 Simona Bene Watts1
Simona Bene Watts1 Barbara Gauthier2,†Anders C. Erickson3,†Julie Morrison4Mavis Sebastian4Lawrence Gillman5Sarah McIntosh5
Barbara Gauthier2,†Anders C. Erickson3,†Julie Morrison4Mavis Sebastian4Lawrence Gillman5Sarah McIntosh5 Connie Ens6Elizabeth Sherwin7Rod McCormick8
Connie Ens6Elizabeth Sherwin7Rod McCormick8 Shubhayan Sanatani6,‡
Shubhayan Sanatani6,‡ Laura Arbour1,5,9*‡
Laura Arbour1,5,9*‡
- 1Island Medical Program, University of British Columbia, Victoria, BC, Canada
- 2Epidemiology and Surveillance Unit, Interior Health Authority, Kelowna, BC, Canada
- 3Health Protection Branch, Ministry of Health, Victoria, BC, Canada
- 4Gitxsan Community Member, Hazelton, BC, Canada
- 5Department of Medical Genetics, University of British Columbia, Vancouver, BC, Canada
- 6Department of Pediatrics, Division of Cardiology, British Columbia Children’s Hospital, Vancouver, BC, Canada
- 7Department of Pediatrics, Children’s National Hospital, Washington, DC, United States
- 8Department of Education and Social Work, Thompson Rivers University, Kamloops, BC, Canada
- 9Division of Medical Sciences, University of Victoria, Victoria, BC, Canada
Introduction: Congenital Long QT Syndrome (LQTS) is common in a First Nations community in Northern British Columbia due to the founder variant KCNQ1 p.V205M. Although well characterized molecularly and clinically in adults, no data have been previously reported on the pediatric population. The phenotype in adults has been shown to be modified by a splice site variant in KCNQ1 (p.L353L). The CPT1A p.P479L metabolic variant, also common in Northern Indigenous populations, is associated with hypoglycemia and infant death. Since hypoglycemia can affect the corrected QT interval (QTc) and may confer risk for seizures (also associated with LQTS), we sought to determine the effect of all three variants on the LQTS phenotype in children within our First Nations cohort.
Methods: As part of a larger study assessing those with LQTS and their relatives in a Northern BC First Nation, we assessed those entering the study from birth to age 18 years. We compared the corrected peak QTc and potential cardiac events (syncope/seizures) of 186 children from birth to 18 years, with and without the KCNQ1 (p.V205M and p.L353L) and CPT1A variants, alone and in combination. Linear and logistic regression and student t-tests were applied as appropriate.
Results: Only the KCNQ1 p.V205M variant conferred a significant increase in peak QTc 23.8 ms (p < 0.001) above baseline, with females increased by 30.1 ms (p < 0.001) and males by 18.9 ms (p < 0.01). There was no evidence of interaction effects with the other two variants studied. Although the p.V205M variant was not significantly associated with syncope/seizures, the odds of having a seizure/syncope were significantly increased for those homozygous for CPT1A p.P479L compared to homozygous wild type (Odds Ratio [OR]3.0 [95% confidence interval (CI) 1.2–7.7]; p = 0.019).
Conclusion: While the KCNQ1 p.V205M variant prolongs the peak QTc, especially in females, the CPT1A p.P479L variant is more strongly associated with loss of consciousness events. These findings suggest that effect of the KCNQ1 p.V205M variant is mild in this cohort, which may have implications for standard management. Our findings also suggest the CPT1A p.P479L variant is a risk factor for seizures and possibly syncope, which may mimic a long QT phenotype.
1 Introduction
Long QT syndrome (LQTS) predisposes those affected to arrhythmia, syncope, seizures, and sudden cardiac death. While generally thought to be rare, affecting ∼1 in 2,000 individuals (1), congenital LQTS affects approximately 1 in 125 individuals in the Gitxsan community (2). The Gitxsan First Nation territory (People of the River Mist) covers a landmass of 33,000 square kilometres within northwest British Columbia (BC), Canada. The Gitxsan community voiced concern about the high rate of LQTS in 2004, and from this a community-based participatory research project was initiated. The project identified the novel pathogenic variant KCNQ1 p.V205M as the primary cause of LQTS (3). Pathogenic variants in KCNQ1 contribute to LQTS type 1 (LQTS1), the most common cause of LQTS. The effect of the p.V205M variant has been well characterized molecularly and within this First Nation adult population (3–5); however, the effect within the pediatric population has not been reported. Understanding the pediatric manifestations of this condition in the Gitxsan is imperative when considering the life course of the condition and relevant management strategies.
In addition to p.V205M, other genetic variants that may influence the presentation of the LQTS phenotype, or may mimic the phenotype, are present within the Gitxsan population. These include KCNQ1 p.L353L, a synonymous splice site variant and modifier of the LQTS phenotype (5), ANK2 p.S646F, a variant associated with structural heart disease and an adult onset form of LQTS (6), and CPT1A p.P479L, a variant associated with infant death (7, 8) and hypoglycemia (9–11). Not only is the CPT1A variant commonly found among Northern Indigenous populations in Canada (12) and BC First Nations (7), but hypoglycemia may present with symptoms (e.g., seizures) that can be difficult to differentiate from LQTS.
The variants that cause the cardiac phenotype of LQTS1 (KCNQ1) are also expressed in pancreatic islet cells and may predispose individuals to hypoglycemia (13). Moreover, beta-blockers are the mainstay treatment for LQTS and may alter the adrenergic response to hypoglycemia (14), and hypoglycemia itself has been observed to prolong the QT interval (15) and trigger arrythmia (16, 17). Over the last decade, at least three First Nations children in Northern BC with LQTS1 have presented with symptomatic hypoglycemia and a reduced level of consciousness (Table 1). All children were either heterozygous or homozygous for CPT1A p.P479L and taking beta-blocker medication, leading to the concern that the combination of the p.V205M and common CPT1A variant when inherited together might present with a more severe or varied phenotype.
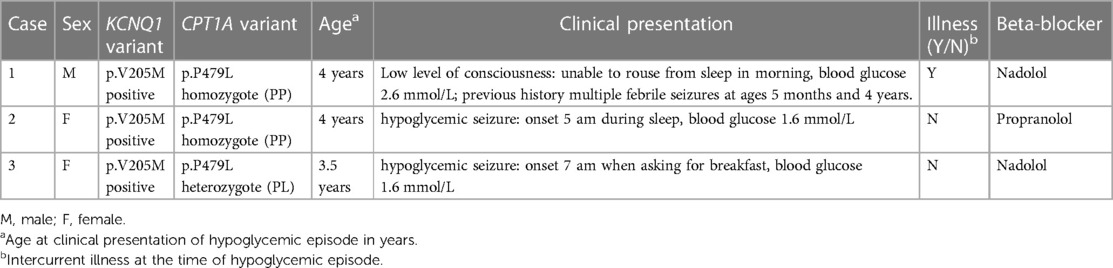
Table 1. Case presentation of three Northern BC First Nations children with symptoms of hypoglycemia and reduced levels of consciousness.
Founder populations provide a unique opportunity to study the effect of genetic variants within a relatively similar population. We sought to understand the effect of the KCNQ1 p.V205M and p.L353L variants within the pediatric population and whether events consistent with a LQTS phenotype were influenced by the presence of the CPT1A p.P479L variant. Because of the unclear cardiac effect of the ANK2 p.S646F variant which is also present in this community, we opted to exclude those with the variant in this analysis.
2 Methods
2.1 Enrollment and data collection
As part of a larger community-based longitudinal research study (3), participants were invited to enroll if they had clinical features of LQTS or were related to an individual with LQTS. Community approval and participant/parent informed consent and assent was obtained. Upon enrollment, a health information questionnaire was administered, medical records reviewed, a comprehensive family history recorded, and a 12-lead electrocardiogram (ECG) performed. Events leading to loss of consciousness (LOC) including syncope or seizure were recorded. Participants from birth until age 18 years of age and enrolled from 2005 to 2019 were included. Ongoing outpatient cardiology visits to BC Children's Hospital or Northern outreach clinics were recorded on all p.V205M positive participants, and negative participants were referred for cardiology related symptoms.
2.1.1 Exclusions
Participants 18 years and older at time of enrollment, without an ECG, missing genotyping data for any of the variants under study (KCNQ1 p.V205M, ANK2 p.S646F or CPT1A p.P479L), ANK2 p.S646F positive, or with a potentially confounding medical diagnosis, were excluded. For the KCNQ1 p.L353L specific analysis, those with unknown variant status were excluded (N = 2). For the QTc analysis, only those related to participants with the p.V205M variant were included as controls to minimize ascertainment bias (78 were excluded because they were not related to a case). Of note, two p.V205M positive females were excluded because their QTcs were outliers potentially skewing results, but the cases are described below.
2.2 Ethics
Research ethics approval was received by the University of British Columbia Clinical Research Ethics Board, Northern Health Authority Research Ethics Board, Island Health Research Ethics Board, and the University of Victoria Research Ethics Board. The Gitxsan Health Society Board of Directors approved the research and a local research review committee participated in the research design and reviewed results. The results were also presented publicly for Gitxsan community members.
2.3 Genotyping
Blood or saliva was collected for DNA sequencing under the stipulation of DNA on Loan (18). Genotyping for the KCNQ1 variants p.V205M and p.L353L was performed at the Molecular Genetics Laboratory at the BC Children's & Women's Hospital via clinical grade Sanger sequencing. Research testing for CPT1A p.P479L and ANK2 p.S646F variants was performed at the Centre for Applied Genomics at The Hospital for Sick Children in Toronto.
2.4 QTc determination
For those p.V205M positive and their 1st to 3rd degree relatives, all available ECGs from birth to their eighteenth birthday were included (ECG upon enrollment and any previous or subsequent ECGs taken during childhood). All ECGs were manually read and the results denoted as mQTc. The QT interval was measured in lead II or V5 using the tangent method (19) by at least one of two pediatric cardiologists (SS and ES) blinded to clinical and variant status. The QT interval corrected for heart rate (QTc) was then calculated using Bazett's formula (20). When the ECG was read by more than one practitioner, the average of the two final QTc measurements was calculated. If the difference between final QTc measurements was greater than 30 ms, the ECG was discussed and the final measurement agreed upon by consensus.
2.5 Statistical analysis
The highest mQTc measured during childhood was identified as the participant's “peak” mQTc. The mean peak mQTc between p.V205M positive cases and negative relatives (see below for description) was compared using descriptive analyses and t-tests. Univariate, bivariate, and multivariate linear regression (Ordinary Least Squares) was then used to explore the association of genetic variants, independently and in combination, on peak mQTc. Sex was also included as a variable.
Additionally, t-tests were performed to compare the highest mQTc recorded from p.V205M positive cases and negative controls within defined age categories (<1 year old, 1–4 years old, 5–9 years old, 10–14 years old, and 15–17 years old). Univariate and multivariate binomial logistic regression analysis were used to calculate the odds ratio [OR] between the genetic variants on LOC events considering the KCNQ1 p.V205M and CPT1A p.P479L variants, independently and in combination. Logistic regression was also used to calculate the odds of LOC events by mQTc. STATA 16-IC and GraphPad Prism were used to perform the statistical analyses.
3 Results
3.1 Participants
A total of 276 pediatric participants were enrolled in the study. Eighty-seven were excluded because they had no ECGs for analysis or genotyping was incomplete. Three children were excluded from the analysis due to confounding medical circumstances. Two of the three children had the p.V205M variant and their QTcs were considered outliers. Of those, one had evidence of left ventricular non-compaction cardiomyopathy (LVNC) and the other had a mixed connective tissue disorder which was complicated by an overdose with a QT prolonging drug (hydroxychloroquine). Their QTc values ranged from 580ms-622 ms and were excluded from the QTc analysis because of the potential of skewing the results. The third exclusion was p.V205M negative and had Wolff-Parkinson-White syndrome.
For the QTc analysis, there were 40 p.V205M heterozygotes (referred to as “V205M positive”) and no p.V205M homozygotes. There were 68 p.V205M negative 1–3rd degree relatives. Of the 40 participants who were p.V205M positive, 38 were confirmed to be positive through family cascade genetic testing, of which three were shown to have a prolonged QTc on ECG prior to genetic testing. Two participants presented symptomatically and had subsequent genetic testing. All 40 participants were advised beta-blocker therapy, with 35/40 prescribed beta-blockers at diagnosis. Compliance of beta-blocker therapy was not assessed in this study. All had a family history of LQTS. For this analysis, a total of 214 ECGs were manually read (mQTc) on 108 participants.
For the LOC analysis, all p.V205M positive cases including the QTc outliers, 67 of their 1–3rd degree relatives, and 78 unrelated individuals who were enrolled in the study were included. Two individuals (one p.V205M positive and one p.V205M negative) were removed due to confounding neurological conditions. Additionally, two individuals reported “possible” seizures and were therefore excluded from the seizure-specific analysis. See Supplementary Figure S2 for details.
The mean age of those p.V205M positive was 7.6 years (SD = 5.8) at enrollment, and they were followed for a mean of 5.8 years (range of follow up 0–13.1 years) by cardiology services. The mean age at enrollment for p.V205M negative participants was 8.7 years (SD = 4.8). Cardiology follow-up was minimal as would be expected (8 months); however, 46 ECGs on p.V205M negative individuals were on record for a period of up to 9.6 years post enrollment. Participant demographics are further described in Tables 2, 3.
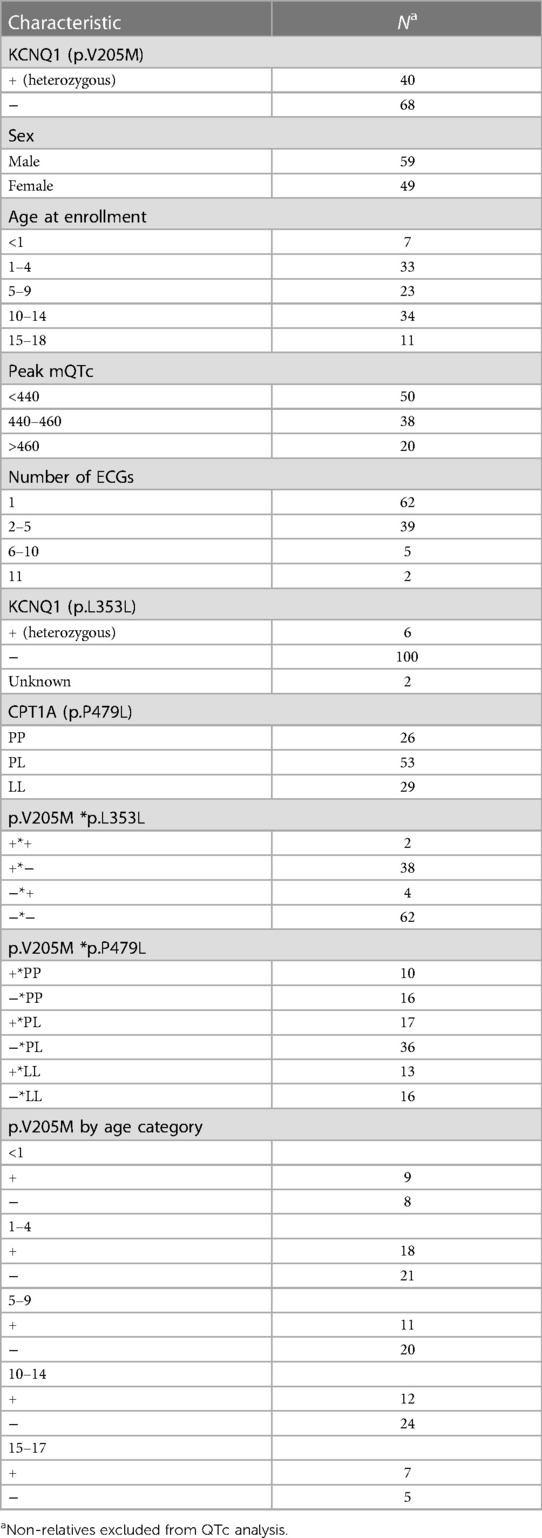
Table 2. Participant demographics of QTc analysis (n = 108).

Table 3. Participant demographics of LOC analysis (n = 186).
3.2 Peak QTc in childhood
Figure 1 shows the distribution of those KCNQ1 p.V205M positive participants compared to those without the p.V205M (mean QTc = 451.5 ms ± 24.3 ms and 427.1 ms ± 18.6 ms respectively p < 0.0001).
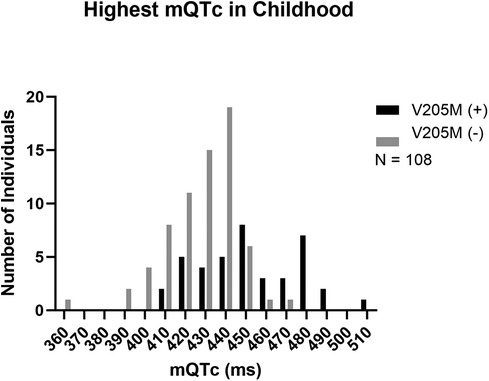
Figure 1. Peak mQTc in childhood. The peak mQTc measured in childhood from p.V205M positive children is depicted in black and the peak mQTc measured in childhood from negative control children is depicted in grey.
Regression analyses (Table 4) showed an average increase of QTc from baseline by 23.8 ms (95% CI: 15.3–32.5, p < 0.001) which was greater in females (30.1 ms) than males (18.9 ms), each significantly higher than baseline (p < 0.001), but not significantly different from each other. When added to the model, the p.L353L variant demonstrated a non-significant association of 15.5 ms among males and 9.1 ms among females. There was no association between QTc and the p.P479L variant (Supplementary Table S1).
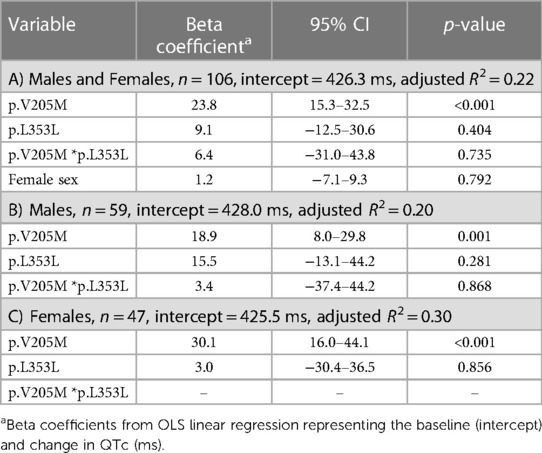
Table 4. Linear regression analysis of variant association on peak QTc in childhood.
3.3 Intra-patient variability
Figure 2 presents manual QTc measurements on repeat ECGs recorded in childhood at different ages for six cases. These cases were chosen as examples to demonstrate intra-patient variability in QTc throughout childhood.
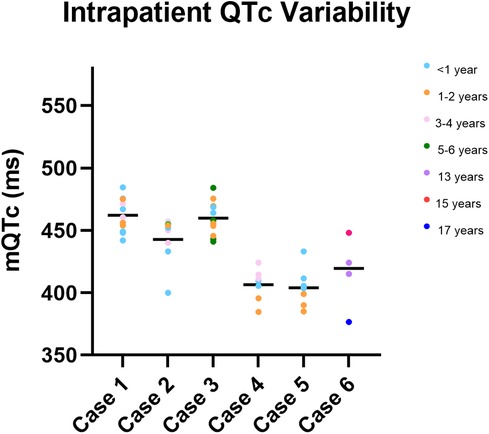
Figure 2. Intra-patient QTc variability in childhood. Depiction of manual QTc measurements recorded in childhood for six children (cases 1–6). Manual QTc measurements are depicted by coloured circles, with the mean mQTc measurement depicted by a black bar. The colour of the circle indicates the age at which the QTc was measured. Significant variability in mQTc measurements throughout childhood are observed in each child.
3.4 Highest QTc by age category
Results comparing the highest QTc obtained from participants within five age categories (males and females combined) are shown in Supplementary Figure S1. In all age categories, the mean QTc was prolonged for those who were p.V205M positive, but statistically significant in the 1–4 year, 5–9 year, and 10–14 year age categories. Although the QTc was generally more prolonged in the remaining two categories, the differences were not statistically significant with smaller sample sizes.
3.5 Syncope and seizures
No statistically significant association between p.V205M and the odds of experiencing at least one LOC event in childhood was observed (OR = 1.3, 95%CI 0.6–2.9) (Table 5A1). However, the odds were 3.0 times higher in children who were homozygous for the CPT1A variant compared to children who were negative (95% CI 1.2–7.7) (Table 5A2). Of note, the odds of experiencing a seizure were >4 for those homozygous for the p.P479L variant compared to those negative (95% CI 1.1–17.2) and 3.0 times higher for those heterozygous for p.P479L with a trend towards statistical significance (95% CI 0.8–10.8) (Table 5C2). When stratified by seizure type, the odds of experiencing a non-febrile seizure were 7.4 (95% CI 0.9–64.3) and febrile seizure were 2.3 (95% CI 0.4–13.2) for those homozygous for the p.P479L variant compared to negative cases (Supplementary Table S2). There was no evidence of interaction between the two variants.
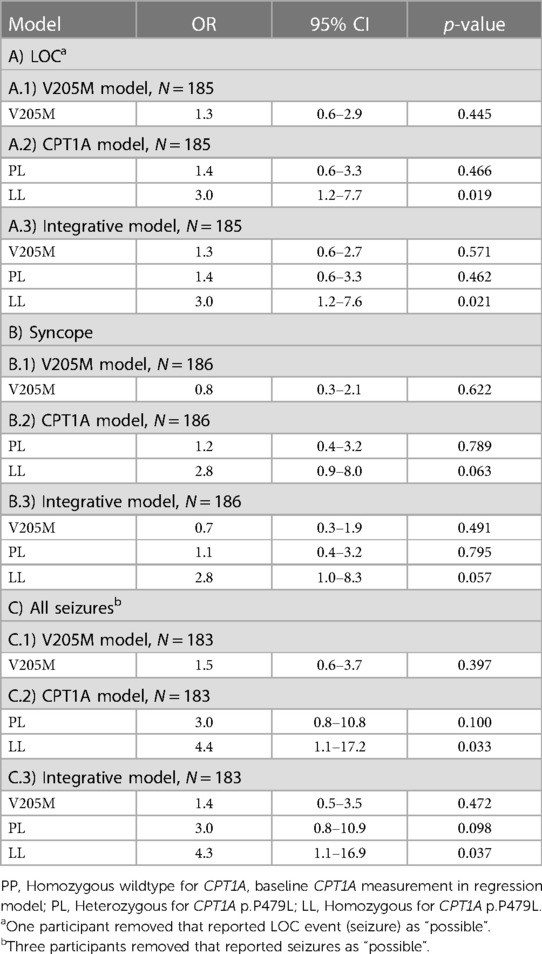
Table 5. Logistic regression analysis of LOC events.
Table 6 presents the number of children who experienced at least one LOC event in childhood stratified by p.V205M and p.P479L genotype. Of those without either variant (p.V205M negative and p.P479L negative), 10% experienced syncope and 3% seizures, which is similar to the general population rates (21, 22).
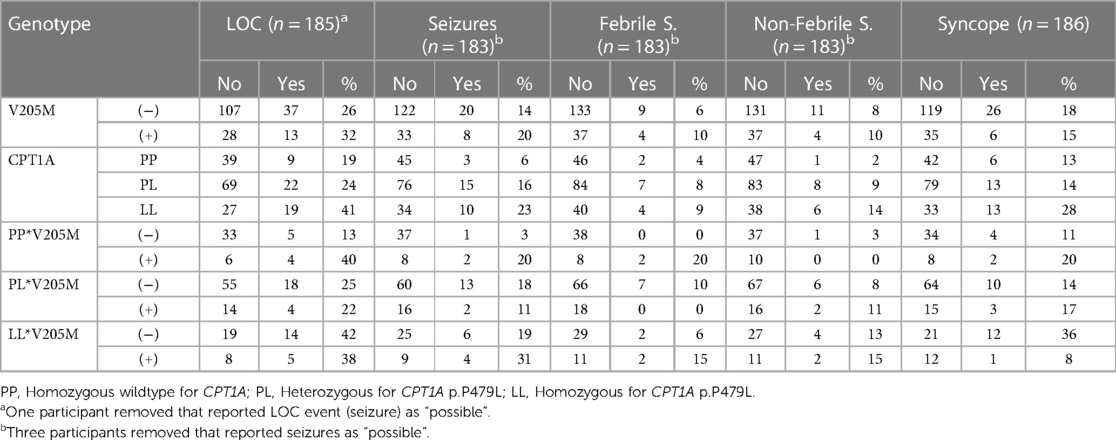
Table 6. Number of participants that experienced at least one LOC event in childhood by genotype.
Supplementary Table S3 depicts odds of LOC events by mQTc length among those p.V205M positive. Although there was an increased OR compared to reference (440 ms or less), there was no statistically significant difference for the categories with an increased mQTc length.
4 Discussion
4.1 Peak QTc
Although our results support that p.V205M significantly prolongs the QTc above baseline, with females notably more prolonged than males, our results suggests that those identified through cascade genetic testing (the majority of our cohort) have a mild phenotype. Only four cases presented prior to family screening. Two of these p.V205M positive cases were extreme outliers for the QTc analysis and thus excluded, including one who suffered a torsades de pointes/ventricular fibrillation cardiac arrest with an overdose of hydroxychloroquine. The second excluded case exhibited a high load of premature ventricular contractions with non-sustained ventricular tachycardia and was noted to have echocardiogram findings consistent with left ventricular non-compaction of unclear etiology (congenital vs. acquired left ventricular remodeling). Broad cardiac panel genetic testing (>130 genes) did not reveal any additional genetic contributors suggesting another diagnosis. For the remaining children, the majority were identified through cascade genetic testing (95%), and the mean QTc of 451.5 ± 24.26 ms is considered within the “borderline” category (440–460 ms) of pediatric QTc intervals (23). Our results suggest that the p.V205M variant has less of an effect in the pediatric age group than reported in our adult cohort. The mean QTc for variant positive adults was 476 ± 35 ms (4), suggesting a “prolonged” QTc (>470 ms) for adults (23). Similar to Gitxsan adults, the p.V205M variant in children was found to have a more clinically relevant effect on females compared to males, and that effect becomes more apparent in the 10–14 year old age category (see Supplementary Figure S1). Our data suggests that the effect of the p.V205M is generally mild in childhood and is corroborated by little evidence of historic pediatric deaths through our pedigree analysis.
A milder phenotype throughout childhood is consistent with the multigenerational presence of the variant. In circumstances with a severe presentation in childhood, reduced survival might impact population frequency of the variant. This phenomenon of a milder phenotype in founder populations in general with LQTS has been previously reported. Similar findings have been shown in Finland, where children with KCNQ1 and KCNH2 founder variants have been associated with significantly fewer cardiac events than children with Finnish non-founder KCNQ1 and KCNH2 variants (24). It is well known that proband presentations are more severe than those detected with cascade genetic testing and this effect can be exhibited in founder populations. Our founder population shows similar characteristics to the Finnish reports.
Although we did not find statistical evidence that the KCNQ1 p.L353L variant prolongs the QTc in childhood, in contrast to our previous studies of adults, the sample size of children with p.L353L was small (N = 6) and likely underpowered to show any effect. Similarly, no effect of p.P479L alone or interaction effect between p.V205M and p.P479L on the QTc was observed. If there is an effect on the QTc due to hypoglycemia, it might be intermittent, and therefore not detected in our results because of the timing of ECGs.
4.2 Syncope, seizure and cardiac arrest
There was no significant increase in odds of syncope or seizures, alone or in combination with the p.V205M variant. We observed the rate of syncope to be low (15%) in those who are positive, which is consistent with most previous literature on the topic (0%–9.3%) (24–26). One study of note demonstrated a rate of 27%, which was observed in a study cohort exclusively composed of children with LQTS1 not taking beta-blockers (27). The majority of studies reporting the rate of cardiac arrest in pediatric LQTS1 patients have found it to be low, ranging from 0 to 1.6% (24–30). There was one exception suggesting an 8% risk of cardiac arrest, although there were only 12 LQTS1 individuals in this study. In that study, high risk infants with LQTS identified with torsades de pointes or heart block as study inclusion criteria were assessed (31). Within our cohort, as mentioned above, one p.V205M positive case with an overdose of a QT prolonging drug experienced torsades de pointes and a ventricular fibrillation arrest (1/42 or 2.4%). Notably, there were no cardiac events with typical daily activities, sleep, or exercise. However, most of our cases were identified through cascade testing and although most were prescribed beta-blockers, adherence was not documented.
Overall report of seizures was common within our cohort, ranging from 14% in the p.V205M negative participants to 20% in those positive (Table 6). Our results showed an association of the CPT1A p.P479L variant with seizures and possibly syncope. Although the variant's impact on fatty acid oxidation predisposing children to hypoglycemia is the most likely explanation, the role of CPT1A in the brain has not been well explored; therefore, other mechanisms cannot be excluded at this time. Previous studies have found an association between p.P479L and hypoglycemia (9–11), and LOC events (most commonly seizures) are a known manifestation of hypoglycemia. To our knowledge, there is only one previous report (a published abstract) of an association between seizures and the p.P479L variant. This study found that p.P479L homozygotes were more likely to experience non-febrile seizures compared to heterozygote and wildtype children (32). Our study corroborates that work, but more research is indicated. We are not aware of previous studies reporting an association between syncopal events and p.P479L. While hypoglycemia is known to be a rare cause of syncope (33), it is also possible the association is due to participants misreporting of the events documented with the medical history questionnaire. Further study and more precise delineation of syncope or seizure phenotypes is indicated in all age groups. Several children were enrolled into the study with suspicion of LQTS because they presented with syncope/seizures. Our evidence suggests that in Northern BC, the CPT1A p.P479L variant is more likely to contribute to syncope and seizures than the p.V205M variant. Given the CPT1A variant is common in BC First Nations and other Northern/Arctic populations, these results may be useful for other northern populations.
4.3 Pediatric LQTS management
Beta-blocker induced hypoglycemia has been observed in LQTS children (14) and hypoglycemic syncope may occur as a rare side-effect which risks being misinterpreted as an arrhythmic event (33). Those with the CPT1A p.P479L variant may be at increased risk for hypoglycemia, especially during illness or prolonged fasting, and beta blocker medication could increase the risk for a hypoglycemic event. Given that those with loss of function variants in KCNQ1 may be prone to post-prandial hypoglycemia due to increased insulin release from the pancreas (13), increased vigilance is warranted when prescribing beta-blockers to those with both KCNQ1 variants and the CPT1A p.P479L variant. In this population, clinicians counsel families about avoiding prolonged periods of fasting and ensuring adequate glucose intake during intercurrent illness.
Given that cardiac arrest is rare in the pediatric population and there is no evidence to suggest the p.V205M variant confers increased risk for seizures/syncope, we suggest that the clinical management of children with p.V205M may not always require beta-blockers. As noted, two of our more severe cases were detected symptomatically; however, those detected through cascade genetic testing showed only mildly increased QTcs. Given that most cases were diagnosed at young ages because of a family member presenting, provided with advice to avoid QT prolonging medications, and prescribed beta-blocker therapy, the management may have mitigated more severe childhood presentations. It is also possible that this population will have a less severe presentation given the milder presentations often documented with founder variants traced back for several generations. In accordance with well-established management standards most children with LQTS1 will be prescribed beta-blocker medications (34, 35). The AHA/ACC/HRS guidelines recommend beta-blockers to all LQTS patients with a QTc ≥470 ms and state that it is reasonable for asymptomatic molecularly diagnosed individuals with a QTc <470 ms to take beta-blockers (class IIa recommendation). The guidelines state that the benefits of beta-blockers are greater than the risks for asymptomatic individuals with a QTc <470 ms (35). However, it has also been suggested that beta-blockers may be unnecessary for some asymptomatic children (24, 36). Waddell-Smith et al. (2015) proposed that beta-blockers may not be essential if: “(1) the QTc is less than 470 ms and, (2) the patient does not have a C-loop LQTS type 1 missense mutation and, (3) the patient does not partake in high risk activities, and (4) the patient is either a preschool boy or prepubertal girl”. Additionally, due to the mild phenotype observed in children with Finnish founder variants, Finnish pediatric cardiologists suggest children with KCNH2 founder variants start beta-blockers later in childhood but before the onset of puberty, provided their QTc is below 470 ms and there is no family history of cardiac arrest (24). Intentional non-therapy has also been proposed for asymptomatic adults with an older age at diagnosis and a QTc <470 ms (37).
In many circumstances, the evidence supporting beta-blocker therapy as a life-saving medication is firmly established (38); however, in circumstances that are less clear, shared-decision making with parents and older children could be considered. Although our sample size is relatively small, we posit that beta-blockers could be optional for p.V205M positive patients if they are asymptomatic, consistently measure a QTc <470 ms (note intra-patient variability of QTc is typical, see Figure 2), do not participate in high-risk activities, do not require treatment with potentially QT prolonging medications, and have no close family history of cardiac arrest. These decisions would of course follow the principles of shared decision making, in which the patient and care providers are fully informed about treatment pathways, risks, and benefits of each option. We also emphasize the importance of a safety plan, which includes supervised swimming, communal CPR awareness, and automated external defibrillator access. It is important to note that in our umbrella study documenting the family histories of more than 150 p.V205M positive cases of all ages, women of childbearing years have been observed to be at highest risk for cardiac arrest. However, only one cardiac arrest before age 18 has been reported to date.
In general, for those identified by family cascade testing with the p.V205M variant, we suggest avoidance of QT prolonging drugs and yearly (or more often as indicated) ECG QTc review (34, 35) by an informed pediatrician or cardiologist. The merits of beta-blocker medications should be discussed for all and recommended for those with a documented QTc greater than 470 ms, identified symptomatically, and participating in competitive sports. Self-awareness of cardiac symptoms (palpitations, syncope) and seeking medical attention as needed will reduce risk of events (34, 35). In addition, replenishing electrolytes in periods of dehydration (such as gastroenteritis and vigorous sports activities) is important to consider.
Although none of the children in this study were homozygous for the p.V205M variant, previous documentation of the clinical phenotype of homozygous cases suggest a more severe phenotype which includes pediatric onset of clinical features (4). As such, we recommend beta-blockers for any children homozygous for the p.V205M variant as well as close clinical monitoring, given additional management (such as implantable cardioverter defibrillator) may be necessary.
Although the CPT1A p.P479L variant is specific to Indigenous populations, our results are a reminder that genetic variants other than those contributing to LQTS may influence the phenotype. Given the high rate of the CPT1A p.P479L in British Columbia First Nations and other Northern Indigenous populations, more study should be carried out to better understand the apparent associated risk of syncope and seizures and consider potential preventative strategies.
5 Conclusion
The KCNQ1 p.V205M variant was found to significantly prolong the QTc in childhood in the Gitxsan population; however, there was no statistical evidence that the p.V205M variant increased the odds of syncope/seizures or other cardiac events in childhood. In contrast, the CPT1A p.P479L variant, which is also common in this population, was found to significantly increase the odds of LOC events in childhood which may be misinterpreted as cardiac events. More research is needed to further elucidate the individual and combined effects of these variants. That most cases with the p.V205M variant are asymptomatic during childhood is reassuring. Although QT prolonging drugs should be avoided for all children with the p.V205M variant, other management strategies for those who are p.V205M positive, such as beta-blocker therapy, could be considered on a case-by-case basis.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving humans were approved by University of British Columbia Clinical Research Ethics Board, Northern Health Authority Research Ethics Board, Island Health Research Ethics Board University of Victoria Research Ethics Board. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin.
Author contributions
SB: Conceptualization, Formal Analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. BG: Conceptualization, Data curation, Methodology, Writing – review & editing. AE: Formal Analysis, Writing – review & editing. JM: Conceptualization, Supervision, Writing – review & editing. MS: Conceptualization, Supervision, Writing – review & editing. LG: Data curation, Project administration, Writing – review & editing. SM: Data curation, Methodology, Project administration, Writing – review & editing. CE: Project administration, Writing – review & editing. ES: Formal Analysis, Methodology, Writing – review & editing. RM: Conceptualization, Supervision, Writing – review & editing. SS: Conceptualization, Supervision, Writing – review & editing, Formal Analysis, Methodology. LA: Conceptualization, Formal Analysis, Methodology, Supervision, Writing – review & editing, Funding acquisition, Project administration, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This research was funded by the Canadian Institutes of Health Research, grant no. 152972 to LA and by a British Columbia Graduate Scholarship to SB. LA is supported through a BC Children’s Hospital Research Institute Investigatorship.
Acknowledgments
We acknowledge our continued partnership with the Gitxsan Research Advisory Committee and Gitxsan Health Society and extend our gratitude to the research participants, local healthcare providers, and wider Gitxsan community. We thank Dr. Carmen Rodriguez de France, Alexa McAdam, Ashleigh Hansen, Dr. Jimena Gonzalez Lema, and Emma Ewasiuk for their support and guidance during this project. This paper was adapted from the Graduate Thesis of SBW published by University of Victoria. A portion of the results in this paper was presented at the Canadian Cardiovascular Congress 2023 and American Society of Human Genetics in 2020.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2024.1394105/full#supplementary-material
References
1. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Alessandra B, Giuliano B, et al. Prevalence of the congenital long-QT syndrome. Circulation. (2009) 120:18. doi: 10.1161/CIRCULATIONAHA.109.863209
2. Jackson H, Huisman L, Sanatani S, Arbour LT. Long QT syndrome. Can Med Assoc J. (2011) 183:11. doi: 10.1503/cmaj.100138/-/DC1
3. Arbour L, Rezazadeh S, Eldstrom J, Weget-Simms G, Rupps R, Dyer Z, et al. A KCNQ1 V205M missense mutation causes a high rate of long QT syndrome in a first nations community of northern British Columbia: a community-based approach to understanding the impact. Genet Med. (2008) 10:7. doi: 10.1097/GIM.0b013e31817c6b19
4. Jackson HA, Mcintosh S, Whittome B, Asuri S, Casey B, Tang A, et al. LQTS In northern BC: homozygosity for KCNQ1 V205M presents with a more severe cardiac phenotype but with minimal impact on auditory function. Clin Genet. (2014) 86:1. doi: 10.1111/cge.12235
5. Kapplinger JD, Erickson A, Asuri S, Tester D, McIntosh S, Kerr C, et al. KCNQ1 P.L353l affects splicing and modifies the phenotype in a founder population with long QT syndrome type 1. J Med Genet. (2017) 54:6. doi: 10.1136/jmedgenet-2016-104153
6. Swayne LA, Murphy NP, Asuri S, Chen L, Xiaoxue X, McIntosh S, et al. Novel variant in the ANK2 membrane-binding domain is associated with ankyrin-B syndrome and structural heart disease in a first nations population with a high rate of long QT syndrome. Circ Cardiovasc Genet. (2017) 10:1. doi: 10.1161/CIRCGENETICS.116.001537
7. Sinclair GB, Collins S, Popescu O, McFadden D, Arbour L, Vallance HD. Carnitine palmitoyltransferase I and sudden unexpected infant death in British Columbia first nations. Pediatrics. (2012) 130:5. doi: 10.1542/peds.2011-2924
8. Collins SA, Surmala P, Osborne G, Greenberg C, Williamson Bathory L, Edmunds-Potvin S, et al. Causes and risk factors for infant mortality in Nunavut, Canada 1999−2011. BMC Pediatr. (2012) 12:1. doi: 10.1186/1471-2431-12-190
9. Greenberg CR, Dilling LA, Thompson GR, Seargeant L, Haworth J, Phillips S, et al. The paradox of the carnitine palmitoyltransferase type ia P479l variant in Canadian aboriginal populations. Mol Genet Metab. (2009) 96:4. doi: 10.1016/j.ymgme.2008.12.018
10. Collins SA, Hildes-Ripstein GE, Thompson JR, Edmunds S, Miners A, Rockman-Greenberg C, et al. Neonatal hypoglycemia and the CPT1A P479l variant in term newborns: a retrospective cohort study of inuit newborns from Kivalliq Nunavut. Paediatr Child Health. (2020) 26:4. doi: 10.1093/pch/pxaa039
11. Gillingham MB, Hirschfeld M, Lowe S, Matern D, Shoemaker J, Lambert W, et al. Impaired fasting tolerance among Alaska native children with a common carnitine palmitoyltransferase 1A sequence variant. Mol Genet Metab. (2011) 104:3. doi: 10.1016/j.ymgme.2011.06.017
12. Collins SA, Sinclair G, McIntosh S, Bamforth F, Thompson R, Sobol I, et al. Carnitine palmitoyltransferase 1A (CPT1A) P479l prevalence in live newborns in Yukon, northwest territories, and Nunavut. Mol Genet Metab. (2010) 101:2–3. doi: 10.1016/j.ymgme.2010.07.013
13. Torekov SS, Iepsen E, Christiansen M, Linneberg A, Pedersen O, Holst J, et al. KCNQ1 long QT syndrome patients have hyperinsulinemia and symptomatic hypoglycemia. Diabetes. (2014) 63:4. doi: 10.2337/db13-1454
14. Poterucha JT, Bos JM, Cannon BC, Ackerman MJ. Frequency and severity of hypoglycemia in children with beta-blocker-treated long QT syndrome. Heart Rhythm. (2015) 12:8. doi: 10.1016/j.hrthm.2015.04.034
15. Suys B, Heuten S, de Wolf D, Verherstraeten M, de Beeck LO, Matthys D, et al. Glycemia and corrected QT interval prolongation in young type 1 diabetic patients: what is the relation? Diabetes Care. (2006) 29:2. doi: 10.2337/diacare.29.02.06.dc05-1450
16. Frier BM, Schernthaner G, Heller SR. Hypoglycemia and cardiovascular risks. Diabetes Care. (2011) 34:S2. doi: 10.2337/dc11-s220
17. Gill GV, Woodward A, Casson IF, Weston PJ. Cardiac arrhythmia and nocturnal hypoglycaemia in type 1 diabetes-the “dead in bed” syndrome revisited. Diabetologia. (2009) 52:1. doi: 10.1007/s00125-008-1177-7
18. Arbour L, Cook D. DNA on loan: issues to consider when carrying out genetic research with aboriginal families and communities. Community Genet. (2006) 9:3. doi: 10.1159/000092651
19. Vink AS, Neumann B, Lieve KVV, Sinner MF, Hofman N, El Kadi S, et al. Determination and interpretation of the QT interval. Circulation. (2018) 138:21. doi: 10.1161/CIRCULATIONAHA.118.033943
20. Bazett HC. An analysis of the time-relations of electrocardiograms. Ann Noninvasive Electrocardiol. (1997) 2:2. doi: 10.1111/j.1542-474x.1997.tb00325.x
21. Friedman MJ, Sharieff GQ. Seizures in children. Pediatr Clin North Am. (2006) 53:2. doi: 10.1016/j.pcl.2005.09.010
22. Friedman KG, Alexander ME. Chest pain and syncope in children: a practical approach to the diagnosis of cardiac disease. J Pediatr. (2013) 163:3. doi: 10.1016/j.jpeds.2013.05.001
23. Goldenberg I, Moss AJ, Zareba W. QT Interval: how to measure it and what is “normal”. J Cardiovasc Electrophysiol. (2006) 17:3. doi: 10.1111/j.1540-8167.2006.00408.x
24. Koponen M, Marjamaa A, Hiippala A, Happonen J-M, Havulinna A, Salomaa V, et al. Follow-Up of 316 molecularly defined pediatric long-QT syndrome patients. Circ Arrhythm Electrophysiol. (2015) 8:4. doi: 10.1161/CIRCEP.114.002654
25. Chambers KD, Ladouceur VB, Alexander ME, Hylind RJ, Belivacqua L, Mah DY, et al. Cardiac events during competitive, recreational, and daily activities in children and adolescents with long QT syndrome. J Am Heart Assoc. (2017) 6:9. doi: 10.1161/JAHA.116.005445
26. Villain E, Denjoy I, Lupoglazoff JM, Guicheney P, Hainque B, Bonnet D. Low incidence of cardiac events with β-blocking therapy in children with long QT syndrome. Eur Heart J. (2004) 25:16. doi: 10.1016/j.ehj.2004.06.016
27. Ozawa J, Ohno S, Hisamatsu T, Hideki I, Makiyama T, Suzuki H, et al. Pediatric cohort with long QT syndrome—kCNH2 mutation carriers present late onset but severe symptoms. Circ J. (2016) 80:3. doi: 10.1253/circj.CJ-15-0933
28. Petko C, Bradley DJ, Tristani-Firouzi M, Cohen M, Sanatani S, Saarel E, et al. Congenital long QT syndrome in children identified by family screening. Am J Cardiol. (2008) 101:12. doi: 10.1016/j.amjcard.2008.02.068
29. Vink AS, Clur SAB, Geskus RB, Blank AC, De Kezel CCA, Yoshinaga M, et al. Effect of age and sex on the QTc interval in children and adolescents with type 1 and 2 long-QT syndrome. Circ Arrhythm Electrophysiol. (2017) 10:4. doi: 10.1161/CIRCEP.116.004645
30. Kwok SY, ChB M, Pflaumer A, Pantaleo S-J, Date E, Jadhav M, et al. Ten-year experience in atenolol use and exercise evaluation in children with genetically proven long QT syndrome. J Arrhythm. (2017) 33:6. doi: 10.1016/j.joa.2017.08.004
31. Moore JP, Gallotti RG, Shannon KM, Bos JM, Sadeghi E, Strasburger JF, et al. Genotype predicts outcomes in fetuses and neonates with severe congenital long QT syndrome. JACC Clin Electrophysiol. (2020) 6:12. doi: 10.1016/J.JACEP.2020.06.001
32. Greenberg C, Stannard K, Webb J. CPT1 P479l variant: clinical significance amongst Inuit children in Canada [abstract]. 4th International Meeting on Indigenous Child Health. Securing our Future: Advancing Circles of Caring (2011).
33. Hussain T, Greenhalgh K, Mcleod KA. Hypoglycaemic syncope in children secondary to beta-blockers. Arch Dis Child. (2009) 94:968–9. doi: 10.1136/adc.2008.145052
34. Schwartz PJ, Ackerman MJ. The long QT syndrome: a transatlantic clinical approach to diagnosis and therapy. Eur Heart J. (2013) 34:40. doi: 10.1093/eurheartj/eht089
35. Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Circulation. (2018) 138:13. doi: 10.1161/CIR.0000000000000549
36. Waddell-Smith KE, Earle N, Skinner JR. Must every child with long QT syndrome take a beta blocker? Arch Dis Child. (2015) 100:3. doi: 10.1136/archdischild-2014-306864
37. MacIntyre CJ, Rohatgi RK, Sugrue AM, Bos JM, Ackerman MJ. Intentional nontherapy in long QT syndrome. Heart Rhythm. (2020) 17:7. doi: 10.1016/J.HRTHM.2020.02.017
Keywords: long QT syndrome (LQTS), First Nations, Indigenous, carnitine palmitoyltransferase 1A, long QT syndrome type 1, KCNQ1
Citation: Bene Watts S, Gauthier B, Erickson AC, Morrison J, Sebastian M, Gillman L, McIntosh S, Ens C, Sherwin E, McCormick R, Sanatani S and Arbour L (2024) A mild phenotype associated with KCNQ1 p.V205M mediated long QT syndrome in First Nations children of Northern British Columbia: effect of additional variants and considerations for management. Front. Pediatr. 12:1394105. doi: 10.3389/fped.2024.1394105
Received: 6 March 2024; Accepted: 16 May 2024;
Published: 31 May 2024.
Edited by:
Roy Morello, University of Arkansas for Medical Sciences, United StatesReviewed by:
Matteo Vatta, Indiana University Bloomington, United StatesYuko Wada, Vanderbilt University Medical Center, United States
© 2024 Bene Watts, Gauthier, Erickson, Morrison, Sebastian, Gillman, McIntosh, Ens, Sherwin, McCormick, Sanatani and Arbour. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Arbour, bGFyYm91ckB1dmljLmNh
†The views and findings presented in this manuscript are the authors’ and do not necessarily represent that of the BC Ministry of Health or Interior Health Authority
‡These authors share senior authorship