 De-lu Chen
De-lu Chen Song Guo
Song Guo Qiu-li Chen
Qiu-li Chen Shan-jiao Qiu
Shan-jiao Qiu Jun Zhang
Jun Zhang Hua-mei Ma
Hua-mei Ma Yan-hong Li
Yan-hong Li- Department of Pediatrics, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
This study represents the first documentation of the coexistence of complete androgen insensitivity syndrome (CAIS) with Müllerian duct remnants (MDRs) in mainland China. Additionally, we provide a comprehensive review of the existing literature concerning CAIS with MDRs resulting from androgen receptor (AR) gene mutations. This study broadens the clinical spectrum of CAIS and offer novel insights for further exploration into Müllerian duct regression. A 14-year-old patient, initially raised as female, presented to the clinic with complaints of “primary amenorrhea.” Physical examination revealed the following: armpit hair (Tanner stage 2), breast development (Tanner stage 4 with bilateral breast nodule diameter of 7 cm), sparse pubic hair (Tanner stage 3), clitoris measuring 0.8 cm × 0.4 cm, separate urethral and vaginal openings, and absence of palpable masses in the bilateral groin or labia majora. The external genital virilization score was 0 points. Serum follicle-stimulating hormone level was 13.43 IU/L, serum luteinizing hormone level was 31.24 IU/L, and serum testosterone level was 14.95 nmol/L. Pelvic magnetic resonance imaging (MRI) did not reveal a uterus or bilateral fallopian tubes, but nodules on both sides of the pelvic wall indicated cryptorchidism. The karyotype was 46,XY. Genetic testing identified a maternal-derived hemizygous variation c.2359C > T (p.Arg787*) in the AR gene. During abdominal exploration, dysplastic testicles and a dysplastic uterus were discovered. Histopathological analysis revealed the presence of fallopian tube-like structures adjacent to the testicles. The CAIS patient documented in this study exhibited concurrent MDRs, thus expanding the spectrum of clinical manifestations of AIS. A review of prior literature suggests that the incidence of CAIS combined with histologically MDRs is not uncommon. Consequently, the identification of MDRs in AIS cases may represent an integral aspect of clinical diagnosis for this condition.
1 Introduction
Androgen insensitivity syndrome (AIS, OMIM #300068) is a rare X-linked recessive genetic disorder. AIS arises from pathogenic mutations in the androgen receptor (AR) gene, leading to complete or partial resistance to androgens, resulting in varying degrees of undervirilization. AIS represents a prevalent 46,XY disorder of sexual development (DSD) (1). In 1953, Morris initially described this syndrome as “testicular feminization syndrome" (2). There are no precise data regarding the prevalence of AIS. Research by Danish Berglund et al. suggested that the prevalence of AIS in 46,XY females was 6.4 per 100,000 live-born females (3). The estimated prevalence ranges from one in 20400 to one in 99,100 genetic males based on a proven molecular diagnosis (1). The AR gene is located at Xq11–12 and contains 8 coding exons, occupying more than 90 kb of genomic DNA (4). The AR protein, encoded by the AR gene, is part of the nuclear receptor superfamily and is also known as NR3C4 (nuclear receptor subfamily 3, Group C, Member 4) (5). The AR protein is composed of 920 amino acids and has a molecular weight of 110 kDa. This protein consists of four structural domains: the N-terminal transcription activation domain (NTD), the DNA-binding domain (DBD), the hinge region, and the ligand-binding domain (LBD) (6). The clinical manifestations and degree of androgen resistance can be classified into three types: complete AIS (CAIS), partial AIS (PAIS), and mild AIS (MAIS). In CAIS, patients exhibit pronounced feminization, characterized by female-appearing external genitalia and a blind-ended vaginal pouch. The testes may be located at any position along the descent pathway, including within the abdominal cavity, the inguinal canal, or the labia majora. The condition may be associated with either unilateral or bilateral inguinal hernias, and patients often present with primary amenorrhea after puberty. During embryonic development, the testes of AIS patients produce anti-Müllerian hormone (AMH), which induces regression of the Müllerian ducts (MDs), the anlage of the female internal reproductive organs (7). Therefore, the MD structure is often absent in AIS (1).
In this study, we report a case of CAIS coexisting with MD remnants (MDRs). Additionally, we provide a comprehensive summary of previously documented cases of CAIS, all confirmed through AR gene sequencing, which exhibited the presence of MDRs. This study was approved by the Medical Ethics Committee of the First Affiliated Hospital, Sun Yat-sen University [Approval No. (2023) 799].
2 Case description
2.1 General information
A 14-year-old Han Chinese girl raised as a female with nonconsanguineous parents was admitted to our clinic on August 1, 2023, with a chief complaint of breast development over the past four years without the onset of menstruation. The child exhibited good academic performance and female-like behavior. Breast development commenced four years prior, yet menarche had not occurred. Approximately one year ago (August 2022), a pelvic ultrasound performed at the primary hospital revealed an immature uterus and small gonads bilaterally. Karyotype analysis demonstrated a 46,XY genotype. For further diagnosis and treatment, the patient was admitted to our center on August 1, 2023. Since the onset of the condition, the patient has not manifested symptoms such as dizziness, headache, polydipsia, polyuria, vision impairment, hearing loss, or olfaction dysfunction.
2.1.1 Past medical history
The patient was generally healthy with no history of exposure to toxins, radioactive substances, or contraceptives. Personal history: The patient was the first pregnancy and birth of the mother and was delivered at a gestational age of 39 weeks through an uneventful vaginal delivery. Her birth weight and length were 3.1 kg and 50 cm, respectively. The mother had an uneventful pregnancy course. Family history: The paternal and maternal heights were 165 cm and 155 cm, respectively. The target height was 153.5 ± 5 cm for the female, and 166.5 ± 5 cm for the male. Both parents had normal development during puberty. The patient's younger sister, aged 4 years and 10 months, exhibited normal growth and development. However, the parents declined to consent to a karyotype analysis for her. Her maternal grandmother and two maternal aunts (II-4, II-5; Figure 1B) had regular menstruation and normal fertility. Four maternal female cousins (III-3, III-4, III-6, III-7; Figure 1B) were unmarried and had never been pregnant but had regular menstruation. Two maternal male cousins (III-5 and III-8; Figure 1B) did not complain of micropenis.
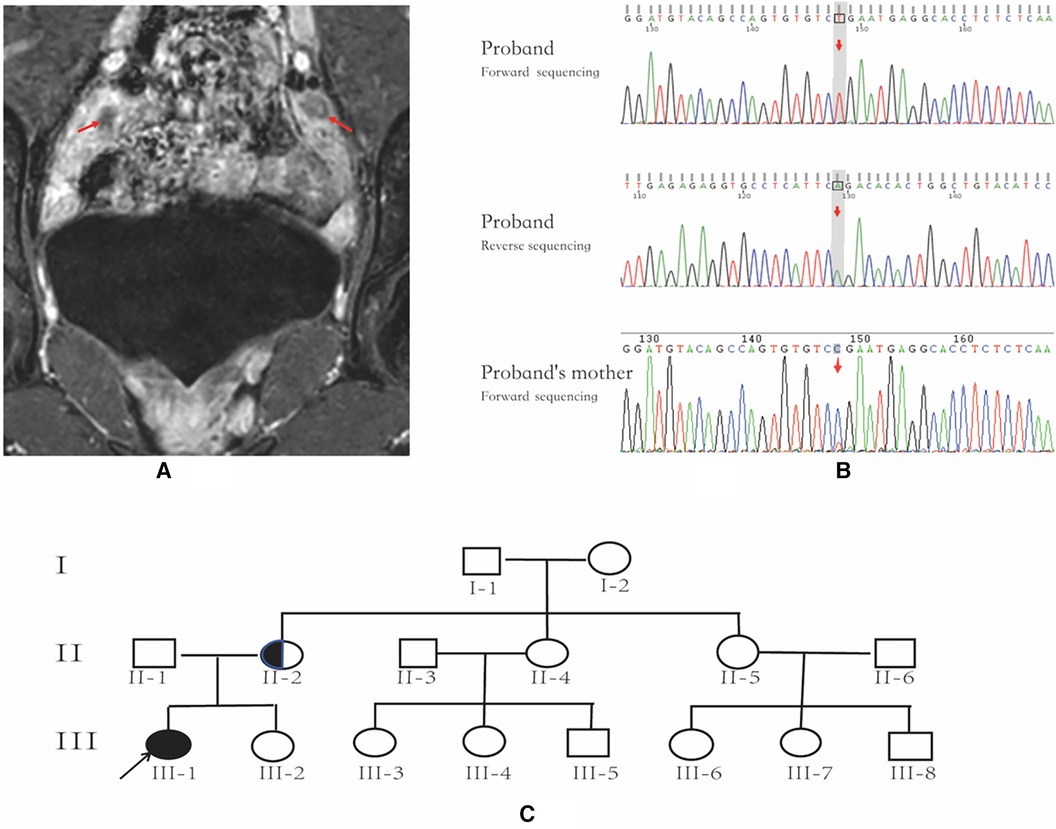
Figure 1. Pelvic MRI, AR gene sequencing chart and family pedigree. (A) Pelvic MR image; the arrows indicate bilateral testicles. (B) AR gene sequencing chart of the patient and the patient's mother; the arrow indicates the location of AR variant. The patient has an AR variant in hemizygosity (c.2359C > T, p.Arg787*), inherited from the mother. (C) family pedigree; the arrow indicates the proband, and the half-black/half-white circle is the carrier.
2.1.2 Physical examination
The patient's attire and mannerisms were feminine. Other physical measurements were recorded as follows: height 163 cm, weight 46 kg, and blood pressure 117/70 mmHg. The skin appeared smooth and devoid of acne. No palpable masses were identified in the inguinal region or labia majora. In terms of Tanner staging, the patient exhibited the following characteristics: Tanner stage 2 for axillary hair, Tanner stage 4 for breast development, with bilateral glandular nodules measuring 7 cm in diameter, and Tanner stage 3 for sparse pubic hair. The clitoris measured 0.8 cm × 0.4 cm, with separate urethral and vaginal openings. The external genitalia displayed a normal female appearance, and the External Masculinization Score (EMS) (8) was recorded as zero.
Further examination conducted at our center revealed that there were no abnormalities in the routine blood and urine tests. Additionally, there were no abnormalities detected in liver function, kidney function, blood glucose, blood lipids, or electrolytes. The 8AM cortisol concentration was 242.83 nmol/L, while the 8AM adrenocorticotropic hormone concentration was 3.77 pmol/L. Thyroid function was found to be within normal limits.
Serum levels of beta-human chorionic gonadotropin, alpha-fetoprotein, and carcinoembryonic antigen were within the normal range. FSH (13.43 IU/L), LH (31.24 IU/L), and T (14.95 nmol/L) were significantly elevated. Particularly noteworthy was the disproportionately higher serum LH level compared to the serum T level (Table 1). DHEAS (12.21 µmol/L), 17-α-OHP (34.55 nmol/L), and A4 (10.8 nmol/L) were also significantly elevated as summarized in Table 1. Her sex hormone-binding globulin (SHBG) concentration was 38.00 nmol/L, with a calculated free androgen index (FAI) of 39%. However, despite these findings, the patient did not exhibit clinical signs of hyperandrogenism, such as hirsutism or acne. Notably, the concentrations of anti-Müllerian hormone (AMH) and inhibin B (INHB) were markedly elevated at 206.51 ng/ml and 250.00 pg/ml, respectively, which are notably high for a female individual.
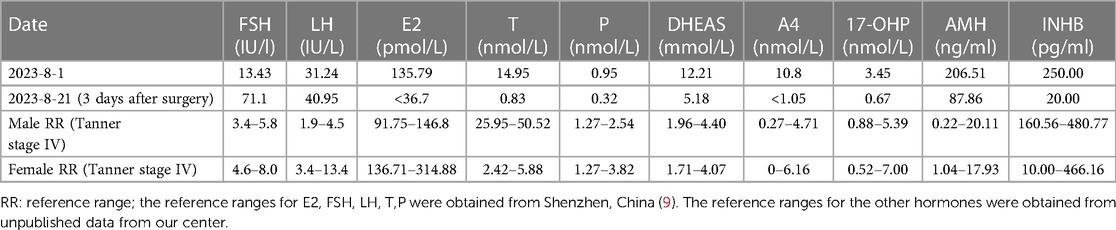
Table 1. Patient sex hormone levels before and after surgery.
The patient exhibited elevated levels of T, INHB, and AMH, indicating the likely presence of testicular tissue. A repeated pelvic ultrasound revealed nodules at the inguinal ring on both sides (approximately 1.2 cm × 0.6 cm on the left and 1.2 cm × 0.7 cm on the right), probably indicating the testes. However, the uterus and bilateral accessories were not visualized. Pelvic magnetic resonance imaging (MRI) showed no evidence of a uterus or bilateral accessories. The nodules observed on both sides of the pelvic wall were identified as testes (size 11 × 18 mm on the left side, size 24 × 11 mm on the right side), with a slightly high signal on T2-weighted imaging (T2WI) and marked enhancement (Figure 1A). Additionally, no abnormalities were detected on pituitary MRI.
Following the acquisition of informed consent from the patient and her parents, whole-exome genetic testing was conducted by Guangzhou Kingmed Center for Clinical Laboratory Co., Ltd. Molecular diagnosis unveiled an AR variant in hemizygosity (c.2359C > T, p.Arg787*), inherited from the mother (Figure 1B,C). Moreover, no aberrations were detected in the AMH and AMHR2 genes. It is noteworthy that several researchers have previously documented the occurrence of the same site mutation in the AR gene among individuals diagnosed with AIS (10, 11).
2.2 Diagnosis and treatment
The patient was raised as a female and exhibited breast development and female external genitalia. Imaging of pelvic MRI suggested the presence of testicular tissue in the body. Elevated levels of T, AMH, and INHB, accompanied by increased FSH and LH levels, were noted. The karyotype was 46,XY. Based on these findings, a preliminary diagnosis of AIS with a completely female external genital phenotype was established. There is an increased risk of gonadal malignancies due to the assignment of a female gender to the patient and the likelihood of an CAIS diagnosis (genetic results were unavailable at the time of surgery).
Following comprehensive communication with the parents and obtaining informed consent, the patient underwent laparoscopic examination. On August 18, 2023, in the pediatric surgery department, bilateral orchiectomy and vaginography procedures were conducted. Under laparoscopic view, the left internal ring was closed, while the right internal ring was still open. No vas deferens-like structures were identified at either internal ring. Gonad-like tissues approximately 2.5 cm × 1.0 cm in size were observed adjacent to the iliac vessels on both sides (Figure 2A). The bilateral gonads were connected to a uterine-like structure (Figure 2B). Subsequently, bilateral gonadal excisions were performed. Vaginography revealed a vaginal length of approximately 8 cm, with no discernible image of the uterus (Figure 2C).
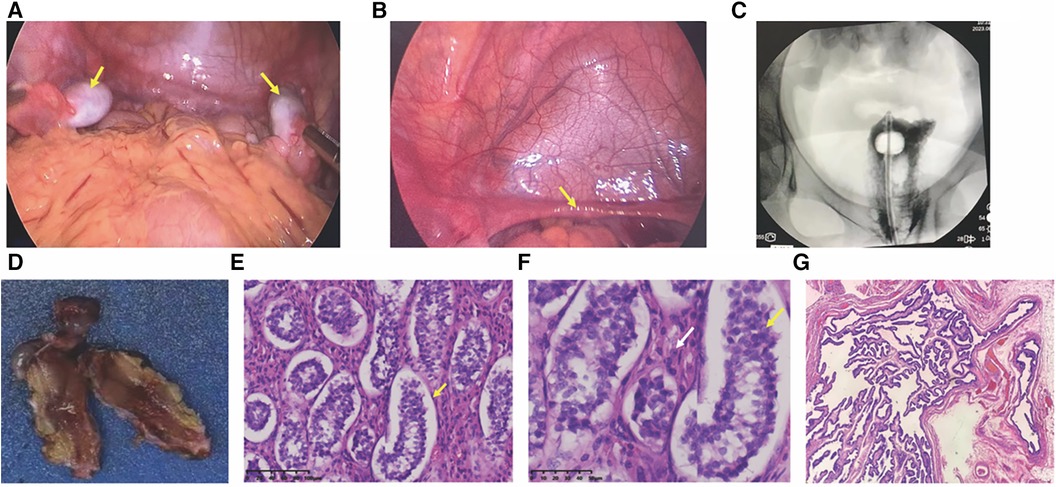
Figure 2. Laparoscopic view and histopathological pictures. (A) Laparoscopic view of bilateral gonads; arrows point to bilateral gonads. (B) Laparoscopic view of the hypoplastic uterus; arrows indicate a hypoplastic uterus. (C) Vaginogram; the patient has a vaginal length of approximately 8 cm, without the uterus. (D) Excised gonadal tissue; macroscopic aspect. (E,F) histopathological pictures. (E) histopathology revealing seminiferous tubules containing only Sertoli cells (×200); (F) Leydig cell hyperplasia indicated by white arrows (×400); (G) tubular structures resembling fallopian tubes (×40).
Histopathology examination revealed bilateral gonadal tissue (Figure 2D) with immature seminiferous tubule-like structures under the microscope. The tubules contained only Sertoli cells (Figure 2E) with an absence of spermatogenic cells. Furthermore, significant proliferation of interstitial Leydig cells was observed (Figure 2F). Additionally, structures resembling fallopian tubes (Figure 2G) and mesenteric cysts were identified.
2.3 Follow-up
Three days post-surgery (August 21, 2023), repeated blood tests revealed the following results: E2 < 36.7 pmol/L, FSH 71.10 IU/L, LH 40.95 IU/L, T 0.83 nmol/L, P 0.32 nmol/L, DHEAS 5.18 µmol/L, A4 < 1.05 nmol/L, 17-OHP 0.67 nmol/L, AMH 87.86 ng/ml, and INHB 20.00 pg/ml (Table 1). Following orchiectomy, there was a notable decrease in E2, T, and androgen precursor hormones, accompanied by significant increases in FSH and LH levels. These findings are indicative of a hypergonadotropic hypogonadism state.
The patient was prescribed estrogen replacement therapy (0.5 mg of estradiol valerate once daily) to maintain secondary sexual characteristics such as breast development and to prevent osteoporosis. Additionally, the parents were counseled to disclose the patient's condition under suitable circumstances, with the provision of psychological support as needed. Furthermore, a plan to perform a hysterectomy for the underdeveloped uterus during the upcoming winter vacation was proposed.
2.4 Genetically confirmed CAIS patients coexisting with MDRs in the literature
The following are the inclusion criteria for CAIS patients in the literature: 1. Genetic confirmation of CAIS. 2. An MDR is present. 3. A comprehensive description of the Müllerian duct's structure and hormone profiles is provided. 4. Research that is available in full text.
Case search strategy: The search was conducted up to November 1, 2023. We searched for ([Androgen Insensitivity Syndrom*(MeSH Terms)] OR [Androgen Resistance Syndrom*(MeSH Terms)] OR [Male Pseudohermaphroditism Due to Androgen Insensitivity(MeSH Terms)] OR [Reifenstei* Syndrome(MeSH Terms)] OR [Testicular Feminizatio*(MeSH Terms)] OR [Androgen Receptor Deficien*(MeSH Terms)] OR [AR Deficien*(MeSH Terms)] OR [DHTR Deficien*(MeSH Terms)] OR [Dihydrotestosterone Receptor Deficien*(MeSH Terms)]) AND (Müllerian) in the PubMed.
There are 127 records according to the case search strategy. We manually screened patients diagnosed with CAIS coexisting with MDRs, resulting in a total of seventeen reports. After excluding six reports that did not undergo genetic testing (12–17) and two reports failed to obtain full text (18, 19), eleven cases remained for further analysis (20–28).
Data form eleven eligible CAIS patients were retrieved from the literature (20–28). Combined with the case reported in this article, a total of 12 patients of CAIS with MDRs have been documented (Table 2). Of these twelve patients, eight had uteruses (20–22, 24, 26, 28), including our patient. Among these individuals, patients 1 (20), 2 (21) and 12 underwent laparoscopy to find the uterus. In patients 4 (22), 11 (28), the uterus is found by MRI or ultrasound. In patients 6 (24), 8 (26), 9 (26), the uterus was confirmed pathologically. The initial clinical presentation, biochemical profile, and gonadal pathology of CAIS patients with MDRs were found to be similar to those without MDRs (1, 29). Patients 8 and 9 were from the same family, and shared identical genetic mutations (26). Among the cases reviewed, six patients were admitted for inguinal hernia, five for primary amenorrhea, and patient 11 for central precocious puberty with cyclical episodes of vaginal bloody spotting. All patients were diagnosed with CAIS confirmed by molecular testing and raised as females, with heights in the medium to upper average range. The age at diagnosis varied widely, spanning from birth to 22 years. Notably, patient 6 was diagnosed with CAIS in utero, and the pregnancy was terminated at 20 weeks of gestation (24). Among post-pubertal patients, varying degrees of breast development and sparse pubic hair growth were observed. Vaginal length ranged from 4 to 8 cm, indicating upper vaginal development from the MD in some patients. Specifically, six of the twelve patients had testes located in the inguinal canal, while five had testes in the abdominal cavity. Among the patients, one had a left testis in the abdominal cavity and another in the inguinal canal. According to the laparoscopic or pathological findings, fallopian structures were observed in 10 out of the 12 patients, uterine structures in 8 patients, and both fallopian and uterine structures in 6 patients. Notably, in patients older than 10 years, the concentration of LH was significantly elevated to >10 IU/L in 6 cases, with a mean value of 26.9 IU/L. Histological examination of the testes revealed Leydig cell hyperplasia in 4 out of 9 patients, while Sertoli cells were either normal or abundantly present. Germ cells were observed in younger patients, specifically patients 3 and 4 (22); In contrast, germ cells were nearly absent in older patients, specifically patients 8 and 9 (26), presenting as only Sertoli cells without germ cells. Genetic analysis identified mutations in the DNA-binding domain (DBD) of the AR gene in two out of the twelve patients, while eight patients (66.7%) in the ligand-binding domain (LBD). Among the twelve patients, one had a nonsense mutation in the AR gene, three had frameshift mutations, and eight (66.7%) had missense mutations.
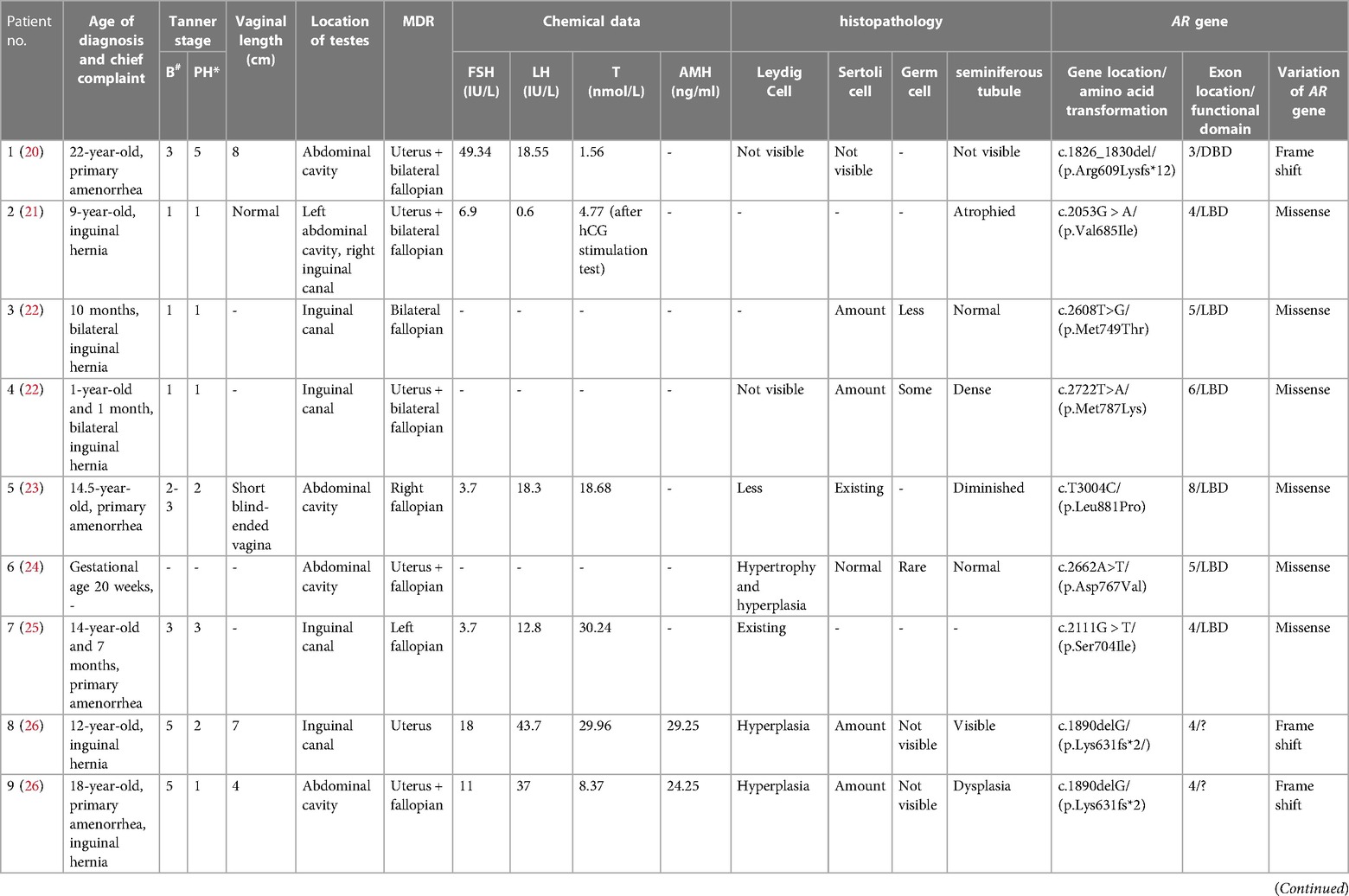
Table 2. Characteristics of CAIS patients with MDRs reported in the literature.
3 Discussion
AIS is a common 46,XY disorder of sexual development. In this study, we reported a case of CAIS attributed to a pathogenic variant in the AR gene coexisting with MDRs. Our findings underscore the importance of considering a diagnosis of CAIS even when MDRs are detected laparoscopically. Additionally, we observed that the initial clinical presentation, biochemical profiles, and gonadal pathology of CAIS patients with MDRs closely resemble those without MDRs. Previous reports indicate that the incidence of histologically MDRs in CAIS patients is approximately one third (30, 31). Consequently, the identification of MDRs should be incorporated into the clinical management strategies for CAIS.
This study reports a case of CAIS featuring a mutation at position c.2359 in the AR gene, resulting in a substitution of cytosine (C) with thymine (T). This alteration converts the arginine residue encoded by codon 787 of exon 6 into a termination codon, thereby prematurely terminating the synthesis of the AR protein. To our knowledge, there are two reported CAIS patients with the same AR gene mutation as our case by Dong et al. (10) and Kharrat et al. (11). Dong et al. (10) presented the presence of fallopian tubes in patients with CAIS(DSD 14 in Table 1 of the reference), while Kharrat et al. (11) reported a CAIS patient without MDRs (Patient 9 in Table 1 of the reference). This finding suggests that the presence of MDRs in CAIS patients may not be strongly related to genotype. Bermudez de la Vega et al. (28) reported that within the same family, CAIS caused by the same AR gene mutation can manifest as gender dysphoria when the patient is raised as a female or as central precocious puberty with periodic vaginal bleeding. This indicates that even the same genotype can have different clinical phenotypes, suggesting the role of environmental factors and epigenetics in the phenotype of CAIS.
This study provides a summary of the clinical data of genetically confirmed cases of CAIS with MDRs. Our findings indicate that the initial clinical presentation, biochemical profiles, and gonadal pathology of CAIS patients with MDRs closely resemble those without MDRs (1, 29).
A few of the patients included in Table 2 still require a great deal of discussion. The patient 1 (20) had a 46,XY karyotype and a complete female phenotype. As compared with serum LH (18.55 IU/L), the serum FSH level (49.34 IU/L) was substantially higher. In testicular histology, seminiferous tubules and sertoli-leydig cells were not present, and serum testosterone level was inadequate (0.45 ng/ml). These clinical signs do not align with the characteristics of classic AIS. Although we included this patient, testicular failure may be the more probable diagnosis. In patient 6 (24), under microscopy, not only MDRs but also Wolffian duct derivatives were discovered. It was hypothesized that elevated local concentrations of testosterone may trigger a reaction from mutant androgen receptors that still exhibited residual function, therefore promoting the development of WD. The AR gene variant of Patient 6 (24) is novel and the same mutation has not been reported previously. But the other variation at the codon 767 of AR gene (i.e., D767E) have been reported and verified to be CAIS in the Androgen Receptor Mutations Database (http://androgendb.mcgill.ca/). It is suggested that codon 767 is a crucial functional location inside the AR gene. Additionally, we used PolyPhen-2 to predict this variant of the AR gene, indicating that this mutation is most likely harmful.
Previous cohort studies have reported that approximately one third of CAIS patients present with coexisting histologically MDRs (30, 31), highlighting that the incidence of such coexisting MDRs in CAIS patients is not low. Although studies specifically addressing the risk of malignancy associated with MDRs in CAIS patients are lacking, existing literature on persistent Müllerian duct syndrome (PMDS) suggests a risk of MD malignancy ranging from 3%–8% (32, 33). Therefore, clinicians should exercise vigilance not only regarding the risk of gonadal malignancy in CAIS patients (34) but also regarding the presence of MDRs and their potential for malignancy.
It's indeed an intriguing phenomenon that MDRs are present alongside CAIS. Typically, in cases of CAIS, AMH released by the testes promotes the regression of MD around 8 weeks of gestation via a paracrine mechanism (35). As a result, Müllerian ducts regress in the majority of clinical instances of CAIS. However, while the testes have the capacity to synthesize AMH, it is plausible that insufficient or untimely AMH production occurred during the critical period of Müllerian duct regression. This possibility warrants further investigation.
Scholars suggested that the persistence of MDRs may be associated with deficiencies in the secretion or function of AMH (17). AMH and AMHR2 gene testing was conducted on the patient as part of our investigation, yielding normal results. Picard JY et al. found that in 12% of patients with PMDS, there was no mutation detected in the AMH or AMHR2 gene (36). This suggests that MD regression is governed by a complex gene regulatory network (GRN) (37). Currently, studies examining the role of the AR gene within this GRN are lacking. To understand why some AIS patients fail to experience the complete regression of MD, more research is necessary.
To the best of knowledge, this study represents the first report of the coexistence of CAIS with MDRs in mainland China. Additionally, we provide a comprehensive summary of the existing literature on CAIS with MDRs due to AR gene mutations. Our findings serve to broaden the clinical spectrum of CAIS and offer novel insights for further investigations into Müllerian duct regression. However, it is essential to acknowledge the limitation of this study, the current lack of a comprehensive explanation for the pathophysiology underlying the presence of MDRs in patients with CAIS, which warrants further research.
In conclusion, the presence of MDRs in children with CAIS expands the spectrum of clinical manifestations of CAIS. Moreover, this study provides an alternative perspective for fundamental research on MD regression, suggesting that the mechanism is not solely reliant on the AMH–AMHR2 signaling pathway. Our research emphasizes the significance of integrating MDRs identification into the overall clinical management of CAIS, since the occurrence of histologically MDRs in CAIS patients is about one third.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Medical Ethics Committee of the First Affiliated Hospital, Sun Yat-sen University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
D-lC: Writing – original draft, Writing – review & editing, Conceptualization. SG: Writing – review & editing, Methodology, Supervision. Q-lC: Writing – review & editing, Conceptualization, Supervision. S-jQ: Supervision, Writing – review & editing. Y-yX: Supervision, Writing – review & editing. JZ: Writing – review & editing, Conceptualization, Methodology, Supervision. H-mM: Writing – review & editing, Methodology, Supervision. Y-hL: Writing – review & editing, Conceptualization, Methodology, Supervision.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors would like to thank pathologist Dr. Cheng Xu for providing histopathological pictures. The authors are indebted to pediatric surgeon Dr. Jun-cheng Liu for providing laparoscopic images. The authors also are grateful to obstetrician Dr. Shao-bin Lin for analyzing the result of gene sequencing. Thanks are given to all the clinicians providing care and management to the patients.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hughes IA, Davies JD, Bunch TI, Pasterski V, Mastroyannopoulou K, MacDougall J. Androgen insensitivity syndrome. Lancet. (2012) 380(9851):1419–28. doi: 10.1016/S0140-6736(12)60071-3
2. Morris JM. The syndrome of testicular feminization in male pseudohermaphrodites. Am J Obstet Gynecol. (1953) 65(6):1192–211. doi: 10.1016/0002-9378(53)90359-7
3. Berglund A, Johannsen TH, Stochholm K, Viuff MH, Fedder J, Main KM, et al. Incidence, prevalence, diagnostic delay, and clinical presentation of female 46,XY disorders of sex development. J Clin Endocrinol Metab. (2016) 101(12):4532–40. doi: 10.1210/jc.2016-2248
4. Brown CJ, Goss SJ, Lubahn DB, Joseph DR, Wilson EM, French FS, et al. Androgen receptor locus on the human X chromosome: regional localization to Xq11-12 and description of a DNA polymorphism. Am J Hum Genet. (1989) 44(2):264–9. PMID: 2563196.2563196
5. Tyutyusheva N, Mancini I, Baroncelli GI, D'Elios S, Peroni D, Meriggiola MC, et al. Complete androgen insensitivity syndrome: from bench to bed. Int J Mol Sci. (2021) 22(3). doi: 10.3390/ijms22031264
6. Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. (2002) 20(13):3001–15. doi: 10.1200/JCO.2002.10.018
7. Behringer RR, Finegold MJ, Cate RL. Mullerian-inhibiting substance function during mammalian sexual development. Cell. (1994) 79(3):415–25. doi: 10.1016/0092-8674(94)90251-8
8. Ahmed SF, Khwaja O, Hughes IA. The role of a clinical score in the assessment of ambiguous genitalia. BJU Int. (2000) 85(1):120–4. doi: 10.1046/j.1464-410x.2000.00354.x
9. Sun L, Li L, Huang B, Fu X, Yang F, Ma D, et al. Establishing reference range of sex hormone of healthy children in Shenzhen area based on chemiluminescence. Chin J Contemp Pediatr. (2017) 19:1257–62. doi: 10.7499/j.issn.1008-8830.2017.12.007
10. Dong Y, Yi Y, Yao H, Yang Z, Hu H, Liu J, et al. Targeted next-generation sequencing identification of mutations in patients with disorders of sex development. BMC Med Genet. (2016) 17(23). doi: 10.1186/s12881-016-0286-2
11. Kharrat M, Tajouri A, Nacef IB, Hizem C, Trabelsi M, Maazoul F, et al. Identification of two additional novel mutations in the AR gene associated with severe forms of androgen insensitivity syndrome. Steroids. (2019) 152:108489. doi: 10.1016/j.steroids.2019.108489
12. Oka M, Katabuchi H, Munemura M, Mizumoto J, Maeyama M. An unusual case of male pseudohermaphroditism: complete testicular feminization associated with incomplete differentiation of the müllerian duct. Fertil Steril. (1984) 41(1):154–6. doi: 10.1016/S0015-0282(16)47559-X
13. Dodge ST, Finkelston MS, Miyazawa K. Testicular feminization with incomplete müllerian regression. Fertil Steril. (1985) 43(6):937–8. doi: 10.1016/S0015-0282(16)48626-7
14. Chen FP. Testicular feminization with incomplete müllerian regression in twin patients: laparoscopic diagnosis and treatment. Acta Obstet Gynecol Scand. (1996) 75(3):304–7. doi: 10.3109/00016349609047109
15. Menakaya UA, Aligbe J, Iribhogbe P, Agoreyo F, Okonofua FE. Complete androgen insensitivity syndrome with persistent mullerian derivatives: a case report. J Obstet Gynaecol. (2005) 25(4):403–5. doi: 10.1080/01443610500143226
16. Kitade M, Takeuchi H, Jinushi M, Kikuchi I, Kumakiri J, Kuroda K. Testicular feminization with persistent wolffian duct and müllerian remnants: similar to mayer-rokitansky-kuster-hauser syndrome. Fertil Steril. (2009) 92(6):2034–6. doi: 10.1016/j.fertnstert.2008.12.126
17. Nichols JL, Bieber EJ, Gell JS. Case of sisters with complete androgen insensitivity syndrome and discordant müllerian remnants. Fertil Steril. (2009) 91(3):932.e15–.e18. doi: 10.1016/j.fertnstert.2008.09.027
18. Swanson ML, Coronel EH. Complete androgen insensitivity with persistent müllerian structures. A case report. J Reprod Med. (1993) 38(7):565–8. PMID: 8410855.8410855
19. Heller DS, Ranzini A, Futterweit W, Dottino P, Deligdisch L. Müllerian remnants in complete androgen insensitivity syndrome. Int J Fertil. (1992) 37(5):283–5. PMID: 1358839.1358839
20. Chen CP, Chen SR, Wang TY, Wang W, Hwu YM. A frame shift mutation in the DNA-binding domain of the androgen receptor gene associated with complete androgen insensitivity, persistent mullerian structures, and germ cell tumors in dysgenetic gonads. Fertil Steril. (1999) 72(1):170–3. doi: 10.1016/S0015-0282(99)00169-7
21. Damiani D, Mascolli MA, Almeida MJ, Jaubert F, Fellous M, Dichtchekenian V, et al. Persistence of mullerian remnants in complete androgen insensitivity syndrome. J Pediatr Endocrinol Metab. (2002) 15(9):1553–6. doi: 10.1515/JPEM.2002.15.9.1553
22. Van YH, Lin JL, Huang SF, Luo CC, Hwang CS, Lo FS. Novel point mutations in complete androgen insensitivity syndrome with incomplete mullerian regression: two Taiwanese patients. Eur J Pediatr. (2003) 162(11):781–4. doi: 10.1007/s00431-003-1301-0
23. Galani A, Sofocleous C, Karahaliou F, Papathanasiou A, Kitsiou-Tzeli S, Kalpini-Mavrou A. Sex-reversed phenotype in association with two novel mutations c.2494delA and c.T3004C in the ligand-binding domain of the androgen receptor gene. Fertil Steril. (2008) 90(5):2008.e1–4. doi: 10.1016/j.fertnstert.2008.01.030
24. Corbetta S, Muzza M, Avagliano L, Bulfamante G, Gaetti L, Eller-Vainicher C, et al. Gonadal structures in a fetus with complete androgen insensitivity syndrome and persistent mullerian derivatives: comparison with normal fetal development. Fertil Steril. (2011) 95(3):1119.e9–14. doi: 10.1016/j.fertnstert.2010.09.028
25. Chin VL, Sheffer-Babila S, Lee TA, Tanaka K, Zhou P. A case of complete androgen insensitivity syndrome with a novel androgen receptor mutation. J Pediatr Endocrinol Metab. (2012) 25(11-12):1145–51. doi: 10.1515/jpem-2012-0135
26. Guven A, Dursun F, Ozkanli S, Gucluer B, Kuru LI. Complete androgen insensitivity syndrome and discordant mullerian remnants: two cases with novel mutation in the androgen receptor. J Pediatr Endocrinol Metab. (2013) 26(9-10):909–14. doi: 10.1515/jpem-2013-0047
27. Grob F, Varela M, Domingo Arce J, Martinez V, Garcia Bruce H. Complete androgen insensitivity syndrome with persistent mullerian remnants. A case report. Endocrinol Nutr. (2013) 60(4):216–8. doi: 10.1016/j.endonu.2012.03.011
28. Bermudez de la Vega JA, Fernandez-Cancio M, Bernal S, Audi L. Complete androgen insensitivity syndrome associated with male gender identity or female precocious puberty in the same family. Sex Dev. (2015) 9(2):75–9. doi: 10.1159/000371617
29. Hannema SE, Scott IS, Rajpert-De Meyts E, Skakkebaek NE, Coleman N, Hughes IA. Testicular development in the complete androgen insensitivity syndrome. J Pathol. (2006) 208(4):518–27. doi: 10.1002/path.1890
30. Cheikhelard A, Morel Y, Thibaud E, Lortat-Jacob S, Jaubert F, Polak M, et al. Long-term followup and comparison between genotype and phenotype in 29 cases of complete androgen insensitivity syndrome. J Urol. (2008) 180(4):1496–501. doi: 10.1016/j.juro.2008.06.045
31. Nakhal RS, Hall-Craggs M, Freeman A, Kirkham A, Conway GS, Arora R, et al. Evaluation of retained testes in adolescent girls and women with complete androgen insensitivity syndrome. Radiology. (2013) 268(1):153–60. doi: 10.1148/radiol.13121068
32. Farikullah J, Ehtisham S, Nappo S, Patel L, Hennayake S. Persistent mullerian duct syndrome: lessons learned from managing a series of eight patients over a 10-year period and review of literature regarding malignant risk from the mullerian remnants. BJU Int. (2012) 110(11 Pt C):E1084–9. doi: 10.1111/j.1464-410X.2012.11184.x
33. Gagliardi F, Lauro A, De Anna L, Tripodi D, Esposito A, Forte F, et al. The risk of malignant degeneration of mullerian derivatives in PMDS: a review of the literature. J Clin Med. (2023) 12(9). doi: 10.3390/jcm12093115
34. King TFJ, Wat WZM, Creighton SM, Conway GS. Bone mineral density in complete androgen insensitivity syndrome and the timing of gonadectomy. Clin Endocrinol (Oxf). (2017) 87(2):136–40. doi: 10.1111/cen.13368
35. Cate RL. Anti-Mullerian hormone signal transduction involved in mullerian duct regression. Front Endocrinol (Lausanne). (2022) 13:905324. doi: 10.3389/fendo.2022.905324
36. Picard JY, Cate RL, Racine C, Josso N. The persistent mullerian duct syndrome: an update based upon a personal experience of 157 cases. Sex Dev. (2017) 11(3):109–25. doi: 10.1159/000475516
Keywords: disorder of sexual development, testicular feminization syndrome, complete androgen insensitivity syndrome, Müllerian duct remnants, Müllerian duct regression
Citation: Chen D-l, Guo S, Chen Q-l, Qiu S-j, Xu Y-y, Zhang J, Ma H-m and Li Y-h (2024) Complete androgen insensitivity syndrome coexisting with müllerian duct remnants: a case report and literature review. Front. Pediatr. 12:1400319. doi: 10.3389/fped.2024.1400319
Received: 13 March 2024; Accepted: 21 May 2024;
Published: 4 June 2024.
Edited by:
Rodolfo A. Rey, Hospital de Niños Ricardo Gutiérrez, ArgentinaReviewed by:
Rafael Loch Batista, University of São Paulo, BrazilJolanta Słowikowska-Hilczer, Medical University of Lodz, Poland
© 2024 Chen, Guo, Chen, Qiu, Xu, Zhang, Ma and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan-hong Li, bHlhbmhAbWFpbC5zeXN1LmVkdS5jbg==; Jun Zhang, emhqdW4yOUBtYWlsLnN5c3UuZWR1LmNu
†These authors have contributed equally to this work and share first authorship