 Maria Fasshauer1
Maria Fasshauer1 Sarah Dinges2
Sarah Dinges2 Olga Staudacher2
Olga Staudacher2 Mirjam Völler2Anna Stittrich3
Mirjam Völler2Anna Stittrich3 Horst von Bernuth2,4,5,6
Horst von Bernuth2,4,5,6 Volker Wahn2,†
Volker Wahn2,† Renate Krüger2*†
Renate Krüger2*†
- 1Immuno Deficiency Center Leipzig, Jeffrey Modell Diagnostic and Research Center for Primary Immunodeficiency Diseases, Hospital St. Georg, Leipzig, Germany
- 2Department of Pediatric Respiratory Medicine, Immunology and Critical Care Medicine, Charité - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, Berlin Institute of Health (BIH), Berlin, Germany
- 3Department of Human Genetics, Labor Berlin - Charité Vivantes GmbH, Berlin, Germany
- 4Department of Immunology, Labor Berlin - Charité VivantesGmbH, Berlin, Germany
- 5Berlin Institute of Health (BIH), Charité - Universitätsmedizin Berlin, Berlin, Germany
- 6Charité - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, Berlin Institute of Health (BIH), Berlin-Brandenburg Center for Regenerative Therapies (BCRT), Berlin, Germany
In patients with severe and recurrent infections, minimal diagnostic workup to test for Inborn Errors of Immunity (IEI) includes a full blood count, IgG, IgA and IgM. Vaccine antibodies against tetanus toxoid are also frequently measured, whereas testing for anti-polysaccharide IgG antibodies and IgG subclasses is not routinely performed by primary care physicians. This basic approach may cause a significant delay in diagnosing monogenic IEI that can present with an impaired IgG response to polysaccharide antigens with or without IgG subclass deficiency at an early stage. Our article reviews genetically defined IEI, that may initially present with an impaired IgG response to polysaccharide antigens, but normal or only slightly decreased IgG levels and normal responses to protein or conjugate vaccine antigens. We summarize clinical, genetic, and immunological findings characteristic for these IEI. This review may help clinicians to identify patients that require extended immunologic and genetic evaluations despite unremarkable basic immunologic findings. We recommend the inclusion of anti-polysaccharide IgG antibodies as part of the initial routine work-up for possible IEI.
Introduction
Inborn Errors of Immunity (IEI), formerly termed primary immunodeficiency diseases (PID), are a heterogeneous group of mainly monogenic diseases characterized by infections and/or immune dysregulation. The absence or impaired function of any immunologic component can determine the clinical presentation of an IEI. The heterogeneity of these diseases makes it difficult to direct routine laboratory work up (1, 2). When IEI with antibody deficiency are suspected (e.g., in patients with recurrent infections of the airways, the ears, the meninges, or the skin), the basic immunologic evaluation recommended by the German AWMF guideline (dating back to 2011), comprises a full blood count and measurement of IgG, IgA, and IgM levels (3). Once B-cell defects are suspected and serum immunoglobulins are detectable, evaluation of the IgG response to protein as well as polysaccharide antigens is recommended (4). To date, most primary care physicians do not routinely determine anti-polysaccharide (e.g., anti-pneumococcal) IgG/IgG2 antibodies or IgG subclasses when an IEI with antibody deficiency is suspected. A growing number of monogenic IEI has been reported, which may initially present with an impaired IgG response to polysaccharide antigens, whereas total IgG levels and response to protein (e.g., anti-tetanus toxoid IgG antibodies) and polysaccharide conjugate vaccines may be normal. Since polysaccharide-specific IgG antibodies fall within the IgG2 subclass fraction, IgG2 levels may also be decreased in these patients (5–7). Without the determination of anti-polysaccharide IgG antibodies and IgG subclasses, the diagnosis of several rare IEI may be missed or delayed. Delayed therapeutic interventions may then result in further infections and irreversible organ damage (e.g., bronchiectasis).
Patients with an impaired IgG response to polysaccharide antigens often suffer from recurrent or severe sinopulmonary infections with encapsulated bacteria that express abundant polysaccharide antigens on their surfaces e.g., Streptococcus (S.) pneumoniae, Haemophilus (H.) influenzae serotype b, and Neisseria (N.) meningitidis (6, 8). Severe invasive infections like meningitis, septicemia, or osteoarticular infections can also occur (7, 9, 10). An impaired IgG response to polysaccharide antigens can be diagnosed if wild-type infections with S. pneumoniae or vaccination with a pure polysaccharide vaccine (e.g., Pneumovax™) do not result in a significant increase of pneumococcal-specific IgG antibodies (11–13). Commercially available tests to assess IgM and IgA pneumococcal polysaccharide specific antibodies before and after pneumococcal polysaccharide based vaccinations are not routinely used but may be helpful in testing patients with IEI under IgG replacement therapy (14–16).
Humoral immune responses to proteins and polysaccharides
Based mainly on studies in mice, Figures 1, 2 illustrate the differences between immune responses against proteins and polysaccharides. Knowledge of these mechanisms is exploited for routine vaccination (17). The main immunological differences between the two mechanisms are summarized in Table 1. It explains the impaired IgG response to polysaccharide antigens described in some patients after polysaccharide vaccinations, usually with a 23-valent pneumococcal polysaccharide vaccine. In natural infections, for instance with pneumococci, protein and polysaccharide responses occur in parallel, in contrast to vaccine responses. The review by Gingerich and Mousa summarizes the structure of the bacteria and the targets of antibody formation (18).
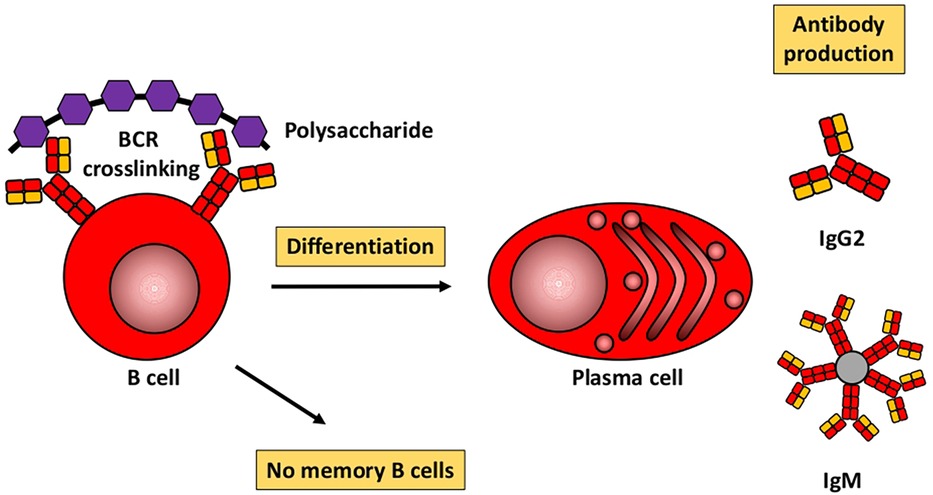
Figure 1. Humoral immune response to polysaccharides. Modified from Pollard et al. (17).
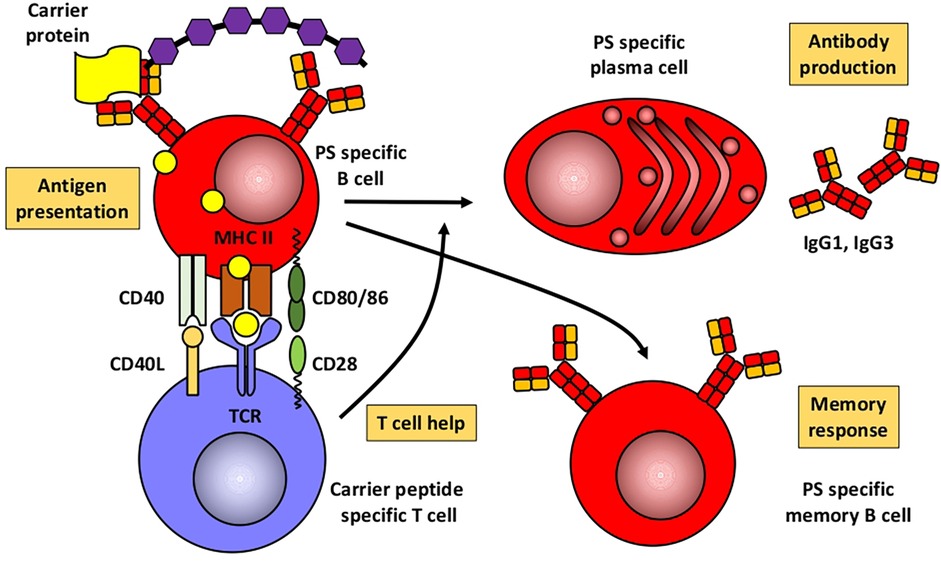
Figure 2. Humoral immune response to conjugated polysaccharides. Modified from Pollard et al. (17). PS, polysaccharide.
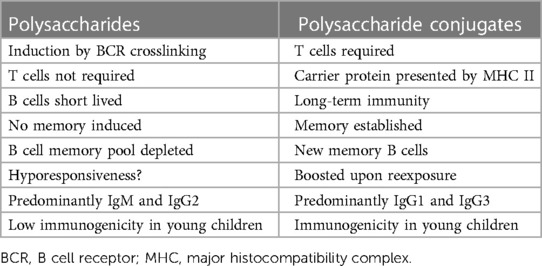
Table 1. From Pollard et al. (17).
Recent literature has focused on the question as to which polysaccharides response is “normal”, the missing standardization of measuring anti-polysaccharide antibodies, the differences in assessment specifically in conjugate-primed and non-primed individuals, excluding other underlying diseases, or the need to verify patient-reported respiratory tract infections in patients with an impaired response to polysaccharide antigens. This shows the importance of measuring anti-polysaccharide antibodies and evaluating an immune response to polysaccharide antigens, as it further enlightens immune response to certain microbes (19, 20). Over the last decade, next-generation sequencing (NGS) for detecting genetic variants in IEI has identified an ever-increasing number of genetic causes and led to more specific diagnoses (21). Some of the monogenic IEI lack anti-polysaccharide antibodies due to an impaired immune response to polysaccharide antigens. However, IgG in these IEI can be normal or slightly decreased and the response to protein (e.g., tetanus toxoid) or conjugate (e.g., polysaccharide-protein) vaccines can also be unimpaired. These IEI -presented below- mostly show other immunologic abnormalities in addition to an impaired IgG response to polysaccharide antigens. Our review is therefore not restricted to IEI with specific antibody deficiency (SAD), since SAD is strictly defined as an IEI with an impaired response to polysaccharide antigens as the only abnormal immunologic finding (13, 20, 22). Following these basic considerations, we will now discuss specific aspects of respective monogenic IEI. Common clinical, genetic, and immunologic findings are also summarized in Table 2.
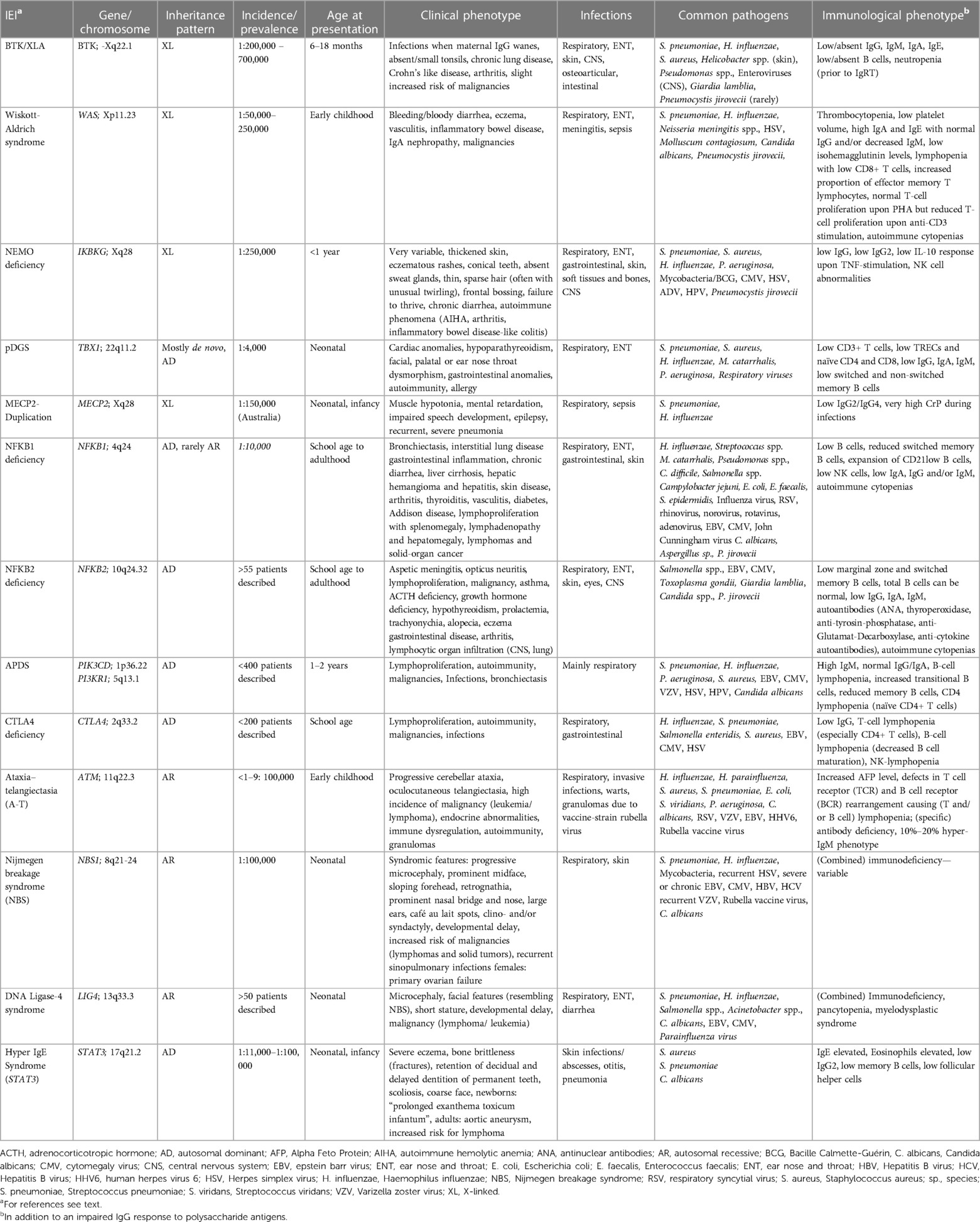
Table 2. Monogenic IEI with impaired IgG response to polysaccharide antigens but normal IgG levels: Common clinical, genetic, and immunologic findings.
Hypomorphic mutations in BTK
X-linked agammaglobulinemia (XLA; M. Bruton) was first described in 1952 (23). It is an IEI with an estimated incidence of 1:200,000–700,000 live births. More than 600 pathogenic variants within BTK (encoding Bruton's Tyrosine Kinase) on chromosome Xq22.1 have been described to date (24). XLA patients have low or absent peripheral B cells and most patients have absent or very low IgG, IgA and IgM. Prior to IgG replacement therapy (IgRT) neutropenia is frequently observed (25–27). Without IgRT patients present with recurrent bacterial and viral infections beyond 3–6 months of age, once maternal IgG levels wane. Otitis media and pneumonia are the most frequent infections, severe invasive infections (meningitis, septic arthritis) as well as skin and gastrointestinal infections can also occur. Airway and invasive infections are mostly caused by S. pneumoniae, H. influenzae and S. aureus. Pseudomonas and Helicobacter species have been reported in chronic skin infections (28, 29). XLA patients are also susceptible to enteroviruses (e.g., poliovirus, coxsackievirus, echovirus) (30), that should be suspected in XLA patients with meningoencephalitis. Giardia lamblia can cause chronic diarrhea. In older children and adult XLA patients, chronic lung disease and autoimmunity (arthritis, enteropathy) is frequently reported, despite adequate IgG levels under IgRT (31, 32). Case studies have described patients with hypomorphic mutations in BTK who have no or only slightly decreased IgG levels, respond well to tetanus-toxoid vaccinations, but cannot mount an IgG response to polysaccharide antigens (33, 34).
Wiskott-Aldrich syndrome
The X-linked Wiskott-Aldrich syndrome (WAS) was first described in 1937 (35), and further delineated by R. Aldrich in 1954 (36). Positional cloning in the 1990s identified mutations in the gene encoding the Wiskott-Aldrich syndrome protein (WASp) as the cause of the disease (37). WASp is a crucial regulator of various functions in hematopoietic and immune cells, including cytoskeletal reorganization, immune synapse formation, and intracellular signaling (38, 39). Genotype-phenotype correlations for more than 150 mutations are sparse. WAS has an estimated incidence of about 4 per million live male births and presents a wide spectrum of clinical phenotypes (40). The disease commonly manifests as a bleeding disorder coupled with increased susceptibility to infections (encapsulated pathogens, viruses, opportunistic pathogens), autoimmunity (e.g., eczema, autoimmune cytopenias, vasculitis, inflammatory bowel disease, IgA nephropathy), and an increased risk of hematological malignancies (41). Typically, both cellular and humoral immunity are affected. Common laboratory findings include thrombocytopenia with reduced platelet volume and lymphopenia, often due to T-cell loss, while B-cell counts usually remain stable. Platelet volume may normalize in patients post splenectomy, so normal platelet volume does not exclude WAS in patient post splenectomy (42). Decreased CD8+ T cell count and function, reduced natural killer (NK) cell cytotoxicity, and impaired regulatory T cell (Treg) cell function contribute to the immunodeficiency and autoimmunity (43). WASp gene mutations disrupt actin polymerization in WAS-deficient T cells, affecting T- and B-cell interactions that are crucial for memory cell development and isotype switching (44). As a result, immunoglobulin levels are often altered (typically elevated IgA and IgE, normal IgG, and decreased IgM), isohemagglutinin titers tend to be low, and the specific IgG antibody response to various antigens is often insufficient (e.g., pneumococcal polysaccharide and tetanus toxoid) (45).
Nuclear factor-kappa B essential modulator (NEMO) deficiency
NEMO deficiency syndrome is a rare, combined immunodeficiency that was concurrently elucidated in three seminal publications in the early 2000s (46–48). It is caused by hypomorphic mutations in the X-linked IKBKG (also known as inhibitor of nuclear factor kappa-B kinase subunit gamma, NEMO) gene. Genetic analysis is complicated through homologous pseudogene sequences. NEMO is a key player in the NF-κB pathway, consequently, crucial for the development and function of the ectoderm, the immune system, and bones. The prevalence of NEMO deficiency is about 1:250,000. The phenotype spectrum in NEMO deficiency is exceedingly broad. 80% of patients exhibit ectodermal dysplasia (EDA) characterized by thickened skin, eczema, conical teeth, absent sweat glands, and hypotrichosis (49). Immune deficiency in NEMO patients renders them susceptible to recurrent respiratory tract infections, encapsulated bacteria, severe viral (CMV) and opportunistic infections (pneumocystis, environmental mycobacteria) (50, 51). Typically, patients present with severe pneumococcal infections despite vaccination, early episodes of meningitis, or deep tissue infections (51). Besides EDA and immune deficiency, a subset of patients also suffers from osteopetrosis, lymphedema, and/or autoimmunity. Characteristic laboratory findings are normal or low IgG (especially low IgG2) levels with elevated or decreased IgM or IgA levels. Immune response to stimulation with LPS, IL-1ß and TNF may be decreased, and IgG response to pneumococcal polysaccharide vaccination is in most cases insufficient. Class-switched B cells tend to be reduced, and/or NK cell activity abnormal (49–54).
DiGeorge syndrome/22q11.2 deletion syndrome
Neonatal co-occurrence of thymic aplasia and hypoparathyroidism was initially described by DiGeorge in 1965 (55) and later called DiGeorge syndrome (DGS). The 22q11.2 microdeletion is both the most common cause of DGS and the most common human chromosomal microdeletion syndrome in general, with an incidence of 1:4000 live births. Over 90% of the cases are de novo mutations causing haploinsufficiency of TBX1, a transcription factor involved in patterning of the third pharyngeal pouch. The genotype-phenotype correlation is weak and there is great clinical variability. While less than 1% of DGS patients are athymic (complete DGS), the majority presents with less severe thymic hypofunction (partial DGS, pDGS). pDGS is defined as <1,500/mm3 CD3+ T cells and at least one of the following: (1) cardiac malformation, which is the main cause of death, (2) hypocalcemia, or (3) facial or palatal dysmorphism. Gastrointestinal anomalies, autoimmunity, mostly cytopenias and thyroid disease, and allergic manifestations are frequent. Patients present with recurrent infections including sinusitis, otitis media, bronchitis, and pneumonia (56). T-cell lymphopenia of varying severity is the most common feature, though 20% of patients have normal T-cell counts (57). Over time, T cells meet levels of age-matched controls due to homeostatic expansion, resulting in decreased T-cell receptor repertoire (56, 58). Lymphoproliferation upon stimulation with mitogens is mostly adequate. Delayed B-cell maturation, reduced numbers of naïve and unswitched memory B cells and peripheral class switched memory B cells, as well as hypogammaglobulinemia and isolated IgA or IgM deficiency may occur (58). Impaired IgG response to polysaccharide antigens was reported in 40 to 55% of patients, most likely due to impaired assistance of thymus-derived T cells to the humoral immune response (56, 59).
Methyl-CpG-Binding protein 2 (MECP2) duplication syndrome
MECP2 duplication syndrome is an X-linked disorder first described in 2005 (60, 61). For a recent review, see (62). In Australia, incidence is estimated at 1: 150,000 live births, respective estimates from Europe are not available. More than 500 patients have been reported to date (reviewed in (62). Penetrance is 100%, affected males present with neonatal muscular hypotonia, severe intellectual disabilities, recurrent and frequent seizures, low sensitivity to pain, as well as severe infections of the lower airways. During infections, hyperinflammation with very high levels of inflammatory markers [e.g., C-reactive protein (CrP)] can be observed. The majority of affected patients die from severe pneumonia in childhood or as young adults. Most patients have low IgG2 and IgG4 subclasses and an impaired response to polysaccharide antigens (60, 63–71).
Nuclear factor-kappa B1 (NFKB1) deficiency
NFKB deficiency is an IEI with immune dysregulation. NFKB1 loss-of-function or hypomorphic variants, first reported in 2015 (72), are inherited autosomal dominant resulting in haploinsufficiency (73). Recently, in 2021, the first autosomal recessive variant leading to NFKB1 deficiency was identified (74). NFKB1 deficiency affects up to 1 in 10,000 individuals. Three quarters of the patients fulfill the criteria of CVID. NFKB1 deficiency accounts for 4 to 5% of genetically resolved CVID cases, thus representing the most common monogenetic etiology of CVID (73, 75). Incomplete penetrance and age-depended disease progression were reported (73). Patients present with respiratory, gastrointestinal, skin, and opportunistic infections, mostly bacterial. Lymphoproliferation and autoimmunity, particularly cytopenias, are characteristic. There is a high frequency of multiorgan autoinflammation, noninfectious enteropathy, hepatopathy, and malignancy. Hypogammaglobulinemia and low IgA and/or IgM are common findings. A relative increase of CD21low B cells was associated with autoimmune and lymphoproliferative phenotypes. CD4 T-cell defects and a TH1 and proinflammatory cytokine predominance are possible. Decreased numbers of circulating NK cells are frequent (75). An impaired IgG response to polysaccharide antigens was described in 53%–65% of patients with NFKB1 deficiency. NF-κB regulates the expression of activation-induced cytidine deaminase, an enzyme mediating class switch recombination, thereby possibly impairing humoral immunity if mutated (76, 77).
NFKB2 deficiency
In 2013, the first patients with NFKB2 mutations were reported with a phenotype of CVID and adrenocorticotropic hormone (ACTH) insufficiency, a syndrome called DAVID (deficient anterior pituitary with CVID) (78). NFKB2 encodes the precursor p100 and is located at 10q24.32. The reported damaging mutations follow an autosomal dominant pattern. To date, 27 cases of DAVID syndrome and a total of at least 50 cases of CVID with and without DAVID syndrome with confirmed pathological variants in the NFKB2 gene have been published (79). Clinical expressivity and penetrance of NFKB2-related diseases are heterogeneous. Of note, inflammatory, autoimmune, and malignant manifestations often go beyond the common spectrum seen in CVID (79). A key feature, often preceding other symptoms, is autoimmune alopecia. Patients may also present initially with respiratory infections, diarrhea, and arthritis. Over time, about 40% of mutation carriers develop ACTH deficiency, sometimes accompanied by growth hormone and thyroid stimulating hormone deficiencies (80). Recurrent and severe infections with Herpesviridae, candida, and opportunists such as Pneumocysits (P.) jivorecii were reported. Most cases present with hypogammaglobulinemia and reduced marginal zone and switched memory B cells. Total B-cell, T-cell and NK-cell counts are often unimpaired. An impaired IgG response to polysaccharide antigens is present in almost half of the patients. The NFKB2 pathway is indispensable for both B-cell maturation, survival and function and T follicular helper cell generation, explaining humoral immunodeficiency (79).
Activated phosphoinositide 3-kinase δ syndrome (APDS)
APDS is caused by either pathogenic gain-of-function variants in PIK3CD (APDS1) or loss-of-function variants in PIK3R1 (APSD2) (81). First described in 2013, APDS results in hyperreactive mTOR signaling and subsequent immune dysregulation (82). Inheritance is typically autosomal dominant, with onset of symptoms in the second year of life. Patients suffer from sinopulmonary infections resulting in bronchiectasis, B-cell lymphoproliferation (non-malignant or malignant), and autoimmune/inflammatory conditions [e.g., autoimmune hemolytic anemia, enteropathy, and immune thrombocytopenia (ITP)]. They are at increased risk of lymphoma, hemophagocytic lymphohistiocytosis (HLH), enteropathy, and occasionally developmental delay. Typical pathogens causing bacterial infections include S. pneumoniae, H. influenza, Pseudomonas (P.) aeruginosa and S. aureus. Viral infections are commonly caused by Herpesviridae (EBV, CMV, VZV, HSV) or HPV, and fungal infections by C. albicans. The immunological phenotype in APDS is characterized by a diminished antibody response to polysaccharide antigen, elevated IgM, normal IgG and IgA, B-cell lymphopenia, increased transitional B cells, reduced memory B cells, and CD4 lymphopenia, especially naïve CD4+ T cells (83). The CD4/CD8 ratio is decreased (81, 84, 85).
Cytotoxic T lymphocyte antigen 4 (CTLA4) deficiency
Heterozygous pathogenic variants in CTLA4 were initially reported in 2014. They lead to CTLA4 deficiency through haploinsufficiency, inadequate dimerization, or insufficient effector binding (86). The dominant inheritance pattern is accompanied by incomplete penetrance. Since CTLA4 plays a vital role in Tregs function and suppression of T-cell response, its deficiency results in immune dysregulation. Symptom onset typically occurs around 11 years of age, with a highly variable presentation. The immunological phenotype of CTLA4 deficiency primarily exhibits hypogammaglobulinemia, with lack of anti-polysaccharide antibodies, accompanied by significant reductions in T-cell lymphopenia, particularly evident in CD4+ T cells. B-cell lymphopenia with impaired B-cell maturation, and reduced NK cells are also notable features. Clinical manifestations encompass lymphoproliferation, autoimmune conditions (predominantly cytopenia or gastrointestinal involvement), increased susceptibility to infections, particularly of the respiratory tract [viral (EBV, CMV, HSV), bacterial (H. influenzae, S. pneumoniae, Salmonella enteritidis, S. aureus) and fungal (Candida species, Aspergillus species)], as well as gastrointestinal and neurological features. Additionally, patients face an increased risk of malignancies such as lymphoma and gastric cancer, often associated with EBV (87).
Ataxia–telangiectasia (A-T)
Ataxia–telangiectasia (A-T; Louis-Bar Syndrome) was first described in 1941 (88) and further characterized by Boder and Segwick 1958 (89). A-T is caused by mutations in the gene ATM (Ataxia Telangiectasia, Mutated) on human chromosome 11 (11q22.3) (90). The estimated incidence ranges from 1:40,000 to 1:300,000 live births with higher incidences in countries with high rates of consanguinity (91, 92). Several hundred pathogenic and likely pathogenic variants as well as >>1,000 variants of unknown significance (VUS) have been documented in the large gene comprising 60 exons, without any hotspots for mutations (see https://www.LOVD.nl/ATM). ATM protein has a crucial role in repair of DNA double strand breaks caused by radiation, oxidative or other genotoxic stress (90, 93). A-T is inherited in an autosomal recessive order. In non-consanguineous families, most A-T patients are compound heterozygote (94). Clinical features can vary but progressive ataxia noted once children start walking and oculocutaneous telangiectasia (in patients ages 5 years and older) are most prominent findings. Frequent sinopulmonary infections due to immunodeficiency, malignancies (in 1/4 of patients) as well as chronic lung and liver disease, gonadal insufficiency/sterility and premature aging are also reported. Life expectancy is limited mostly due to lung disease and malignancies [for review see (95, 96)]. Approximately 95% of individuals with A-T have marked elevated serum alpha-fetoprotein (AFP) levels (97). Some degree of immunodeficiency is present in approximately 2/3 of A-T patients, with lymphopenia, decreased levels of IgG, IgA, IgM, IgE and/or IgG subclasses and/or an impaired IgG response following vaccinations or infections being observed (98, 99). A-T patients may also may present with an hyper-IgM phenotype (100). A hyper-IgM phenotype or IgA deficiency indicate a poor prognosis (101). Of note, approximately 50% of A-T patients are detected by the newborn screening for severe combined immunodeficiency (SCID) based on low numbers of naïve T cells (102). Immunological impairment in most A-T patients sustains over time, thus frequent reevaluations of immunologic parameters are mostly not indicated (103). Immune dysregulation and autoimmunity in A-T such as immune thrombocytopenia (ITP), arthritis, and vitiligo is also observed [reviewed in (95)]. Chronic granuloma caused by live rubella vaccine virus have been described predominantly in patients with A-T and other DNA repair disorders [reviewed in (104, 105)].
Nijmegen breakage syndrome (NBS)
NBS is a rare autosomal recessive disease that was first described by researchers at the University of Nijmegen (106, 107). NBS is caused by mutations in NBN (mapped to human chromosome 8q21.3) that encodes the protein Nibrin, also termed NBS1 or p95 (108, 109). Nibrin plays an important role in DNA damage response and DNA repair (110). The majority of patients affected by NBS are of Slavic origin due to a founder mutation which leads to protein truncation (109). Prevalence is estimated to approximate 1:100,000 live births (110, 111). The main features of NBS include microcephaly, usually since birth, typical facial appearance with prominent midface, sloping forehead, retrognathia, prominent nasal bridge and nose, large ears and mild growth retardation. Some patients may also have café au lait spots, clinodactyly and syndactyly. Psychomotor development may initially be normal or mildly impaired, but intellectual abilities often decline. Female NBS patients are characterized by primary ovarian failure. Patients may suffer from recurrent respiratory and urinary tract infections, gastroenterocolitis, autoimmune diseases, and have a pronounced predisposition to malignancy due to chromosomal instability and radiation hypersensitivity, with more than 40% of NBS patients developing a malignancy (mainly lymphomas) by the age of 20 years (111–113). Immunological abnormalities found in most patients but are highly variable: T-cell lymphopenia (CD4+ T cells and/or CD8+ T cells) and elevated number of NK cells have been reported. IgG and IgA deficiency can occur, an impaired IgG response to pneumococcal polysaccharide antigens is observed in the majority of patients (75%), often combined with decreased levels of IgG2 and IgG4 subclasses (114). In contrast to A-T, immunodeficiency in NBS patients may progress over time (111, 112). Despite confirmed immunodeficiency, some NBS-patients do not suffer from frequent infections and do not require prophylactic antibiotics or IgRT (111, 112, 114).
DNA ligase 4 (LIG4) syndrome
DNA Ligase IV (LIG4) syndrome, also known as DNA Ligase IV deficiency or Ligase 4 syndrome, is a very rare autosomal recessive disorder that also belongs to the group of hereditary diseases associated with defects in cellular responses to DNA damage. The syndrome results from pathogenic variants in the gene encoding DNA ligase IV (LIG4) that was mapped to chromosome 13q33.3. LIG4 syndrome is caused by homozygous or, more often, compound heterozygous hypomorphic mutations in the LIG4 gene. A genotype–phenotype correlation is suspected for single truncating mutations (115). The presentation of LIG4 syndrome is very heterogenous. Some individuals carrying a deleterious variant were reported to be asymptomatic (116). Common clinical findings are microcephaly, severe growth retardation, developmental delay and dysmorphic facial features, chronic liver disease and malignancy predisposition due to pronounced radiosensitivity (117, 118). Patients with hip dysplasia and other skeletal malformations were also reported. Immunologic findings comprise low IgG, variable immunodeficiency, and pancytopenia. Manifestation early in life has been reported for some patients, who presented with the above-mentioned clinical complex as well as severe combined immunodeficiency, radiosensitivity, chronic liver disease, and progressive bone marrow failure. Immunological findings include hypogammaglobulinemia and very low B cells with primarily IgG deficiencies causing sinopulmonary infections. Hypogammaglobulinemia can be accompanied by a decrease in IgA and IgM levels as well as T-cell abnormalities (117, 118). Patients with distal truncating pathogenic variants typically have a milder phenotype and may present later in life with hematological neoplasia with poor response to chemo- and radiotherapy (116, 117). Live expectancy is limited due to hematologic neoplasia, bone marrow failure, and chronic liver disease (117, 118).
Autosomal dominant hyper IgE syndrome (HIES) caused by dominant negative mutations in STAT3
First described as “Job's Syndrome”, the term “Hyper-IgE Syndrome” (HIES) was later introduced because of high IgE-levels (119, 120). Several genetic variants associated with Hyper-IgE have been identified. Autosomal dominant HIES (AD-HIES) is characterized by (1) pulmonary infections, mainly due to S. aureus and S. pneumoniae, (2) bone brittleness and retention of decidual and delayed dentition of permanent teeth, (3) severe eczematous dermatitis and skin infections with S. aureus and Candida spp. (121). The identification of heterozygous, almost exclusively dominant-negative mutations in the gene that encodes the Signal transducer and activator of transcription 3 (STAT3), as genetic cause for AD-HIES explains the syndromic nature of HIES, affecting not only the immune system, but multiple further tissues (122). The reported prevalence among populations varies between 1 and 9: 100,000. (https://www.orpha.net/).
As in atopic dermatitis, the predominant immunological feature of AD-HIES is marked hypereosinophilia and elevated IgE (121, 123). It was shown early and repeatedly thereafter that IgG-responses against pneumococci and levels of IgG subclasses may be low, too (124–127). Thus, in patients with severe eczema normal levels of IgG subclasses and normal responses against pneumococci do not exclude AD-HIES, but low levels of IgG subclasses and/or impaired responses against polysaccharides are compatible with AD-HIES. Low IgG subclasses and/ or impaired humoral responses against pneumococci are probably caused by combined effects of less follicular helper T cells (producers of IL-21), impaired IL-6 signalling (and consequently low IL-21 production) and impaired responses to IL-21, that finally lead to less class-switched memory B cells in many patients with HIES caused by dominant negative mutations in STAT3 (128–134). IgG2 subclass deficiency was described very early in AD-STAT3-HIES (125) and may contribute to the increased susceptibility to pneumococcal infections.
Other rare IEI with an impaired IgG response against polysaccharide antigens
Other rare IEI may present with an impaired IgG response to polysaccharide antigens as a predominant finding (135), e.g., Syk (136) and MAGT1-deficiency (137); but larger case series or cohort studies are lacking.
Discussion
Severe or recurrent infections with encapsulated bacteria (e.g., S. pneumoniae, H. influenzae, N. meningitis species) should prompt clinicians to evaluate patients for IEI, in particular when affected patients had been vaccinated against respective bacterial antigens. Apart from antibody deficiency, differential diagnostic considerations should include complement deficiency, asplenia or splenic impairment as in sickle cell disease, and defects in innate immunity such as Toll-like/IL1R defects e.g., IRAK4/MyD88 deficiency (12, 13). The majority of patients with recurrent or severe infections due to antibody deficiency (e.g., CVID, XLA) will be detected with the basic immunologic tests recommended by German AWMF guidelines (3). However, IgG, IgA, IgM, and even IgG responses to protein-based vaccines may (initially) be normal in patients with monogenic IEI with an impaired response to polysaccharide antigens. Thus, we would like to challenge current German guidelines because these IEI may be missed or the diagnosis delayed. An impaired response to polysaccharide antigens can be diagnosed once polysaccharide-specific antibodies do not increase following polysaccharide-based vaccinations or wild-type infections. In wild-type infections with S. pneumoniae protein as well as polysaccharide-specific antibodies to respective strains should be detectable in high/normal concentrations. Thus, low or absent pneumococcal specific IgG after microbiology-proven pneumococcal infection should prompt for further immunological and genetic workup. Commercially available tests for polysaccharide-specific total and IgG2 antibodies are mostly based on pneumococcal antigens. Patients with recurrent or severe bacterial infections and low specific pneumococcal antibodies should receive a “diagnostic” polysaccharide-based vaccination (e.g., Pneumomvax™) followed by measurement of the IgG vaccine response ≥4 weeks. Unfortunately, various serological and opsonophagocytic assays exist with limited comparability of the results. An international consensus for a standardized approach would be highly desirable. Determination of IgG subclasses may also be helpful, since polysaccharide-specific IgG antibodies are found within the IgG2 subclass fraction, thus IgG2 levels are often below age dependent normal ranges in patients with an impaired response to polysaccharide antigens (138). Of note, children below the age of 2 years respond poorly to polysaccharide antigens, therefore (diagnostic) vaccination with Pneumovax™ is not approved in children <2 years of age. Also, IgG2 and IgG4 levels are frequently low in infancy and are likely increase to normal levels within the first two years of live (138, 139). Diagnosing an impaired IgG response to polysaccharide antigens is therefore more challenging in children younger than 2 years than in older children and adults.
In this review, we listed respective IEI to be considered once an impaired IgG response to polysaccharide antigens has been diagnosed. The clinical characteristics shown in Table 2 imply that
(a) syndromic aspects as in DGS, MECP2-Duplication syndrome, A-T, NBS and Lig4-Deficiency,
(b) male gender (XLA, WAS, MECP2 Duplication syndrome),
(c) a positive family history for infections or autoimmunity, and
(d) consanguinity
in the context of recurrent infections should alert physicians to perform immunologic (and genetic) testing beyond basic evaluations for IEI. In most listed IEI, phenotypic presentations can vary significantly within a pedigree, thus a selectively impaired response to polysaccharide antigens may be present in relatives from patients with more pronounced immunologic findings.
Genetic testing has become more available and less expensive over the last years. Therefore, Whole Exome or Whole Genome Sequencing (WES, WGS) has become a standard diagnostic procedure when IEI is suspected based on clinical and/or immunological findings in high income countries (140). We assume that a growing number of patients with an impaired IgG response to polysaccharide antigens or strictly defined specific antibody deficiency (SAD, see below) will show pathogenic variants in one of the listed genes. Family members of patients with respective IEI should be offered genetic testing to allow close follow-up and early therapeutic interventions when pathogenic variants are identified. Genetic diagnosis of IEI in young children also permits genetic counseling of parents with a desire to have more children.
Delayed diagnosis of IEI with antibody deficiency is common (141), often patients show permanent organ damage due to recurrent sinopulmonary infections when diagnosed: hearing impairment following recurrent otitis, sinus surgery in patients with chronic or recurrent rhinosinusitis, bronchiectasis/chronic lung disease due to recurrent pneumonia (142).
Early diagnosis and treatment of IEI with an impaired IgG response to polysaccharide antigens listed in this review cannot only prevent further infections and organ damage, but can also prevent specific short and long term complications and risks in specific IEI [i.e., bleeding in WAS, risk of malignancy due to exposure to ionizing radiation as well as risk of rubella-induced granuloma following live-rubella vaccination in IEI with DNA repair disorders (A-T, NBS, LIG4 deficiency)] (104, 143).
Treatment of patients with an impaired IgG response to polysaccharide antigens depends on clinical symptoms, however, patients with recurrent bacterial infections will benefit from antibiotic treatment/prophylaxis and/or IgRT. Of note, in patients with normal IgG levels and an impaired IgG response to polysaccharide antigens, IgG dosing and intervals should not be guided by through levels of total IgG but by (a) the clinical response and (b) pneumococcal specific antibodies and/or IgG2 levels (if decreased prior IgRT). In patients with a normal IgG response to protein-based vaccines (tetanus toxoid), non-live protein-based vaccinations are advisable irrespective of IgRT when protective antibody titers cannot be guaranteed in IgG preparations (in particular: Meningococci A, B, C, W, Y). The add-on effect of such vaccinations, however, has not been scientifically evaluated.
Inhalant antibiotics may be an option in individual patients with recurrent pneumonia and bronchiectasis despite IgRT and systemic antibiotic prophylaxis. It has been successfully applied in a patient with MECP2-Duplication syndrome (144) as well as in CVID patients (145). However, there are no controlled trials on inhalant antibiotics in pediatric IEI patients.
HSCT has become an option for a growing number of patients with IEI, the outcome is generally better when patients are transplanted before chronic infections occur, thus, patients with a clear indication for a curative HSCT (e.g., patients with WAS) will benefit from early diagnosis (146). HSCT has been successfully performed in selected cases with DNA repair disorders, mainly because of malignancies (147, 148). Early diagnosis of a DNA repair disorder prior to HSCT enables adjustment of conditioning regimen and minimization of radiation exposure.
More specific treatment options in order to prevent disease progression, organ damage and/or to improve quality of life that are available for distinct IEI mentioned in the article: Recent clinical observations show impressive improvement of dermatitis and almost complete control of skin infections in patients with HIES on Dupilumab [subcutaneously (s.c.)]—an antibody that blocks the IL4-receptor alpha chain (149–155). Abatacept (s.c.), a CTLA4-immunoglobulin fusion drug, has shown excellent treatment results with a good tolerability and safety profile in patients with CTLA4 and LRBA deficiency (156, 157). This has also been shown for the orally available Leniolisib, a small molecule selective inhibitor of PI3Kδ, in patients with APDS (81, 84, 85). In A-T, antisense oligonucleotide treatment has shown amelioration of neurological disease progression in patients with selected ATM mutations (158, 159). This therapy has to be initiated at an early disease state, underscoring the importance of timely diagnosis. Gene therapy may become an option for some of the listed IEI in the future (160, 161).
In this article, we purposely discussed patients that do not fulfill the strict criteria for SAD with a diminished antibody response to polysaccharide antigens following vaccination or wild-type infection as the only immunologic pathology (normal levels of IgG, IgA, IgM and IgG subclasses) (12, 13, 20, 22) for the following reasons: SAD is not a genetically distinct IEI. Indeed, genetic testing of patients with SAD may reveal mutations in various IEI genes described in this article. Also, IgG subclasses are not routinely tested in all countries, it is therefore unclear if patients with unknown IgG subclass levels do have strictly defined SAD. Furthermore, all patients with an impaired IgG response to polysaccharide antigens and frequent or severe bacterial infections will benefit from therapy, irrespective of additional immunologic abnormalities.
Lastly, this review underscores the importance of microbiology testing in patients with recurrent bacterial infections prior to antibiotic therapy. Once encapsulated bacteria are repeatedly detected, immunodeficiency with an impaired response to polysaccharide antigens may be suspected and diagnosed. Microbiology testing also helps to avoid non-effective antibiotic regimens due to resistances in patients that had multiple courses of antibiotics and select an effective prophylactic regimen if required.
Apart from monogenic IES listed in this manuscript, testing for polysaccharide specific antibodies is also helpful in patients with CVID and unclassified antibody deficiencies without an underlying genetic defect identified. Absent or low specific antibodies with no increase following a polysaccharide based vaccination should prompt clinicians to consider antibiotic prophylaxis or IgRT.
Conclusions
In patients with suspected IEI due to severe or recurrent bacterial infections who have normal IgG levels and a normal IgG response to protein-based vaccines, testing for polysaccharide-specific antibodies and IgG subclasses can help to identify rare monogenic IEI early, initiate treatment earlier and, thus, prevent morbidity, mortality, and organ damage. In our opinion, new guidelines on diagnostic measures in IEI should include the determination of specific IgG antibodies and IgG subclasses.
Author contributions
MF: Writing – original draft, Writing – review & editing. SD: Writing – original draft, Writing – review & editing. OS: Writing – original draft, Writing – review & editing. MV: Writing – original draft, Writing – review & editing. AS: Writing – original draft, Writing – review & editing. HvB: Writing – original draft, Writing – review & editing. VW: Conceptualization, Writing – original draft, Writing – review & editing. RK: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article.
We acknowledge financial support from the Open Access Publication Fund of Charité - Universitätsmedizin Berlin.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ACTH, adrenocorticotropic hormone; AFP, alpha-fetoprotein; APDS, activated phosphoinositide 3-kinase δ syndrome; A-T, ataxia telangiectasia; BTK, Bruton's tyrosine kinase; CVID, common variable immunodeficiency; DAVID, deficit in anterior pituitary function and variable immune deficiency; DGS, DiGeorge syndrome; ED, ectodermal dysplasia; HLH, hemophagocytic lymphohistiocytosis; HSCT, hematopoietic stem cell transplantation; ID, immunodeficiency; IEI, Inborn Errors of Immunity; IgRT, immunoglobulin replacement therapy; ITP, immune thrombocytopenia; LIG4, DNA Ligase 4; LPS, lipopolysaccharides; NEMO, nuclear factor-kappa B essential modulator; NBS, Nijmegen Breakage syndrome; NGS, next generation sequencing; NFKB, nuclear factor KappaB; pDGS, partial DiGeorge syndrome; PHA, phytohemagglutinin; PS, polysaccharide; SAD, specific antibody deficiency; SCID, severe combined immunodeficiency; VUS, variant of unknown significance; WAS, Wiskott-Aldrich syndrome; XLA, X-linked agammaglobulinemia.
References
1. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol. (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
2. Pieniawska-Śmiech K, Pasternak G, Lewandowicz-Uszyńska A, Jutel M. Diagnostic challenges in patients with inborn errors of immunity with different manifestations of immune dysregulation. J Clin Med. (2022) 11(14):4220. doi: 10.3390/jcm11144220
3. Farmand S, Baumann U, von Bernuth H, Borte M, Foerster-Waldl E, Franke K, et al. Interdisciplinary AWMF guideline for the diagnostics of primary immunodeficiency. Klin Padiatr. (2011) 223(6):378–85. doi: 10.1055/s-0031-1287835
4. Knight V, Heimall JR, Chong H, Nandiwada SL, Chen K, Lawrence MG, et al. A toolkit and framework for optimal laboratory evaluation of individuals with suspected primary immunodeficiency. J Allergy Clin Immunol Pract. (2021) 9(9):3293–307.e6. doi: 10.1016/j.jaip.2021.05.004
5. Umetsu DT, Ambrosino DM, Quinti I, Siber GR, Geha RS. Recurrent sinopulmonary infection and impaired antibody response to bacterial capsular polysaccharide antigen in children with selective IgG-subclass deficiency. N Engl J Med. (1985) 313(20):1247–51. doi: 10.1056/NEJM198511143132002
6. Epstein MM, Gruskay F. Selective deficiency in pneumococcal antibody response in children with recurrent infections. Ann Allergy Asthma Immunol. (1995) 75(2):125–31. PMID: 7648376
7. Lopez B, Boucher A, Bahuaud M, Mortuaire G, Melliez H, Launay D, et al. Specific polysaccharide antibody deficiency revealed by severe bacterial infections in adulthood: a report on 11 cases. Clin Infect Dis. (2017) 65(2):328–31. doi: 10.1093/cid/cix284
8. Carr TF, Koterba AP, Chandra R, Grammer LC, Conley DB, Harris KE, et al. Characterization of specific antibody deficiency in adults with medically refractory chronic rhinosinusitis. Am J Rhinol Allergy. (2011) 25(4):241–4. doi: 10.2500/ajra.2011.25.3653
9. French MA, Denis KA, Dawkins R, Peter JB. Severity of infections in IgA deficiency: correlation with decreased serum antibodies to pneumococcal polysaccharides and decreased serum IgG2 and/or IgG4. Clin Exp Immunol. (1995) 100(1):47–53. doi: 10.1111/j.1365-2249.1995.tb03602.x
10. Ruuskanen O, Nurkka A, Helminen M, Viljanen MK, Kayhty H, Kainulainen L. Specific antibody deficiency in children with recurrent respiratory infections: a controlled study with follow-up. Clin Exp Immunol. (2013) 172(2):238–44. doi: 10.1111/cei.12053
11. Orange JS, Ballow M, Stiehm ER, Ballas ZK, Chinen J, De La Morena M, et al. Use and interpretation of diagnostic vaccination in primary immunodeficiency: a working group report of the basic and clinical immunology interest section of the American academy of allergy, asthma & immunology. J Allergy Clin Immunol. (2012) 130(3 Suppl):S1–24. doi: 10.1016/j.jaci.2012.07.002
12. Perez E, Bonilla FA, Orange JS, Ballow M. Specific antibody deficiency: controversies in diagnosis and management. Front Immunol. (2017) 8:586. doi: 10.3389/fimmu.2017.00586
13. Perez EE, Ballow M. Diagnosis and management of specific antibody deficiency. Immunol Allergy Clin North Am. (2020) 40(3):499–510. doi: 10.1016/j.iac.2020.03.005
14. Cavaliere FM, Milito C, Martini H, Schlesier M, Dräger R, Schütz K, et al. Quantification of IgM and IgA anti-pneumococcal capsular polysaccharides by a new ELISA assay: a valuable diagnostic and prognostic tool for common variable immunodeficiency. J Clin Immunol. (2013) 33(4):838–46. doi: 10.1007/s10875-012-9856-z
15. Pulvirenti F, Milito C, Cavaliere FM, Mezzaroma I, Cinetto F, Quinti I. IGA antibody induced by immunization with pneumococcal polysaccharides is a prognostic tool in common variable immune deficiencies. Front Immunol. (2020) 11:1283. doi: 10.3389/fimmu.2020.01283
16. Janssen LMA, Heron M, Murk JL, Leenders A, Rijkers GT, de Vries E. The clinical relevance of IgM and IgA anti-pneumococcal polysaccharide ELISA assays in patients with suspected antibody deficiency. Clin Exp Immunol. (2021) 205(2):213–21. doi: 10.1111/cei.13605
17. Pollard AJ, Perrett KP, Beverley PC. Maintaining protection against invasive bacteria with protein-polysaccharide conjugate vaccines. Nat Rev Immunol. (2009) 9(3):213–20. doi: 10.1038/nri2494
18. Gingerich AD, Mousa JJ. Diverse mechanisms of protective anti-pneumococcal antibodies. Front Cell Infect Microbiol. (2022) 12:824788. doi: 10.3389/fcimb.2022.824788
19. Bernth-Jensen JM, Sogaard OS. Polysaccharide responsiveness is not biased by prior pneumococcal-conjugate vaccination. PLoS One. (2013) 8(10):e75944. doi: 10.1371/journal.pone.0075944
20. Sorensen RU. A critical view of specific antibody deficiencies. Front Immunol. (2019) 10:986. doi: 10.3389/fimmu.2019.00986
21. Rojas-Restrepo J, Caballero-Oteyza A, Huebscher K, Haberstroh H, Fliegauf M, Keller B, et al. Establishing the molecular diagnoses in a cohort of 291 patients with predominantly antibody deficiency by targeted next-generation sequencing: experience from a monocentric study. Front Immunol. (2021) 12:786516. doi: 10.3389/fimmu.2021.786516
22. Lawrence MG, Borish L. Specific antibody deficiency: pearls and pitfalls for diagnosis. Ann Allergy Asthma Immunol. (2022) 129(5):572–8. doi: 10.1016/j.anai.2022.05.028
24. Valiaho J, Smith CI, Vihinen M. BTKbase: the mutation database for X-linked agammaglobulinemia. Hum Mutat. (2006) 27(12):1209–17. doi: 10.1002/humu.20410
25. Farrar JE, Rohrer J, Conley ME. Neutropenia in X-linked agammaglobulinemia. Clin Immunol Immunopathol. (1996) 81(3):271–6. doi: 10.1006/clin.1996.0188
26. Kanegane H, Taneichi H, Nomura K, Futatani T, Miyawaki T. Severe neutropenia in Japanese patients with x-linked agammaglobulinemia. J Clin Immunol. (2005) 25(5):491–5. doi: 10.1007/s10875-005-5370-x
27. Lanlokun M, Borden A, Nieves D, Walter JE, Albright D. X-Linked agammaglobulinemia presenting as neutropenia: case report and an overview of literature. Front Pediatr. (2021) 9:633692. doi: 10.3389/fped.2021.633692
28. Inoue K, Sasaki S, Yasumi T, Imai K, Kusunoki T, Morio T, et al. Helicobacter cinaedi-associated refractory cellulitis in patients with X-linked agammaglobulinemia. J Clin Immunol. (2020) 40(8):1132–7. doi: 10.1007/s10875-020-00830-6
29. Demirdag YY, Gupta S. Update on infections in primary antibody deficiencies. Front Immunol. (2021) 12:634181. doi: 10.3389/fimmu.2021.634181
30. Bearden D, Collett M, Quan PL, Costa-Carvalho BT, Sullivan KE. Enteroviruses in X-linked agammaglobulinemia: update on epidemiology and therapy. J Allergy Clin Immunol Pract. (2016) 4(6):1059–65. doi: 10.1016/j.jaip.2015.12.015
31. Lougaris V, Soresina A, Baronio M, Montin D, Martino S, Signa S, et al. Long-term follow-up of 168 patients with X-linked agammaglobulinemia reveals increased morbidity and mortality. J Allergy Clin Immunol. (2020) 146(2):429–37. doi: 10.1016/j.jaci.2020.03.001
32. Qing-Qi R, Ya-Wen L, Huan C, Yu Z, Yun-Fei A, Xue-Mei T, et al. Retrospective study of 98 patients with X-linked agammaglobulinemia complicated with arthritis. Clin Rheumatol. (2022) 41(6):1889–97. doi: 10.1007/s10067-022-06095-1
33. Wood PM, Mayne A, Joyce H, Smith CI, Granoff DM, Kumararatne DS. A mutation in Bruton’s tyrosine kinase as a cause of selective anti-polysaccharide antibody deficiency. J Pediatr. (2001) 139(1):148–51. doi: 10.1067/mpd.2001.115970
34. Kruger R, Baumann U, Borte S, Kolsch U, Lorenz MR, Keller B, et al. Impaired polysaccharide responsiveness without agammaglobulinaemia in three patients with hypomorphic mutations in Bruton tyrosine kinase-No detection by newborn screening for primary immunodeficiencies. Scand J Immunol. (2020) 91(1):e12811. doi: 10.1111/sji.12811
36. Aldrich RA, Steinberg AG, Campbell DC. Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea. Pediatrics. (1954) 13(2):133–9. doi: 10.1542/peds.13.2.133
37. Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. (1994) 78(4):635–44. doi: 10.1016/0092-8674(94)90528-2
38. Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. (2007) 8(1):37–48. doi: 10.1038/nrm2069
39. Notarangelo LD, Miao CH, Ochs HD. Wiskott-Aldrich syndrome. Curr Opin Hematol. (2008) 15(1):30–6. doi: 10.1097/MOH.0b013e3282f30448
40. Perry GS 3rd, Spector BD, Schuman LM, Mandel JS, Anderson VE, McHugh RB, et al. The Wiskott-Aldrich syndrome in the United States and Canada (1892–1979). J Pediatr. (1980) 97(1):72–8. doi: 10.1016/S0022-3476(80)80133-8
41. Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr. (1994) 125(6 Pt 1):876–85. doi: 10.1016/S0022-3476(05)82002-5
42. Litzman J, Jones A, Hann I, Chapel H, Strobel S, Morgan G. Intravenous immunoglobulin, splenectomy, and antibiotic prophylaxis in Wiskott-Aldrich syndrome. Arch Dis Child. (1996) 75(5):436–9. doi: 10.1136/adc.75.5.436
43. Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. (2006) 117(4):725–38. quiz 39. doi: 10.1016/j.jaci.2006.02.005
44. Jin Y, Mazza C, Christie JR, Giliani S, Fiorini M, Mella P, et al. Mutations of the Wiskott-Aldrich syndrome protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood. (2004) 104(13):4010–9. doi: 10.1182/blood-2003-05-1592
45. Ochs HD, Slichter SJ, Harker LA, Von Behrens WE, Clark RA, Wedgwood RJ. The Wiskott-Aldrich syndrome: studies of lymphocytes, granulocytes, and platelets. Blood. (1980) 55(2):243–52. doi: 10.1182/blood.V55.2.243.243
46. Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am J Hum Genet. (2000) 67(6):1555–62. doi: 10.1086/316914
47. Döffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. (2001) 27(3):277–85. doi: 10.1038/85837
48. Jain A, Ma CA, Liu S, Brown M, Cohen J, Strober W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol. (2001) 2(3):223–8. doi: 10.1038/85277
49. Orange JS, Jain A, Ballas ZK, Schneider LC, Geha RS, Bonilla FA. The presentation and natural history of immunodeficiency caused by nuclear factor kappaB essential modulator mutation. J Allergy Clin Immunol. (2004) 113(4):725–33. doi: 10.1016/j.jaci.2004.01.762
50. Hanson EP, Monaco-Shawver L, Solt LA, Madge LA, Banerjee PP, May MJ, et al. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol. (2008) 122(6):1169–77.e16. doi: 10.1016/j.jaci.2008.08.018
51. Salt BH, Niemela JE, Pandey R, Hanson EP, Deering RP, Quinones R, et al. IKBKG (nuclear factor-kappa B essential modulator) mutation can be associated with opportunistic infection without impairing toll-like receptor function. J Allergy Clin Immunol. (2008) 121(4):976–82. doi: 10.1016/j.jaci.2007.11.014
52. Orange JS, Brodeur SR, Jain A, Bonilla FA, Schneider LC, Kretschmer R, et al. Deficient natural killer cell cytotoxicity in patients with IKK-gamma/NEMO mutations. J Clin Invest. (2002) 109(11):1501–9. doi: 10.1172/JCI0214858
53. Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clin Microbiol Rev. (2011) 24(3):490–7. doi: 10.1128/CMR.00001-11
54. Heller S, Kolsch U, Magg T, Kruger R, Scheuern A, Schneider H, et al. T cell impairment is predictive for a severe clinical course in NEMO deficiency. J Clin Immunol. (2020) 40(3):421–34. doi: 10.1007/s10875-019-00728-y
55. DiGeorge A. Discussion on a new concept of the cellular immunology. J Pediatr. (1965) 67:907–8. doi: 10.1016/S0022-3476(65)82145-X
56. Giardino G, Radwan N, Koletsi P, Morrogh DM, Adams S, Ip W, et al. Clinical and immunological features in a cohort of patients with partial DiGeorge syndrome followed at a single center. Blood. (2019) 133(24):2586–96. doi: 10.1182/blood.2018885244
57. McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine (Baltimore). (2011) 90(1):1–18. doi: 10.1097/MD.0b013e3182060469
58. Maggadottir SM, Sullivan KE. The diverse clinical features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome). J Allergy Clin Immunol Pract. (2013) 1(6):589–94. doi: 10.1016/j.jaip.2013.08.003
59. Schubert MS, Moss RB. Selective polysaccharide antibody deficiency in familial DiGeorge syndrome. Ann Allergy. (1992) 69(3):231–8. PMID: 1524280
60. Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. (2005) 77(3):442–53. doi: 10.1086/444549
61. Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, et al. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of rett syndrome. J Med Genet. (2005) 42(2):e12. doi: 10.1136/jmg.2004.023804
62. Gottschalk I, Kolsch U, Wagner DL, Kath J, Martini S, Kruger R, et al. IRAK1 Duplication in MECP2 duplication syndrome does not increase canonical NF-kappaB-induced inflammation. J Clin Immunol. (2023) 43(2):421–39. doi: 10.1007/s10875-022-01390-7
63. Friez MJ, Jones JR, Clarkson K, Lubs H, Abuelo D, Bier JA, et al. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. (2006) 118(6):e1687–95. doi: 10.1542/peds.2006-0395
64. Kirk EP, Malaty-Brevaud V, Martini N, Lacoste C, Levy N, Maclean K, et al. The clinical variability of the MECP2 duplication syndrome: description of two families with duplications excluding L1CAM and FLNA. Clin Genet. (2009) 75(3):301–3. doi: 10.1111/j.1399-0004.2008.01102.x
65. Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A. (2010) 152A(5):1079–88. doi: 10.1002/ajmg.a.33184
66. Budisteanu M, Papuc SM, Tutulan-Cunita A, Budisteanu B, Arghir A. Novel clinical finding in MECP2 duplication syndrome. Eur Child Adolesc Psychiatry. (2011) 20(7):373–5. doi: 10.1007/s00787-011-0184-2
67. Bauer M, Kolsch U, Kruger R, Unterwalder N, Hameister K, Kaiser FM, et al. Infectious and immunologic phenotype of MECP2 duplication syndrome. J Clin Immunol. (2015) 35(2):168–81. doi: 10.1007/s10875-015-0129-5
68. Lim Z, Downs J, Wong K, Ellaway C, Leonard H. Expanding the clinical picture of the MECP2 duplication syndrome. Clin Genet. (2017) 91(4):557–63. doi: 10.1111/cge.12814
69. Giudice-Nairn P, Downs J, Wong K, Wilson D, Ta D, Gattas M, et al. The incidence, prevalence and clinical features of MECP2 duplication syndrome in Australian children. J Paediatr Child Health. (2019) 55(11):1315–22. doi: 10.1111/jpc.14399
70. Sugitate R, Muramatsu K, Ogata T, Goto M, Hayashi S, Sawaura N, et al. Recurrent pneumonia in three patients with MECP2 duplication syndrome with aspiration as the possible cause. Brain Dev. (2022) 44(7):486–91. doi: 10.1016/j.braindev.2022.03.005
71. Ta D, Downs J, Baynam G, Wilson A, Richmond P, Leonard H. Medical comorbidities in MECP2 duplication syndrome: results from the international MECP2 duplication database. Children (Basel). (2022) 9(5):633. doi: 10.3390/children9050633
72. Fliegauf M, Bryant VL, Frede N, Slade C, Woon ST, Lehnert K, et al. Haploinsufficiency of the NF-κB1 subunit p50 in common variable immunodeficiency. Am J Hum Genet. (2015) 97(3):389–403. doi: 10.1016/j.ajhg.2015.07.008
73. Li J, Lei WT, Zhang P, Rapaport F, Seeleuthner Y, Lyu B, et al. Biochemically deleterious human NFKB1 variants underlie an autosomal dominant form of common variable immunodeficiency. J Exp Med. (2021) 218(11):e20210566. doi: 10.1084/jem.20210566
74. Mandola AB, Sharfe N, Nagdi Z, Dadi H, Vong L, Merico D, et al. Combined immunodeficiency caused by a novel homozygous NFKB1 mutation. J Allergy Clin Immunol. (2021) 147(2):727–33.e2. doi: 10.1016/j.jaci.2020.08.040
75. Lorenzini T, Fliegauf M, Klammer N, Frede N, Proietti M, Bulashevska A, et al. Characterization of the clinical and immunologic phenotype and management of 157 individuals with 56 distinct heterozygous NFKB1 mutations. J Allergy Clin Immunol. (2020) 146(4):901–11. doi: 10.1016/j.jaci.2019.11.051
76. Schipp C, Nabhani S, Bienemann K, Simanovsky N, Kfir-Erenfeld S, Assayag-Asherie N, et al. Specific antibody deficiency and autoinflammatory disease extend the clinical and immunological spectrum of heterozygous NFKB1 loss-of-function mutations in humans. Haematologica. (2016) 101(10):e392–e6. doi: 10.3324/haematol.2016.145136
77. Tuovinen EA, Kuismin O, Soikkonen L, Martelius T, Kaustio M, Hamalainen S, et al. Long-term follow up of families with pathogenic NFKB1 variants reveals incomplete penetrance and frequent inflammatory sequelae. Clin Immunol. (2023) 246:109181. doi: 10.1016/j.clim.2022.109181
78. Quentien MH, Delemer B, Papadimitriou DT, Souchon PF, Jaussaud R, Pagnier A, et al. Deficit in anterior pituitary function and variable immune deficiency (DAVID) in children presenting with adrenocorticotropin deficiency and severe infections. J Clin Endocrinol Metab. (2012) 97(1):E121–8. doi: 10.1210/jc.2011-0407
79. Klemann C, Camacho-Ordonez N, Yang L, Eskandarian Z, Rojas-Restrepo JL, Frede N, et al. Clinical and immunological phenotype of patients with primary immunodeficiency due to damaging mutations in NFKB2. Front Immunol. (2019) 10:297. doi: 10.3389/fimmu.2019.00297
80. Mac TT, Castinetti F, Bar C, Julia S, Pasquet M, Romanet P, et al. Deficient anterior pituitary with common variable immune deficiency (DAVID syndrome): a new case and literature reports. J Neuroendocrinol. (2023) 35(6):e13287. doi: 10.1111/jne.13287
81. Rao VK, Webster S, Sediva A, Plebani A, Schuetz C, Shcherbina A, et al. A randomized, placebo-controlled phase 3 trial of the PI3Kdelta inhibitor leniolisib for activated PI3Kdelta syndrome. Blood. (2023) 141(9):971–83. doi: 10.1182/blood.2022018546
82. Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. (2013) 342(6160):866–71. doi: 10.1126/science.1243292
83. Jamee M, Moniri S, Zaki-Dizaji M, Olbrich P, Yazdani R, Jadidi-Niaragh F, et al. Clinical, immunological, and genetic features in patients with activated PI3Kdelta syndrome (APDS): a systematic review. Clin Rev Allergy Immunol. (2020) 59(3):323–33. doi: 10.1007/s12016-019-08738-9
84. Cant AJ, Chandra A, Munro E, Rao VK, Lucas CL. PI3Kdelta Pathway dysregulation and unique features of its inhibition by leniolisib in activated PI3Kdelta syndrome and beyond. J Allergy Clin Immunol Pract. (2024) 12(1):69–78. doi: 10.1016/j.jaip.2023.09.016
85. Rao VK, Kulm E, Sediva A, Plebani A, Schuetz C, Shcherbina A, et al. Interim analysis: open-label extension study of leniolisib for patients with APDS. J Allergy Clin Immunol. (2024) 153(1):265–74.e9. doi: 10.1016/j.jaci.2023.09.032
86. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. (2014) 345(6204):1623–7. doi: 10.1126/science.1255904
87. Hoshino A, Toyofuku E, Mitsuiki N, Yamashita M, Okamoto K, Yamamoto M, et al. Clinical courses of IKAROS and CTLA4 deficiencies: a systematic literature review and retrospective longitudinal study. Front Immunol. (2021) 12:784901. doi: 10.3389/fimmu.2021.784901
88. Louis-Bar D. Sur un syndrome progressif comprenant des télangiectasies capillaires cutanées et conjonctivales symétriques, à disposition naevoïde et des troubles cérébelleux. Confin Neurol. (1941) 4:32–42. doi: 10.1159/000106149
89. Boder E, Sedgwick RP. Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics. (1958) 21(4):526–54. doi: 10.1542/peds.21.4.526
90. Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. (1995) 268(5218):1749–53. doi: 10.1126/science.7792600
91. Swift M, Morrell D, Cromartie E, Chamberlin AR, Skolnick MH, Bishop DT. The incidence and gene frequency of ataxia-telangiectasia in the United States. Am J Hum Genet. (1986) 39(5):573–83. PMID: 3788973; PMCID: PMC1684065.3788973
92. Perlman SL, Boder Deceased E, Sedgewick RP, Gatti RA. Ataxia-telangiectasia. Handb Clin Neurol. (2012) 103:307–32. doi: 10.1016/B978-0-444-51892-7.00019-X
93. Gatti RA, Berkel I, Boder E, Braedt G, Charmley P, Concannon P, et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature. (1988) 336(6199):577–80. doi: 10.1038/336577a0
94. Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N Engl J Med. (2012) 366(7):636–46. doi: 10.1056/NEJMra1006610
95. Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: a review. Orphanet J Rare Dis. (2016) 11(1):159. doi: 10.1186/s13023-016-0543-7
96. McGrath-Morrow SA, Rothblum-Oviatt CC, Wright J, Schlechter H, Lefton-Greif MA, Natale VA, et al. Multidisciplinary management of ataxia telangiectasia: current perspectives. J Multidiscip Healthc. (2021) 14:1637–44. doi: 10.2147/JMDH.S295486
97. Stray-Pedersen A, Borresen-Dale AL, Paus E, Lindman CR, Burgers T, Abrahamsen TG. Alpha fetoprotein is increasing with age in ataxia-telangiectasia. Eur J Paediatr Neurol. (2007) 11(6):375–80. doi: 10.1016/j.ejpn.2007.04.001
98. Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr. (2004) 144(4):505–11. doi: 10.1016/j.jpeds.2003.12.046
99. Chopra C, Davies G, Taylor M, Anderson M, Bainbridge S, Tighe P, et al. Immune deficiency in ataxia-telangiectasia: a longitudinal study of 44 patients. Clin Exp Immunol. (2014) 176(2):275–82. doi: 10.1111/cei.12262
100. Noordzij JG, Wulffraat NM, Haraldsson A, Meyts I, van't Veer LJ, Hogervorst FB, et al. Ataxia-telangiectasia patients presenting with hyper-IgM syndrome. Arch Dis Child. (2009) 94(6):448–9. doi: 10.1136/adc.2008.149351
101. Zielen S, Duecker RP, Woelke S, Donath H, Bakhtiar S, Buecker A, et al. Simple measurement of IgA predicts immunity and mortality in ataxia-telangiectasia. J Clin Immunol. (2021) 41(8):1878–92. doi: 10.1007/s10875-021-01090-8
102. Mallott J, Kwan A, Church J, Gonzalez-Espinosa D, Lorey F, Tang LF, et al. Newborn screening for SCID identifies patients with ataxia telangiectasia. J Clin Immunol. (2013) 33(3):540–9. doi: 10.1007/s10875-012-9846-1
103. Driessen GJ, Ijspeert H, Weemaes CM, Haraldsson A, Trip M, Warris A, et al. Antibody deficiency in patients with ataxia telangiectasia is caused by disturbed B- and T-cell homeostasis and reduced immune repertoire diversity. J Allergy Clin Immunol. (2013) 131(5):1367–75.e9. doi: 10.1016/j.jaci.2013.01.053
104. Perelygina L, Faisthalab R, Abernathy E, Chen MH, Hao L, Bercovitch L, et al. Rubella virus infected macrophages and neutrophils define patterns of granulomatous inflammation in inborn and acquired errors of immunity. Front Immunol. (2021) 12:796065. doi: 10.3389/fimmu.2021.796065
105. Baumann U, Schulte JH, Groß JP, Beier R, Ludwig M, Wahn V, et al. Case report: rubella virus-induced cutaneous granulomas in two pediatric patients with DNA double strand breakage repair disorders—outcome after hematopoietic stem cell transplantation. Front Immunol. (2022) 13:886540. doi: 10.3389/fimmu.2022.886540
106. Hustinx TW, Scheres JM, Weemaes CM, ter Haar BG, Janssen AH. Karyotype instability with multiple 7/14 and 7/7 rearrangements. Hum Genet. (1979) 49(2):199–208. doi: 10.1007/BF00277643
107. Weemaes CM, Hustinx TW, Scheres JM, van Munster PJ, Bakkeren JA, Taalman RD. A new chromosomal instability disorder: the Nijmegen Breakage syndrome. Acta Paediatr Scand. (1981) 70(4):557–64. doi: 10.1111/j.1651-2227.1981.tb05740.x
108. Matsuura S, Weemaes C, Smeets D, Takami H, Kondo N, Sakamoto S, et al. Genetic mapping using microcell-mediated chromosome transfer suggests a locus for Nijmegen Breakage syndrome at chromosome 8q21-24. Am J Hum Genet. (1997) 60(6):1487–94. doi: 10.1086/515461
109. Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen Breakage syndrome. Cell. (1998) 93(3):467–76. doi: 10.1016/S0092-8674(00)81174-5
110. Syed A, Tainer JA. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu Rev Biochem. (2018) 87:263–94. doi: 10.1146/annurev-biochem-062917-012415
111. Varon R, Demuth I, Chrzanowska KH. Nijmegen Breakage syndrome. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, editors. GeneReviews®. Seattle, WA: University of Washington (1993). p. 1993–2024.
112. Chrzanowska KH, Gregorek H, Dembowska-Baginska B, Kalina MA, Digweed M. Nijmegen Breakage syndrome (NBS). Orphanet J Rare Dis. (2012) 7:13. doi: 10.1186/1750-1172-7-13
113. Yilmaz Demirdag Y, Gupta S. Infections in DNA repair defects. Pathogens. (2023) 12(3):440. doi: 10.3390/pathogens12030440
114. Gregorek H, Chrzanowska KH, Michalkiewicz J, Syczewska M, Madalinski K. Heterogeneity of humoral immune abnormalities in children with Nijmegen Breakage syndrome: an 8-year follow-up study in a single centre. Clin Exp Immunol. (2002) 130(2):319–24. doi: 10.1046/j.1365-2249.2002.01971.x
115. Murray JE, Bicknell LS, Yigit G, Duker AL, van Kogelenberg M, Haghayegh S, et al. Extreme growth failure is a common presentation of ligase IV deficiency. Hum Mutat. (2014) 35(1):76–85. doi: 10.1002/humu.22461
116. Felgentreff K, Baxi SN, Lee YN, Dobbs K, Henderson LA, Csomos K, et al. Ligase-4 deficiency causes distinctive immune abnormalities in asymptomatic individuals. J Clin Immunol. (2016) 36(4):341–53. doi: 10.1007/s10875-016-0266-5
117. Altmann T, Gennery AR. DNA Ligase IV syndrome; a review. Orphanet J Rare Dis. (2016) 11(1):137. doi: 10.1186/s13023-016-0520-1
118. Sun B, Chen Q, Wang Y, Liu D, Hou J, Wang W, et al. LIG4 syndrome: clinical and molecular characterization in a Chinese cohort. Orphanet J Rare Dis. (2020) 15(1):131. doi: 10.1186/s13023-020-01411-x
119. Davis SD, Schaller J, Wedgwood RJ. Job’s syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet. (1966) 1(7445):1013–5. doi: 10.1016/S0140-6736(66)90119-X
120. Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics. (1972) 49(1):59–70. doi: 10.1542/peds.49.1.59
121. Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, et al. Hyper-IgE syndrome with recurrent infections–an autosomal dominant multisystem disorder. N Engl J Med. (1999) 340(9):692–702. doi: 10.1056/NEJM199903043400904
122. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. (2007) 448(7157):1058–62. doi: 10.1038/nature06096
123. Hagl B, Heinz V, Schlesinger A, Spielberger BD, Sawalle-Belohradsky J, Senn-Rauh M, et al. Key findings to expedite the diagnosis of hyper-IgE syndromes in infants and young children. Pediatr Allergy Immunol. (2016) 27(2):177–84. doi: 10.1111/pai.12512
124. Sheerin KA, Buckley RH. Antibody responses to protein, polysaccharide, and phi X174 antigens in the hyperimmunoglobulinemia E (hyper-IgE) syndrome. J Allergy Clin Immunol. (1991) 87(4):803–11. doi: 10.1016/0091-6749(91)90126-9
125. Ishizaka A, Kojima K, Sakiyama Y, Matsumoto S, Kuwajima K, Wagatsuma Y, et al. Hyper-response of serum IgG1 to Staphylococcus aureus peptidoglycan in patients with hyper-IgE syndrome. Clin Exp Immunol. (1992) 87(1):53–7. doi: 10.1111/j.1365-2249.1992.tb06412.x
126. Chandesris MO, Melki I, Natividad A, Puel A, Fieschi C, Yun L, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore). (2012) 91(4):e1–e19. doi: 10.1097/MD.0b013e31825f95b9
127. Melo KM, Alves LM, Valente CFC, Tavares FS. One-year intravenous immunoglobulin replacement therapy: efficacy in reducing hospital admissions in pediatric patients with inborn errors of immunity. J Pediatr (Rio J). (2022) 98(2):190–5. doi: 10.1016/j.jped.2021.05.011
128. Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med. (2010) 207(1):155–71. doi: 10.1084/jem.20091706
129. Diehl SA, Schmidlin H, Nagasawa M, Blom B, Spits H. IL-6 triggers IL-21 production by human CD4+ T cells to drive STAT3-dependent plasma cell differentiation in B cells. Immunol Cell Biol. (2012) 90(8):802–11. doi: 10.1038/icb.2012.17
130. Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood. (2012) 119(17):3997–4008. doi: 10.1182/blood-2011-11-392985
131. Berglund LJ, Avery DT, Ma CS, Moens L, Deenick EK, Bustamante J, et al. IL-21 signalling via STAT3 primes human naive B cells to respond to IL-2 to enhance their differentiation into plasmablasts. Blood. (2013) 122(24):3940–50. doi: 10.1182/blood-2013-06-506865
132. Mazerolles F, Picard C, Kracker S, Fischer A, Durandy A. Blood CD4+CD45RO+CXCR5+ T cells are decreased but partially functional in signal transducer and activator of transcription 3 deficiency. J Allergy Clin Immunol. (2013) 131(4):1146–56. 56.e1–5. doi: 10.1016/j.jaci.2012.12.1519
133. Rodriguez-Bayona B, Ramos-Amaya A, Lopez-Blanco R, Campos-Caro A, Brieva JA. STAT-3 activation by differential cytokines is critical for human in vivo-generated plasma cell survival and ig secretion. J Immunol. (2013) 191(10):4996–5004. doi: 10.4049/jimmunol.1301559
134. van de Veen W, Kratz CE, McKenzie CI, Aui PM, Neumann J, van Noesel CJM, et al. Impaired memory B-cell development and antibody maturation with a skewing toward IgE in patients with STAT3 hyper-IgE syndrome. Allergy. (2019) 74(12):2394–405. doi: 10.1111/all.13969
135. Edwards ESJ, Bosco JJ, Ojaimi S, O'Hehir RE, van Zelm MC. Beyond monogenetic rare variants: tackling the low rate of genetic diagnoses in predominantly antibody deficiency. Cell Mol Immunol. (2021) 18(3):588–603. doi: 10.1038/s41423-020-00520-8
136. Wang L, Aschenbrenner D, Zeng Z, Cao X, Mayr D, Mehta M, et al. Gain-of-function variants in SYK cause immune dysregulation and systemic inflammation in humans and mice. Nat Genet. (2021) 53(4):500–10. doi: 10.1038/s41588-021-00803-4
137. Rowane MJ, Stewart-Bates BC, Doll RJ, Meyerson HJ, Venglarcik JS 3rd, Callahan M, et al. CD5 B-Cell predominant primary immunodeficiency: part of the spectrum of MAGT1 deficiency. Ther Adv Allergy Rhinol. (2023) 14:27534030231199675. doi: 10.1177/27534030231199675
138. Wahn V, von Bernuth H. Igg subclass deficiencies in children: facts and fiction. Pediatr Allergy Immunol. (2017) 28(6):521–4. doi: 10.1111/pai.12757
139. Yel L. Selective IgA deficiency. J Clin Immunol. (2010) 30(1):10–6. doi: 10.1007/s10875-009-9357-x
140. Chinn IK, Orange JS. A 2020 update on the use of genetic testing for patients with primary immunodeficiency. Expert Rev Clin Immunol. (2020) 16(9):897–909. doi: 10.1080/1744666X.2020.1814145
141. Branch A, Modi B, Bahrani B, Hildebrand KJ, Cameron SB, Junker AK, et al. Diverse clinical features and diagnostic delay in monogenic inborn errors of immunity: a call for access to genetic testing. Pediatr Allergy Immunol. (2021) 32(8):1796–803. doi: 10.1111/pai.13571
142. Anderson JT, Cowan J, Condino-Neto A, Levy D, Prusty S. Health-related quality of life in primary immunodeficiencies: impact of delayed diagnosis and treatment burden. Clin Immunol. (2022) 236:108931. doi: 10.1016/j.clim.2022.108931
143. Buchbinder D, Hauck F, Albert MH, Rack A, Bakhtiar S, Shcherbina A, et al. Rubella virus-associated cutaneous granulomatous disease: a unique complication in immune-deficient patients, not limited to DNA repair disorders. J Clin Immunol. (2019) 39(1):81–9. doi: 10.1007/s10875-018-0581-0
144. Bauer M, Kruger R, Kolsch U, Unterwalder N, Meisel C, Wahn V, et al. Antibiotic prophylaxis, immunoglobulin substitution and supportive measures prevent infections in MECP2 duplication syndrome. Pediatr Infect Dis J. (2018) 37(5):466–8. doi: 10.1097/INF.0000000000001799
145. Akhter J, Lefaiver CA, Scalchunes C, DiGirolamo M, Warnatz K. Immunologist’s perspectives on assessment and management of lung disease in CVID: a survey of the membership of the clinical immunology society and the European society for immunodeficiencies. J Clin Immunol. (2018) 38(3):237–46. doi: 10.1007/s10875-018-0488-9
146. Mallhi KK, Petrovic A, Ochs HD. Hematopoietic stem cell therapy for Wiskott-Aldrich syndrome: improved outcome and quality of life. J Blood Med. (2021) 12:435–47. doi: 10.2147/JBM.S232650
147. Albert MH, Gennery AR, Greil J, Cale CM, Kalwak K, Kondratenko I, et al. Successful SCT for Nijmegen Breakage syndrome. Bone Marrow Transplant. (2010) 45(4):622–6. doi: 10.1038/bmt.2009.207
148. Wolska-Kusnierz B, Pastorczak A, Fendler W, Wakulinska A, Dembowska-Baginska B, Heropolitanska-Pliszka E, et al. Hematopoietic stem cell transplantation positively affects the natural history of cancer in Nijmegen Breakage syndrome. Clin Cancer Res. (2021) 27(2):575–84. doi: 10.1158/1078-0432.CCR-20-2574
149. Levy R, Beziat V, Barbieux C, Puel A, Bourrat E, Casanova JL, et al. Efficacy of dupilumab for controlling severe atopic dermatitis in a patient with hyper-IgE syndrome. J Clin Immunol. (2020) 40(2):418–20. doi: 10.1007/s10875-020-00751-4
150. Sogkas G, Hirsch S, Jablonka A, Witte T, Schmidt RE, Atschekzei F. Dupilumab to treat severe atopic dermatitis in autosomal dominant hyper-IgE syndrome. Clin Immunol. (2020) 215:108452. doi: 10.1016/j.clim.2020.108452
151. Dixit C, Thatayatikom A, Pappa H, Knutsen AP. Treatment of severe atopic dermatitis and eosinophilic esophagitis with dupilumab in a 14-year-old boy with autosomal dominant hyper-IgE syndrome. J Allergy Clin Immunol Pract. (2021) 9(11):4167–9. doi: 10.1016/j.jaip.2021.06.049
152. Joshi TP, Anvari S, Gupta MR, Davis CM, Hajjar J. Case report: dupilumab successfully controls severe eczema in a child with elevated IgE levels and recurrent skin infections. Front Pediatr. (2021) 9:646997. doi: 10.3389/fped.2021.646997
153. Matucci-Cerinic C, Viglizzo G, Pastorino C, Corcione A, Prigione I, Bocca P, et al. Remission of eczema and recovery of Th1 polarization following treatment with dupilumab in STAT3 hyper IgE syndrome. Pediatr Allergy Immunol. (2022) 33(4):e13770. doi: 10.1111/pai.13770
154. Nihal A, Comstock JR, Holland KE, Singh AM, Seroogy CM, Arkin LM. Clearance of atypical cutaneous manifestations of hyper-IgE syndrome with dupilumab. Pediatr Dermatol. (2022) 39(6):940–2. doi: 10.1111/pde.15072
155. Staudacher O, Kruger R, Kolsch U, Thee S, Gratopp A, Wahn V, et al. Relieving job: dupilumab in autosomal dominant STAT3 hyper-IgE syndrome. J Allergy Clin Immunol Pract. (2022) 10(1):349–51.e1. doi: 10.1016/j.jaip.2021.08.042
156. Schwab C, Gabrysch A, Olbrich P, Patino V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. (2018) 142(6):1932–46. doi: 10.1016/j.jaci.2018.02.055
157. Taghizade N, Babayeva R, Kara A, Karakus IS, Catak MC, Bulutoglu A, et al. Therapeutic modalities and clinical outcomes in a large cohort with LRBA deficiency and CTLA4 insufficiency. J Allergy Clin Immunol. (2023) 152(6):1634–45. doi: 10.1016/j.jaci.2023.08.004
158. Kim J, Woo S, de Gusmao CM, Zhao B, Chin DH, DiDonato RL, et al. A framework for individualized splice-switching oligonucleotide therapy. Nature. (2023) 619(7971):828–36. doi: 10.1038/s41586-023-06277-0
159. Zhao B, Nguyen MA, Woo S, Kim J, Yu TW, Lee EA. Contribution and therapeutic implications of retroelement insertions in ataxia telangiectasia. Am J Hum Genet. (2023) 110(11):1976–82. doi: 10.1016/j.ajhg.2023.09.008
160. Liu X, Li G, Liu Y, Zhou F, Huang X, Li K. Advances in CRISPR/cas gene therapy for inborn errors of immunity. Front Immunol. (2023) 14:1111777. doi: 10.3389/fimmu.2023.1111777
Keywords: IEI, primary immunodeficiency, polysaccharide, vaccines, pneumococcal infections, vaccination
Citation: Fasshauer M, Dinges S, Staudacher O, Völler M, Stittrich A, von Bernuth H, Wahn V and Krüger R (2024) Monogenic Inborn Errors of Immunity with impaired IgG response to polysaccharide antigens but normal IgG levels and normal IgG response to protein antigens. Front. Pediatr. 12:1386959. doi: 10.3389/fped.2024.1386959
Received: 16 February 2024; Accepted: 21 May 2024;
Published: 12 June 2024.
Edited by:
Sara Sebnem Kilic, Bursa Uludağ University, TürkiyeReviewed by:
Federica Pulvirenti, Accademic Hospital Policlinico Umberto, Italy© 2024 Fasshauer, Dinges, Staudacher, Völler, Stittrich, von Bernuth, Wahn and Krüger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renate Krüger, ci5rcnVlZ2VyQGNoYXJpdGUuZGU=
†These authors have contributed equally to this work