HJ, Bezerra GA, Marcero JR, Padhi S, Foster WR, Rembeza E, et al. Human aminolevulinate synthase structure reveals a eukaryotic-specific autoinhibitory loop regulating substrate binding and product release. Nat Commun. (2020) 11:2813. doi: 10.1038/s41467-020-16586-x
17. Shoolingin-Jordan PM, Al-Daihan S, Alexeev D, Baxter RL, Bottomley SS, Kahari ID, et al. 5-Aminolevulinic Acid synthase: mechanism, mutations and medicine. Biochim Biophys Acta. (2003) 1647:361–6. doi: 10.1016/s1570-9639(03)00095-5
18. Méndez M, Moreno-Carralero MI, Morado-Arias M, Fernández-Jiménez MC, de la Iglesia Iñigo S, Morán-Jiménez MJ. Sideroblastic anemia: functional study of two novel missense mutations in ALAS2. Mol Genet Genomic Med. (2016) 4:273–82. doi: 10.1002/mgg3.202
19. Taylor JL, Brown BL. Structural basis for dysregulation of aminolevulinic acid synthase in human disease. J Biol Chem. (2022) 298:101643. doi: 10.1016/j.jbc.2022.101643
20. Furuyama K, Kaneko K. Iron metabolism in erythroid cells and patients with congenital sideroblastic anemia. Int J Hematol. (2018) 107:44–54. doi: 10.1007/s12185-017-2368-0
21. Ono K, Fujiwara T, Saito K, Nishizawa H, Takahashi N, Suzuki C, et al. Congenital sideroblastic anemia model due to ALAS2 mutation is susceptible to ferroptosis. Sci Rep. (2022) 12:9024. doi: 10.1038/s41598-022-12940-9
22. Ma Z, Li D, Yang X, Liang J, Zhu Y. Case report: an infant boy with X-linked sideroblastic anaemia successfully treated by umbilical cord blood haematopoietic stem cell transplantation. Front Genet. (2022) 13:1009988. doi: 10.3389/fgene.2022.1009988
23. Kubasch AS, Fenaux P, Platzbecker U. Development of luspatercept to treat ineffective erythropoiesis. Blood Adv. (2021) 5:1565–75. doi: 10.1182/bloodadvances.2020002177
24. Komrokji RS, Platzbecker U, Fenaux P, Zeidan AM, Garcia-Manero G, Mufti GJ, et al. Luspatercept for myelodysplastic syndromes/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis. Leukemia. (2022) 36:1432–5. doi: 10.1038/s41375-022-01521-4
Keywords: congenital, sideroblastic anemia, ALAS2, in silico analysis, pyridoxine
Citation: Cai J, Liu T, Huang Y, Chen H, Yu M, Zhang D and Huang Z (2024) A novel and apparent de novo ALAS2 missense variant associated with congenital sideroblastic anemia. Front. Pediatr. 12:1411676. doi: 10.3389/fped.2024.1411676
Received: 3 April 2024; Accepted: 19 August 2024;
Published: 29 August 2024.
Edited by:
Luigia De Falco, AMES, centro Polidiagnostico Strumentale, srl, ItalyReviewed by:
Sylvia S. Bottomley, University of Oklahoma Health Sciences Center, United StatesSarah Ducamp, Boston Children's Hospital and Harvard Medical School, United States
Elena Di Pierro, IRCCS Ca 'Granda Foundation Maggiore Policlinico Hospital, Italy
Copyright: © 2024 Cai, Liu, Huang, Chen, Yu, Zhang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongqing Zhang, emhhbmdkb25ncWluZzE5NzZAMTYzLmNvbQ==; Zhanqin Huang, enFodWFuZ0BzdHUuZWR1LmNu
†These authors share first authorship