
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 28 June 2024
Sec. Pediatric Immunology
Volume 12 - 2024 | https://doi.org/10.3389/fped.2024.1384550
This article is part of the Research TopicReviews in Pediatric Primary ImmunodeficienciesView all 11 articles
 Olga Staudacher1
Olga Staudacher1 Horst von Bernuth1,2,3,4*
Horst von Bernuth1,2,3,4*
Chronic granulomatous disease (CGD) is caused by an impaired respiratory burst reaction in phagocytes. CGD is an X-linked (XL) (caused by pathogenic variants in CYBB) or autosomal recessive inborn error of immunity (caused by pathogenic variants in CYBA, NCF1, NCF2, or CYBC1). Female carriers of XL-CGD and unfavorable lyonization may present with the partial or full picture of CGD. Patients with CGD are at increased risk for invasive bacterial and fungal infections of potentially any organ, but especially the lymph nodes, liver, and lungs. Pathogens most frequently isolated are S. aureus and Aspergillus spp. Autoinflammation is difficult to control with immunosuppression, and patients frequently remain dependent on steroids. To diagnose CGD, reactive oxygen intermediates (O2− or H2O2) generated by the NADPH oxidase in peripheral blood phagocytes are measured upon in vitro activation with either phorbol-12-myristate-13-acetate (PMA) and/or TLR4 ligands (E. coli or LPS). Conservative treatment requires strict hygienic conduct and adherence to antibiotic prophylaxis against bacteria and fungi, comprising cotrimoxazole and triazoles. The prognosis of patients treated conservatively is impaired: for the majority of patients, recurrent and/or persistent infections, autoinflammation, and failure to thrive remain lifelong challenges. In contrast, cellular therapies (allogeneic stem cell transplantation or gene therapy) can cure CGD. Optimal outcomes in cellular therapies are observed in individuals without ongoing infections or inflammation. Yet cellular therapies are the only curative option for patients with persistent fungal infections or autoinflammation.
The first clinical accounts of CGD date back to 1954 and 1957, when children with increased susceptibility to bacterial infections in the lungs, lymph nodes, and skin and granulomatous lesions were described. These patients showed pigmented lipid histiocytes in granulomas of visceral tissues and hypergammaglobulinemia in peripheral blood. The disease was named “fatal granulomatous disease of childhood,” later changed to its current name, “chronic granulomatous disease” (CGD) (1–3). Antibiotic treatment and surgical drainage of abscesses improved life expectancy from less than 10 years to less than 20 years. Until the late 1960s, the presence of chronic granulomas with typical lipid-laden histiocytes remained the only way to diagnose CGD. In 1967 Robert Baehner and David Nathan described how “intact leucocytes of two children with chronic granulomatous disease fail to reduce nitroblue tetrazolium (NBT) during phagocytosis.” They also established this failure of phagocytes to reduce NBT as the first screening test for CGD (4, 5). At the same time, clinical observations discovered CGD not only in boys but also in girls, suggesting X-linked as well as autosomal recessive inheritance for CGD (6, 7). In the 1970s it became evident that phagocytes from patients with CGD lack a functional NADPH oxidase and that these phagocytes cannot form superoxide upon activation (8–10). Aspergillosis, BCGitis upon vaccination with BCG, and non-infectious (auto)inflammatory manifestations of CGD were clinically relevant issues that were first addressed 20 years after the initial descriptions of mainly bacterial infections (11, 12). Life expectancy improved further upon the introduction of antibiotic prophylaxis directed against bacteria with cotrimoxazole (13), and against aspergillosis with itraconazole (14, 15). Early observations indicated that CGD can be cured through allogeneic bone marrow transplantation. However, initial results were not considered sufficiently encouraging to offer this option upfront to every individual with CGD (16–18).
Pathogenic variants in genes encoding subunits or regulatory proteins of the phagosomal NADPH oxidase complex cause CGD. The NADPH oxidase core complex consists of six proteins: the membrane-bound catalytic subunit gp91phox (coded by CYBB), the membrane-bound activator p22phox (coded by CYBA), the regulatory subunits p47phox (coded by NFC1), p67phox (coded by NCF2), p40phox (coded by NCF4), and the GTPase RAC (in 96% of cases RAC2, RAC1 is also possible) (19). EROS (coded by CYBC1) is a chaperone protein for the dimerization of gp91phox and p22phox (20, 21). Upon activation, the phagosomal NADPH oxidase catalyzes the reaction of NADPH to form a cationic NADP+, a proton (H+), and two electrons. The electrons then enter the phagolysosome. There, oxygen molecules are reduced to two superoxide anions (O2−) (NADPH + 2O2⇔ NADP+ + 2O2− + H+). A subsequent reaction catalyzed by a superoxide dismutase (SOD) [or alternatively myeloperoxidase (MPO)] produces superoxide peroxide (H2O2). The superoxide peroxide then either spontaneously decomposes into two superoxide ions (OH−) or, in a second reaction catalyzed by MPO, combines with a chloride molecule to form hypochlorite (ClO−). Both reactive oxygen species (ROS) not only lyse bacteria but also cause a pH-dependent influx of K+, which subsequently activates proteases, crucial for the destruction of bacteria (Figure 1) (22). X-linked pathogenic variants in CYBB or autosomal recessive pathogenic variants in CYBA, NCF1, NCF2, and CYBC1 lead to absent or severely reduced production of superoxide in all phagocytes (neutrophils as well as monocytes, macrophages, and eosinophils) (23–25).
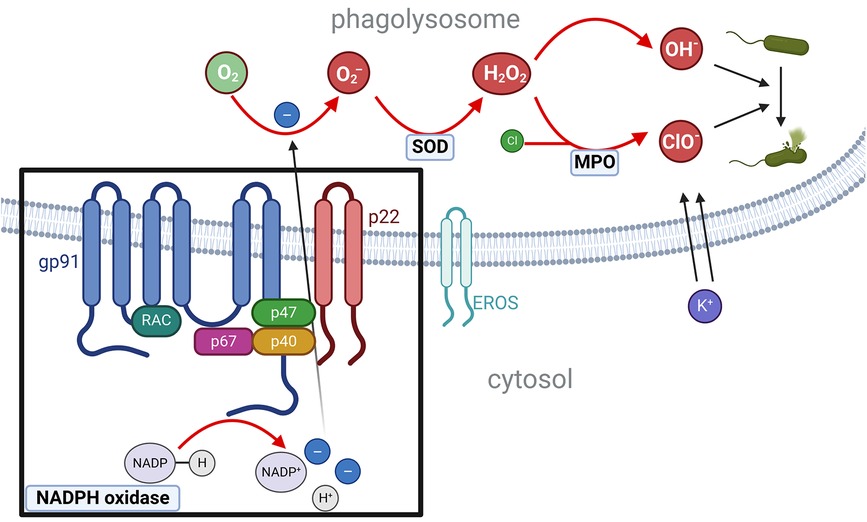
Figure 1 NADPH oxidase complex and the production of ROS.
Due to a large deletion in the X chromosome, patients with the X-linked form of the disease may display a contiguous gene defect causing the McLeod phenotype in erythrocytes, as well as Duchenne muscular dystrophy or retinitis pigmentosa. Patients with the McLeod phenotype must not receive repeated transfusions with erythrocytes expressing K20 and XK1. Acanthocytosis of erythrocytes in the peripheral blood may be indicative of the McLeod phenotype, but its absence does not rule it out (26–28).
ROS serve immunoregulatory functions in addition to their antimicrobial effect. The activation of ataxia-telangiectasia-mutated (ATM) kinase, for instance, requires NADPH oxidase (29). NADPH-deficient phagocytes show autophagic dysfunction, increased production of IL-1β upon activation with LPS, and an altered interferon signature. CGD patients show impaired apoptosis of peripheral blood neutrophils, yet not of monocytes (30–33).
P40phox deficiency, caused by autosomal recessive pathogenic variants in NCF4, is a similar but distinct disease. Neutrophils with p40phox deficiency exhibit impaired burst activity, while macrophages are less affected and demonstrate a greater capacity for producing reactive oxygen intermediates than in patients with bona fide CGD. As a result, patients with p40phox deficiency exhibit a far milder clinical phenotype. To date, no patients with p40phox deficiency have been described as suffering from invasive bacterial and fungal infections (34–36). Dominant negative mutations in RAC2 result in a phenotype resembling leukocyte adhesion deficiency with an impaired oxidative burst, as RAC2 is not only crucial for NADPH oxidase but also controls cytoskeleton formation and cell adhesion (37).
Patients with CGD are at increased risk for invasive bacterial and fungal infections. Symptoms usually start in infancy, with a median age at diagnosis of CGD between 2.5 and 3 years. However, some patients are not identified until adolescence or adulthood (38–40). The infectious phenotype may differ significantly depending on local climate and antimicrobial resistance patterns. The most common pathogens are (from most to least common) Staphylococcus aureus (S. aureus), Aspergillus spp., Burkholderia cepacia, Serratia spp., Nocardiae, and Salmonella spp (38, 39, 41–45). Staphylococcal infections mostly affect the skin, lymph nodes, rectum, and brain. Burkholderia cepacia manifests in the lungs and can cause the life-threatening Cepacia syndrome, a condition similar to a cytokine storm in macrophage activation syndrome. Serratia and Proteus spp. cause liver abscess, and infections from Nocardia most often affect the lungs (38, 39, 41, 44–46). Molds, such as e.g., Histoplasma spp., Phellinus spp., Rasamsonia spp., Rhizopus spp., and Trichosporon spp., and, far less commonly, Mucormucosis, pose particular threats (47–52). Invasive mold infections mainly affect the lungs, brain, bones (as osteomyelitis), and nasal cavities (Figure 2) (53, 54). BCGitis is another common presenting symptom in regions where infants are routinely vaccinated with BCG (55, 56). Yet neither mycobacteria other than tuberculosis (MOTT) nor bona fide tuberculosis infections are common in CGD (38, 39, 45, 57, 58).
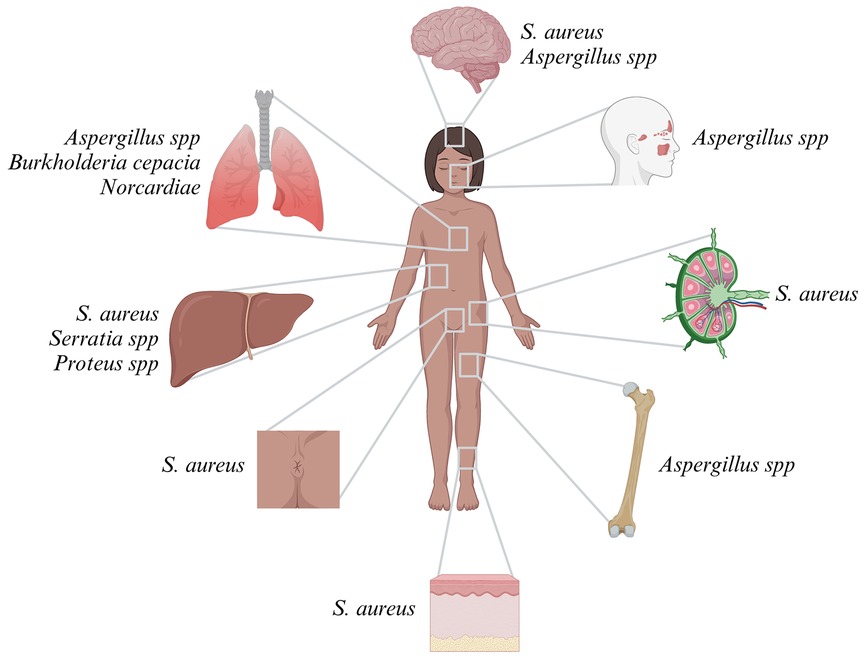
Figure 2 Infection sites and most common pathogens in CGD.
The eponymous granulomas of CGD are often found in the urinary tract, intestines, or lungs. They can lead to stenosis in hollow organs, or fibrosis. Granuloma formation can be seen as a mechanism by which the immune system contains infections, yet it is noteworthy that many granulomas found in patients with CGD are sterile and of autoinflammatory origin (59–61). About half the patients with CGD not only suffer from infections but also develop autoinflammation or immune dysregulation. Inflammatory bowel disease is the most common manifestation and may resemble Crohn's disease (62). Histology, however, may differ from Crohn's disease by pigmented lipid histiocytes, microgranulomas, and eosinophilic abscesses (63, 64). Further organs potentially affected by autoinflammation are the urinary tract/bladder and the lungs, but also sites like the brain, joints, and retina. Incident patients with CGD and systemic conditions resembling systemic lupus erythematosus, vasculitis, sarcoidosis, and thrombocytopenia were also described (59, 65–68) (Figure 3).
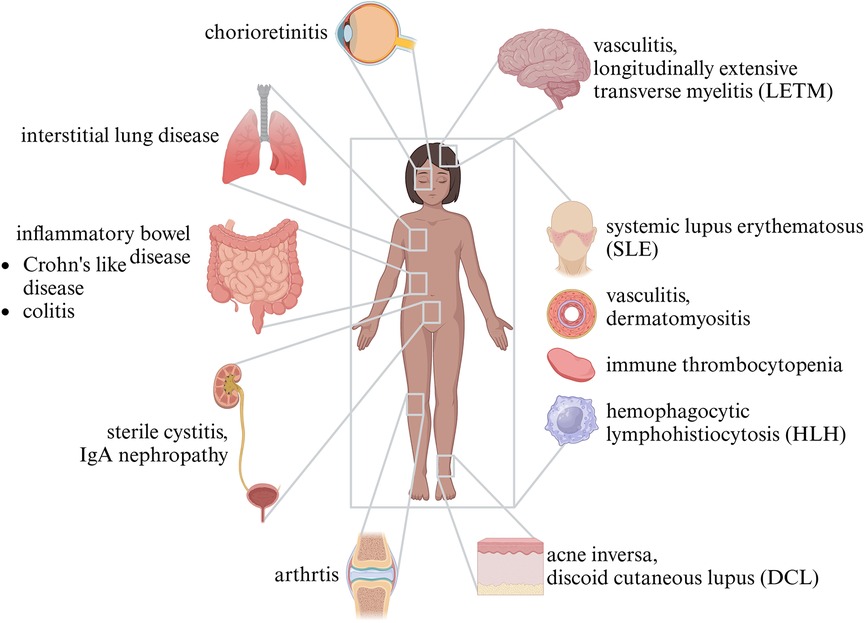
Figure 3 Autoinflammation in CGD.
It is important to consider CGD in patients presenting with infections at any of the above sites. But in countries with endemic tuberculosis, the presence of difficult-to-treat or disseminated tuberculosis should prompt an evaluation for CGD (69). As Crohn's-like disease is common in CGD and may develop before the onset of invasive fungal or bacterial infections, CGD must be ruled out in any patient with Crohn's disease (64, 70–72). Similarly, CGD should be ruled out in patients with sterile granuloma formation (73–75).
Important differential diagnoses of CGD are autosomal dominant hyper-IgE syndrome caused by dominant negative mutations in STAT3 (“DN-STAT3-HIES”) and MyD88/IRAK4 deficiency. DN-STAT3-HIES is often associated with abscesses and pneumonia caused by S. aureus. Infections with Aspergillus spp. are much less common, and invasive aspergillosis is extremely rare in DN-STAT3-HIES, but aspergillomas may occur in the context of characteristic pneumatoceles, which are not typically seen in CGD (76). In contrast, chronic mucocutaneous candidiasis is common. In addition, DN-STAT3-HIES is associated with skeletal abnormalities and severe dermatitis, both of which are not features of CGD (77–79). On the other hand, patients with MyD88/IRAK4 deficiency often experience invasive infections with Streptococcus pneumoniae, S. aureus, and Pseudomonas aeruginosa, but fungal infections are rare (80–82). In contrast to patients with CGD, patients with DN-STAT3-HIES and MyD88/IRAK4 deficiency have impaired IL-6 signaling or production. This leads to cold abscesses in DN-STAT3-HIES and to low CrP despite invasive infections in MyD88/IRAK4 deficiency (77, 82–85).
To diagnose CGD, ROS (O2− or H2O2) generated by the NADPH oxidase in phagocytes of peripheral blood are measured upon in vitro activation with either phorbol-12-myristate-13-acetate (PMA) and/ or TLR4-ligands (E. coli or LPS). We suggest that the assay be repeated at least once and that two different stimuli for the induction of the respiratory burst be used. ROS can be detected by different assays. O2− can be measured by chemiluminescence or the seminal NBT assay and H2O2 by the FACS-based DiHydroRhodamine (DHR) assay (Figure 4) (86, 87). While it was once considered the gold standard for the diagnosis of CGD, NBT has since been surpassed by the flow-cytometry-based DHR assay in terms of time effectiveness, sensitivity, and quantification. However, the NBT assay remains a cost-effective and relatively easy-to-perform option, requiring only a microscope, a stimulant, and NBT. Especially in low-resource settings, the NBT assay is still a highly effective diagnostic tool (88–90).
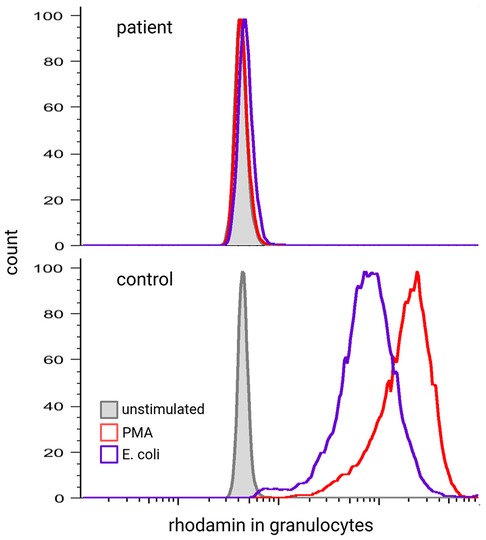
Figure 4 Respiratory burst of a healthy control and patient with CGD measured by DHR assay.
Functional assays that measure the respiratory burst in phagocytes may be false positive in patients treated with paracetamol/acetaminophen, metamizole, or mesalazine (5-ASA) (91–93). Furthermore, CGD must be distinguished from MPO deficiency, which is not considered an inborn error of immunity. In CGD, the oxygen burst reaction is impaired in all phagocytes, whereas in MPO deficiency, the oxygen burst reaction is only impaired in neutrophils (94).
In patients with a repeatedly impaired respiratory burst, at least the seven genes CYBA, CYBB, NCF1, NCF2, NCF4 CYBC1, and RAC2 should be analyzed for causative mutations.
Upon diagnosis, all patients with CGD should receive conservative treatment with antibacterial and antifungal prophylaxis with cotrimoxazole and triazoles. Cotrimoxazole significantly reduces the incidence of bacterial infections while being relatively inexpensive (95). Itraconazole is still the most widely used agent to prevent invasive Aspergillus and other mold infections, but posaconazole may be a choice to consider depending on local resistance patterns (15). Female carriers with unfavorable lyonization of pathogenic variants in the X-linked CYBB may develop partial or complete clinical manifestation of CGD (38, 96–98). Given the documented occurrence of serious bacterial infections in patients with ROS below 10%, we propose considering prophylactic treatment against bacteria when ROS levels are below 20% and against molds when ROS levels are below 5% (38, 96, 98). We recommend regularly assessing the respiratory burst of carriers, with a suggested interval of 5 years (96).
Especially in North America, but less so in Europe, a significant number of patients are treated with IFN-γ (99). It has been shown to improve the splicing efficiency of CYBB (100). A recent meta-analysis showed a significant reduction in the likelihood of infection, but to date there is insufficient evidence of clinical improvement in patients with CGD on IFN-γ and data on long-term effects are lacking (101). The question of whether there is a general benefit of IFN-γ for all patients with CGD remains unanswered. Treatment is costly and not without side effects (mainly fever, but also mental impairment). In our opinion, IFN-γ can be considered in patients with particular pathogenic variants of CYBB.
Hygienic conduct includes avoiding exposure to molds. Patients with CGD should therefore abstain from doing agricultural work and gardening (including composting, mucking out stables, working in barns, etc.) as well as demolition of moist walls. Alternatively, patients must wear an FFP3 mask to prevent mold from entering their airways (102). We do not advise people with CGD to keep pets, although the risk may be manageable if there is strict adherence to hygiene (103). Furthermore, patients undergoing conservative treatment derive substantial benefits from regular follow-up by physicians experienced in CGD care (102). In our European setting, we strive for intervals of 3 months between visits. Although anti-infective prophylaxis greatly reduces mortality, infections still occur at a rate of 0.26–0.64 per patient-year with a cumulative lifetime risk of 20%–40% for aspergillosis, which remains the leading cause of death (39, 43).
Immunosuppressive therapy can become necessary to control autoinflammation and immune dysregulation (59, 104). For mild IBD in CGD, sulfasalazine or alternative aminosalicylates are commonly used as initial treatments (105). Recently, monoclonal antibodies targeting pro-inflammatory cytokines (such as infliximab, anakinra, adalimumab, and ustekinumab) have been explored (106–109). However, the available evidence is limited and ustekinumab is the only treatment that has shown somewhat favorable outcomes (109). Despite their known adverse effects, patients often remain dependent on corticosteroids to control IBD.
In retrospective studies, the median life expectancy of conservatively treated CGD patients is between 30 and 40 years. Additionally, quality of life and academic and professional achievements are severely impaired on conservative treatment (39, 110, 111). In contrast, allogenic hematopoietic stem cell transplantation (HSCT) can potentially cure CGD. Because transplantation-related mortality was originally at 15%–50%, HSCT was historically considered a salvage therapy only for patients with recurrent infections or refractory inflammation (112, 113). Improved HLA matching, fludarabine-based reduced-toxicity conditioning, and the accumulation of clinical experience in guiding patients with CGD through allogenic HSCT have reduced treatment-related morbidity and mortality (Table 1) (115–121). Over the past decade, transplant series have reported survival rates of 83%–96% in CGD patients following matched donor transplantation (115, 116). Several studies have compared the prognosis of CGD patients treated conservatively with those treated with HSCT. A Swedish study of 41 patients reported a superior outcome with HSCT (93% vs. 74% survival) (114). Other studies, including our own retrospective European study, still failed to describe a clearly better survival after HSCT, with survival rates ranging from 76%–90% in both cohorts (41, 43, 45, 115, 118). However, significant reductions in infectious episodes and catch-up growth after HSCT are clearly evident in data from the UK, US, and Europe (43, 45, 115). In particular, patients between the ages of 5 and 14 without active complications at the time of HSCT have excellent outcomes (45, 115, 119, 120). Therefore, all young CGD patients with a ≥9/10 HLA-matched available donor should be considered for HSCT early in life, before chronic sequelae caused by infections and/or autoinflammation occur. Patients for whom no ≥9/10 HLA-matched donor is available can be offered haploidentical HSCT from an unaffected parent [published experience reviewed in (122)]. The idea of early transplantation also holds true for symptomatic X-linked carriers of mutations in CYBB and an unfavorable lyonization (96, 121). HSCT is also possible in older patients after recurrent or persistent infection or autoinflammation and may be the only life-saving option (96, 116, 117).
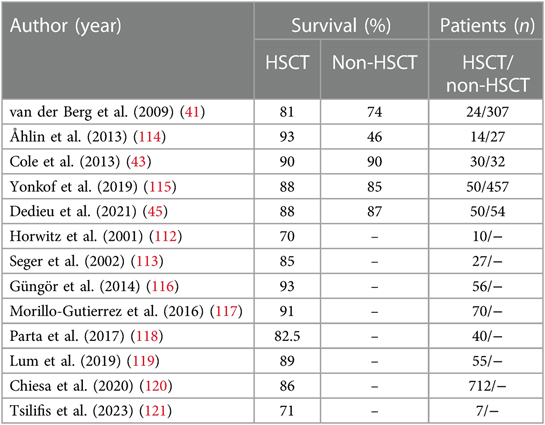
Table 1 Survival rates in CGD patients treated with HSCT vs. patients on conservative treatment (non-HSCT).
Gene therapy for CGD remains limited to trials in only a few highly specialized centers around the world. It is often mentioned as a future alternative cellular therapy “for patients without a suitable donor” (123, 124). But at least most children can be treated by HSCT from a haploidentical parent (122, 125–127). Gene therapy is also promoted for patients with a “high disease burden,” as conditioning is milder than with HSCT (123). However, in the most recent trials, the mortality rates for gene therapy are still higher than those for HSCT. This is presumably due to the disease burden of the patients enrolled, but it may also be because of a high rate of graft rejection in gene therapy (120, 124, 128). To date, there is a lack of long-term data and head-to-head trials, or at least meta-analyses, comparing gene therapy with haploidentical HSCT in patients for whom a matched donor is not available (129). Nevertheless, gene therapy may emerge as an alternative to HSCT in selected situations (124, 130).
Chronic granulomatous disease (CGD) is caused by an impaired respiratory burst reaction of all phagocytes rather than an impaired burst in neutrophilic granulocytes only. CGD is an X-linked (caused by pathogenic variants in CYBB) or autosomal recessive inborn error of immunity (caused by pathogenic variants in CYBA, NCF1, NCF2, or CYBC1). Patients with CGD are at increased risk for bacterial and/or fungal invasive infections of any organ, but mainly the lymph nodes, liver, and lungs. The leading pathogens isolated are S. aureus and Aspergillus spp. But Serratia, Proteus spp., Burkholderia cepacia, Nocardia spp., and Salmonella spp. are still often isolated, and infections with almost any intracellular bacteria and fungi are possible. Infections with Aspergillus spp. and Burkholderia cepacia remain the major cause of morbidity and mortality in patients on conservative treatment. Patients often develop skin infections by S. aureus. Autoinflammation, and inflammatory bowel disease in particular, is difficult to control by immunosuppression, and patients frequently remain dependent on steroids. Female carriers of pathogenic variants in CYBB and unfavorable lyonization may present with the partial or even full picture of CGD. For the diagnosis of CGD, reactive oxygen intermediates (O2− or H2O2) generated by the NADPH oxidase in phagocytes of peripheral blood are measured upon in vitro activation with either PMA and/ or TLR4 ligands (E. coli or LPS). These assays may be false positives in patients treated with paracetamol, metamizol or mesalazine (5-ASA). Conservative treatment must adhere to a strict hygienic conduct and antibiotic prophylaxis against bacteria and fungi. Cotrimoxazole and triazoles that work against intracellular bacteria and Aspergillus spp. are the mainstay of the latter. With ongoing advancements in diagnostics, prophylaxis, and therapeutic modalities, it is plausible that life expectancy may surpass the age range of 30–40 years in conservatively treated patients. Most patients with CGD who receive conservative treatment, however, face lifelong challenges such as recurrent and/or persistent infections as well as steroid-dependent autoinflammation and subsequently failure to thrive. Overall, this leads to an unfavorable psychosocial prognosis. In contrast, cellular therapies (allogenic HSCT from a healthy donor or autologous gene therapy-modified cells) can cure CGD. HSCT in individuals without ongoing infections or inflammation offers a fair, yet unfortunately still far from completely event-free prognosis and chance for overall survival. But neither persistent infections nor refractory autoinflammation are a contraindication against HSCT; rather, they are an indication to proceed to a definite cure through cellular therapy. If no HLA-matched donor is available, most infants and children can be transplanted from a haploidentical parent. If no such donor is available, gene therapy may be an alternative option.
OS: Visualization, Writing – review & editing. HB: Conceptualization, Writing – original draft, Writing – review & editing, Supervision.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We would like to thank Uwe Koelsch, Nadine Unterwalder, and Christian Meisel, Department of Immunology, Labor Berlin, for the reliable functional diagnostic workup and Anna Stittrich, Department of Genetics, Labor Berlin, for the genetic workup of our patients. Thanks go to past and present members of Pediatric Pneumology and Immunology (Sabine Belard, Sarah Dinges, Cinzia Dedieu, Mirjam Voeller, Renate Krueger, Sybille Landwehr-Kenzel, Luise Martin, Volker Wahn), of Pediatric Stem Cell Transplantation (Sandra Cyrull, Jan Dörr, Angelika Eggert, Wolfram Ebell, Jörn-Sven Kühl, Annette Künkele, Michel Lauenspach, Lena Oevermann, Julia Scheiermann, Arend von Stackelberg, Felix Zirngibl), and of Pediatric Gastroenterology (Philip Bufler, Stephan Henning, Christian Hudert, Johanna Overberg, Benedikt Weber) for their dedicated patient care. We are indebted to the nurses and staff of wards 29 and 39i at the Children's Hospital Charité in Berlin. We thank Michael Albert, Fabian Hauck (Hauner Children's Hospital, Ludwig Maximilian University of Munich), Ansgar Schulz, Manfred Hönig (Children's Hospital, University of Ulm), Ulrich Baumann (Children's Hospital, Medizinische Hochschule Hannover), Christian Hedrich, Jochen Roesler, Catharina Schütz (Children's Hospital, Technical University Dresden), Nizar Mahlahoui, Stephan Blanche, Alain Fischer (Hôpital Necker-Enfants Malades, Paris), and Steven Holland (NIH, Bethesda, Maryland) for their clinical and scientific cooperation over many years. We thank the patients and their families for their trust in our care. Our figures were created using biorender. We acknowledge financial support from the Open Access Publication Fund of Charité—Universitätsmedizin Berlin.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Janeway CA, Craig J, Davidson M, Downey W, Gitlin D, Sullivan JC. Hypergammaglobulinemia associated with severe, recurrent and chronic non-specific infection. Am J Dis Child. (1954) 88:388–92.
2. Landing BH, Shirkey HS. A syndrome of recurrent infection and infiltration of viscera by pigmented lipid histiocytes. Pediatrics. (1957) 20:431–8. doi: 10.1542/peds.20.3.431
3. Bridges RA, Berendes H, Good RA. A fatal granulomatous disease of childhood; the clinical, pathological, and laboratory features of a new syndrome. AMA J Dis Child. (1959) 97:387–408. doi: 10.1001/archpedi.1959.02070010389004
4. Baehner RL, Nathan DG. Leukocyte oxidase: defective activity in chronic granulomatous disease. Science. (1967) 155:835–6. doi: 10.1126/science.155.3764.835
5. Baehner RL, Nathan DG. Quantitative nitroblue tetrazolium test in chronic granulomatous disease. N Engl J Med. (1968) 278:971–6. doi: 10.1056/NEJM196805022781801
6. Quie PG, Kaplan EL, Page AR, Gruskay FL, Malawista SE. Defective polymorphonuclear-leukocyte function and chronic granulomatous disease in two female children. N Engl J Med. (1968) 278:976–80. doi: 10.1056/NEJM196805022781802
7. Azimi PH, Bodenbender JG, Hintz RL, Kontras SB. Chronic granulomatous disease in three female siblings. JAMA. (1968) 206:2865–70. doi: 10.1001/jama.1968.03150130023004
8. Curnutte JT, Whitten DM, Babior BM. Defective superoxide production by granulocytes from patients with chronic granulomatous disease. N Engl J Med. (1974) 290:593–7. doi: 10.1056/NEJM197403142901104
9. Hohn DC, Lehrer RI. NADPH oxidase deficiency in X-linked chronic granulomatous disease. J Clin Invest. (1975) 55:707–13. doi: 10.1172/JCI107980
10. Segal AW, Jones OT. Novel cytochrome b system in phagocytic vacuoles of human granulocytes. Nature. (1978) 276:515–7. doi: 10.1038/276515a0
11. Esterly JR, Sturner WQ, Esterly NB, Windhorst DB. Disseminated BCG in twin boys with presumed chronic granulomatous disease of childhood. Pediatrics. (1971) 48:141–4. doi: 10.1542/peds.48.1.141
12. Raubitschek AA, Levin AS, Stites DP, Shaw EB, Fudenberg HH. Normal granulocyte infusion therapy for aspergillosis in chronic granulomatous disease. Pediatrics. (1973) 51:230–3. doi: 10.1542/peds.51.2.230
13. Philippart AI, Colodny AH, Baehner RL. Continuous antibiotic therapy in chronic granulomatous disease: preliminary communication. Pediatrics. (1972) 50:923–5. doi: 10.1542/peds.50.6.923
14. Petropoulou T, Liese J, Tintelnot K, Gahr M, Belohradsky BH. Long-term treatment of patients with itraconazole for the prevention of aspergillus infections in patients with chronic granulomatous disease (CGD). Mycoses. (1994) 37(Suppl 2):64–9. 7609746.7609746
15. Gallin JI, Alling DW, Malech HL, Wesley R, Koziol D, Marciano B, et al. Itraconazole to prevent fungal infections in chronic granulomatous disease. N Engl J Med. (2003) 348:2416–22. doi: 10.1056/NEJMoa021931
16. Delmas Y, Goudemand J, Farriaux JP. Letter: chronic familial granulomatosis. Treatment using bone marrow graft (1 case). Nouv Presse Med. (1975) 4:2334–5. 788668.1101217
17. Foroozanfar N, Hobbs JR, Hugh-Jones K, Humble JG, James DCO, Selwyn S, et al. Bone-marrow transplant from an unrelated donor for chronic granulomatous disease. Lancet. (1977) 1:210–3.64747
18. Hobbs JR, Monteil M, McCluskey DR, Jurges E, el Tumi M. Chronic granulomatous disease 100% corrected by displacement bone marrow transplantation from a volunteer unrelated donor. Eur J Pediatr. (1992) 151:806–10. doi: 10.1007/BF01957929
19. Noreng S, Ota N, Sun Y, Ho H, Johnson M, Arthur CP, et al. Structure of the core human NADPH oxidase NOX2. Nat Commun. (2022) 13:6079. doi: 10.1038/s41467-022-33711-0
20. Thomas DC, Clare S, Sowerby JM, Pardo M, Juss JK, Goulding DA, et al. Eros is a novel transmembrane protein that controls the phagocyte respiratory burst and is essential for innate immunity. J Expl Med. (2017) 214:1111–28. doi: 10.1084/jem.20161382
21. Arnadottir GA, Norddahl GL, Gudmundsdottir S, Agustsdottir AB, Sigurdsson S, Jensson BO, et al. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat Commun. (2018) 9:4447. doi: 10.1038/s41467-018-06964-x
22. Reeves EP, Lu H, Jacobs HL, Messina CGM, Bolsover S, Gabella G, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. (2002) 416:291–7. doi: 10.1038/416291a
23. Thomas DC, Charbonnier L-M, Schejtman A, Aldhekri H, Coomber EL, Dufficy ER, et al. EROS/CYBC1 mutations: decreased NADPH oxidase function and chronic granulomatous disease. J Allergy Clin Immunol. (2019) 143:782–5.e1. doi: 10.1016/j.jaci.2018.09.019
24. Roos D, van Leeuwen K, Hsu AP, Priel DL, Begtrup A, Brandon R, et al. Hematologically important mutations: x-linked chronic granulomatous disease (fourth update). Blood Cells Mol Dis. (2021) 90:102587. doi: 10.1016/j.bcmd.2021.102587
25. Batlle-Masó L, Rivière JG, Franco-Jarava C, Martín-Nalda A, Garcia-Prat M, Parra-Martínez A, et al. Molecular challenges in the diagnosis of X-linked chronic granulomatous disease: CNVs, intronic variants, skewed X-chromosome inactivation, and gonosomal mosaicism. J Clin Immunol. (2023) 43:1953–63. doi: 10.1007/s10875-023-01556-x
26. Giblett ER, Klebanoff SJ, Pincus SH. Kell phenotypes in chronic granulomatous disease: a potential transfusion hazard. Lancet. (1971) 1:1235–6. doi: 10.1016/s0140-6736(71)91738-7
27. Symmans WA, Shepherd CS, Marsh WL, Oyen R, Shohet SB, Linehan BJ. Hereditary acanthocytosis associated with the McLeod phenotype of the Kell blood group system. Br J Haematol. (1979) 42:575–83. doi: 10.1111/j.1365-2141.1979.tb01170.x
28. Lhomme F, Peyrard T, Babinet J, Abou-Chahla W, Durieu I, Moshous D, et al. Chronic granulomatous disease with the McLeod phenotype: a French national retrospective case series. J Clin Immunol. (2020) 40:752–62. doi: 10.1007/s10875-020-00791-w
29. Harbort CJ, Soeiro-Pereira PV, von Bernuth H, Kaindl AM, Costa-Carvalho BT, Condino-Neto A, et al. Neutrophil oxidative burst activates ATM to regulate cytokine production and apoptosis. Blood. (2015) 126:2842–51. doi: 10.1182/blood-2015-05-645424
30. Kasahara Y, Iwai K, Yachie A, Ohta K, Konno A, Seki H, et al. Involvement of reactive oxygen intermediates in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood. (1997) 89:1748–53. doi: 10.1182/blood.V89.5.1748
31. von Bernuth H, Kulka C, Roesler J, Gahr M, Rösen-Wolff A. NADPH oxidase is not required for spontaneous and staphylococcus aureus-induced apoptosis of monocytes. Ann Hematol. (2004) 83:206–11. doi: 10.1007/s00277-003-0837-4
32. de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci USA. (2014) 111:3526–31. doi: 10.1073/pnas.1322831111
33. Kelkka T, Kienhöfer D, Hoffmann M, Linja M, Wing K, Sareila O, et al. Reactive oxygen species deficiency induces autoimmunity with type 1 interferon signature. Antioxid Redox Signal. (2014) 21:2231–45. doi: 10.1089/ars.2013.5828
34. van de Geer A, Nieto-Patlán A, Kuhns DB, Tool AT, Arias AA, Bouaziz M, et al. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J Clin Invest. (2018) 128:3957–75. doi: 10.1172/JCI97116
35. Reis-Melo A, Espinheira MC do, Pinto-Pais I, Bonito Vitor A, Bustamante J, Trindade E. Perianal disease and granulomas: think out of the box…. GE Port J Gastroenterol (2020) 27:119–23. doi: 10.1159/000502358
36. Neehus A-L, Fusaro M, NCF4 consortium, Lévy R, Bustamante J. A new patient with p40phox deficiency and chronic immune thrombocytopenia. J Clin Immunol (2023) 43:1173–7. doi: 10.1007/s10875-023-01498-4
37. Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. PNAS USA. (2000) 97:4654–9. doi: 10.1073/pnas.080074897
38. Winkelstein JA, Marino MC, Johnston RB, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore). (2000) 79:155–69. doi: 10.1097/00005792-200005000-00003
39. Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T, Finocchi A, et al. Clinical features, long-term follow-up and outcome of a large cohort of patients with chronic granulomatous disease: an Italian multicenter study. Clin Immunol. (2008) 126:155–64. doi: 10.1016/j.clim.2007.09.008
40. Roesler J, Segerer F, Morbach H, Kleinert S, Thieme S, Rösen-Wolff A, et al. P67-phox (NCF2) lacking exons 11 and 12 is functionally active and leads to an extremely late diagnosis of chronic granulomatous disease (CGD). PLoS One. (2012) 7:e34296. doi: 10.1371/journal.pone.0034296
41. van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PLoS One. (2009) 4:e5234. doi: 10.1371/journal.pone.0005234
42. Falcone EL, Holland SM. Invasive fungal infection in chronic granulomatous disease: insights into pathogenesis and management. Curr Opin Infect Dis. (2012) 25:658–69. doi: 10.1097/QCO.0b013e328358b0a4
43. Cole T, Pearce MS, Cant AJ, Cale CM, Goldblatt D, Gennery AR. Clinical outcome in children with chronic granulomatous disease managed conservatively or with hematopoietic stem cell transplantation. J Allergy Clin Immunol. (2013) 132:1150–5. doi: 10.1016/j.jaci.2013.05.031
44. Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, Osgood S, et al. Common severe infections in chronic granulomatous disease. Clin Infect Dis. (2015) 60:1176–83. doi: 10.1093/cid/ciu1154
45. Dedieu C, Albert MH, Mahlaoui N, Hauck F, Hedrich C, Baumann U, et al. Outcome of chronic granulomatous disease: conventional treatment vs. stem cell transplantation. Pediatr Allergy Immunol. (2021) 32:576–85. doi: 10.1111/pai.13402
46. Galluzzo ML, Hernandez C, Davila MTG, Pérez L, Oleastro M, Zelazko M. Clinical and histopathological features and a unique spectrum of organisms significantly associated with chronic granulomatous disease osteomyelitis during childhood. Clin Infect Dis. (2008) 46:745–9. doi: 10.1086/527446
47. Dotis J, Pana ZD, Roilides E. Non-aspergillus fungal infections in chronic granulomatous disease. Mycoses. (2013) 56:449–62. doi: 10.1111/myc.12049
48. Al-Otaibi AM, Al-Shahrani DA, Al-Idrissi EM, Al-Abdely HM. Invasive mucormycosis in chronic granulomatous disease. Saudi Med J. (2016) 37:567–9. doi: 10.15537/smj.2016.5.14239
49. Haidar G, Zerbe CS, Cheng M, Zelazny AM, Holland SM, Sheridan KR. Phellinus species: an emerging cause of refractory fungal infections in patients with X-linked chronic granulomatous disease. Mycoses. (2017) 60:155–60. doi: 10.1111/myc.12573
50. Babiker A, Gupta N, Gibas CFC, Wiederhold NP, Sanders C, Mele J, et al. Rasamsonia sp: an emerging infection amongst chronic granulomatous disease patients. A case of disseminated infection by a putatively novel rasamsonia argillacea species complex involving the heart. Med Mycol Case Rep. (2019) 24:54–7. doi: 10.1016/j.mmcr.2019.04.002
51. Eshaghi H, Moradi L, Adimi P, Gharagozlou M, Movahedi M, Parvaneh N. Invasive rasamsonia argillacea infection in chronic granulomatous disease: report of a new case and literature review. J Mycol Med. (2021) 31:101106. doi: 10.1016/j.mycmed.2020.101106
52. Fogel LA, Grady RM, SLCH Consortium, He M, Kitcharoensakkul M. Steroid-responsive pulmonary hypertension in a pediatric patient with chronic granulomatous disease and histoplasmosis. J Clin Immunol (2023) 43:1118–21. doi: 10.1007/s10875-023-01473-z
53. Blumental S, Mouy R, Mahlaoui N, Bougnoux M-E, Debré M, Beauté J, et al. Invasive mold infections in chronic granulomatous disease: a 25-year retrospective survey. Clin Infect Dis. (2011) 53:e159–69. doi: 10.1093/cid/cir731
54. Desai JV, Lionakis MS. The role of neutrophils in host defense against invasive fungal infections. Curr Clin Microbiol Rep. (2018) 5:181–9. doi: 10.1007/s40588-018-0098-6
55. Conti F, Lugo-Reyes SO, Blancas Galicia L, He J, Aksu G, Borges de Oliveira E, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol. (2016) 138:241–8.e3. doi: 10.1016/j.jaci.2015.11.041
56. Zeng Y, Ying W, Wang W, Hou J, Liu L, Sun B, et al. Clinical and genetic characteristics of BCG disease in Chinese children: a retrospective study. J Clin Immunol. (2023) 43:756–68. doi: 10.1007/s10875-022-01422-2
57. Rawat A, Vignesh P, Sudhakar M, Sharma M, Suri D, Jindal A, et al. Clinical, immunological, and molecular profile of chronic granulomatous disease: a multi-centric study of 236 patients from India. Front Immunol. (2021) 12:625320. doi: 10.3389/fimmu.2021.625320
58. Abd Elaziz D, El Hawary R, Meshaal S, Alkady R, Lotfy S, Eldash A, et al. Chronic granulomatous disease: a cohort of 173 patients-10-years single center experience from Egypt. J Clin Immunol. (2023) 43:1799–811. doi: 10.1007/s10875-023-01541-4
59. von Bernuth H, Knöchel B, Wendisch J, Bergert R, Winkler U, Hahn G, et al. Klinisches Bild, Diagnostik und Therapie granulomatöser Entzündungen ohne Erregernachweis bei 6 Patienten mit septischer Granulomatose (CGD). Monatsschr Kinderheilkd. (2003) 151:49–56. doi: 10.1007/s00112-001-0387-6
60. Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJB, Sewell GW, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med. (2009) 206:1883–97. doi: 10.1084/jem.20091233
61. Kuijpers T, Lutter R. Inflammation and repeated infections in CGD: two sides of a coin. Cell Mol Life Sci. (2012) 69:7–15. doi: 10.1007/s00018-011-0834-z
62. Ament ME, Ochs HD. Gastrointestinal manifestations of chronic granulomatous disease. N Engl J Med. (1973) 288:382–7. doi: 10.1056/NEJM197302222880802
63. Marks DJB, Miyagi K, Rahman FZ, Novelli M, Bloom SL, Segal AW. Inflammatory bowel disease in CGD reproduces the clinicopathological features of Crohn’s disease. Am J Gastroenterol. (2009) 104:117–24. doi: 10.1038/ajg.2008.72
64. Alimchandani M, Lai J-P, Aung PP, Khangura S, Kamal N, Gallin JI, et al. Gastrointestinal histopathology in chronic granulomatous disease: a study of 87 patients. Am J Surg Pathol. (2013) 37:1365–72. doi: 10.1097/PAS.0b013e318297427d
65. Schuetz C, Hoenig M, Schulz A, Lee-Kirsch MA, Roesler J, Friedrich W, et al. Successful unrelated bone marrow transplantation in a child with chronic granulomatous disease complicated by pulmonary and cerebral granuloma formation. Eur J Pediatr. (2007) 166:785–8. doi: 10.1007/s00431-006-0317-7
66. Henrickson SE, Jongco AM, Thomsen KF, Garabedian EK, Thomsen IP. Noninfectious manifestations and complications of chronic granulomatous disease. J Pediatric Infect Dis Soc. (2018) 7:S18–24. doi: 10.1093/jpids/piy014
67. Tanaka M, Taniguchi K, Miki S, Iwanari S, Ikeda M, Hasui M, et al. Rapidly progressive IgA vasculitis-associated nephritis successfully treated with immunosuppressive therapy in an adolescent with chronic granulomatous disease. CEN Case Rep. (2021) 10:461–7. doi: 10.1007/s13730-021-00586-x
68. Chiriaco M, De Matteis A, Cifaldi C, Di Matteo G, Rivalta B, Passarelli C, et al. Characterization of AR-CGD female patient with a novel homozygous deletion in CYBC1 gene presenting with unusual clinical phenotype. Clin Immunol. (2023) 251:109316. doi: 10.1016/j.clim.2023.109316
69. Vignesh P, Sil A, Aggarwal R, Laha W, Mondal S, Dhaliwal M, et al. Tuberculosis and bacillus calmette-guérin disease in patients with chronic granulomatous disease: an experience from a tertiary care center in north India. J Clin Immunol. (2023) 43:2049–61. doi: 10.1007/s10875-023-01581-w
70. Lekstrom-Himes JA, Gallin JI. Immunodeficiency diseases caused by defects in phagocytes. New Engl J Med. (2000) 343:1703–14. doi: 10.1056/NEJM200012073432307
71. De Ravin SS, Naumann N, Robinson MR, Barron KS, Kleiner DE, Ulrick J, et al. Sarcoidosis in chronic granulomatous disease. Pediatrics. (2006) 117:e590–5. doi: 10.1542/peds.2005-1349
72. Facco PU, Barsioti M, Martin C, Sementilli A, Rocha LF, Ciaccia MCC, et al. 2017 LASID meeting abstracts. J Clin Immunol. (2017) 37(Suppl 1):1–74. doi: 10.1007/s10875-017-0442-2
73. Huang JS, Noack D, Rae J, Ellis BA, Newbury R, Pong AL, et al. Chronic granulomatous disease caused by a deficiency in p47(phox) mimicking Crohn’s disease. Clin Gastroenterol Hepatol. (2004) 2:690–5. doi: 10.1016/s1542-3565(04)00292-7
74. Ramanuja S, Wolf KM, Sadat MA, Mahoney SJ, Dinauer MC, Nelson RP. Newly diagnosed chronic granulomatous disease in a 53-year-old woman with Crohn disease. Ann Allergy Asthma Immunol. (2005) 95:204–9. doi: 10.1016/S1081-1206(10)61212-4
75. LaBere B, Gutierrez MJ, Wright H, Garabedian E, Ochs HD, Fuleihan RL, et al. Chronic granulomatous disease with inflammatory bowel disease: clinical presentation, treatment, and outcomes from the USIDNET registry. J Allergy Clin Immunol Pract. (2022) 10:1325–33.e5. doi: 10.1016/j.jaip.2021.12.035
76. Grenier PA, Brun AL, Longchampt E, Lipski M, Mellot F, Catherinot E. Primary immunodeficiency diseases of adults: a review of pulmonary complication imaging findings. Eur Radiol. (2023) 34(6):4142–54. doi: 10.1007/s00330-023-10334-7
77. Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, et al. Hyper-IgE syndrome with recurrent infections: an autosomal dominant multisystem disorder. New Engl J Med. (1999) 340:692–702. doi: 10.1056/NEJM199903043400904
78. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. (2007) 448:1058–62. doi: 10.1038/nature06096
79. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. (2007) 357:1608–19. doi: 10.1056/NEJMoa073687
80. Picard C, Puel A, Bonnet M, Ku C-L, Bustamante J, Yang K, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. (2003) 299:2076–9. doi: 10.1126/science.1081902
81. von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku C-L, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. (2008) 321:691–6. doi: 10.1126/science.1158298
82. Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore). (2010) 89:403–25. doi: 10.1097/MD.0b013e3181fd8ec3
83. Enders A, Pannicke U, Berner R, Henneke P, Radlinger K, Schwarz K, et al. Two siblings with lethal pneumococcal meningitis in a family with a mutation in interleukin-1 receptor-associated kinase 4. J Pediatr. (2004) 145:698–700. doi: 10.1016/j.jpeds.2004.06.065
84. von Bernuth H, Puel A, Ku C-L, Yang K, Bustamante J, Chang H-H, et al. Septicemia without sepsis: inherited disorders of nuclear factor-kappa B-mediated inflammation. Clin Infect Dis. (2005) 41(Suppl 7):S436–9. doi: 10.1086/431994
85. Kallinich T, Kölsch U, Lieber M, Unterwalder N, Spors B, Lorenz M, et al. Septic arthritis or juvenile idiopathic arthritis: the case of a 2 year old boy. Pediatr Allergy Immunol. (2015) 26:389–91. doi: 10.1111/pai.12373
86. Nathan DG, Baehner RL, Weaver DK. Failure of nitro blue tetrazolium reduction in the phagocytic vacuoles of leukocytes in chronic granulomatous disease. J Clin Invest. (1969) 48:1895–904. doi: 10.1172/JCI106156
87. Emmendörffer A, Nakamura M, Rothe G, Spiekermann K, Lohmann-Matthes ML, Roesler J. Evaluation of flow cytometric methods for diagnosis of chronic granulomatous disease variants under routine laboratory conditions. Cytometry. (1994) 18:147–55. doi: 10.1002/cyto.990180306
88. Blancas-Galicia L, Santos-Chávez E, Deswarte C, Mignac Q, Medina-Vera I, León-Lara X, et al. Genetic, immunological, and clinical features of the first Mexican cohort of patients with chronic granulomatous disease. J Clin Immunol. (2020) 40:475–93. doi: 10.1007/s10875-020-00750-5
89. Qureshi S, Mir F, Junejo S, Saleem K, Zaidi S, Naveed AB, et al. The spectrum of primary immunodeficiencies at a tertiary care hospital in Pakistan. World Allergy Organ J. (2020) 13:100133. doi: 10.1016/j.waojou.2020.100133
90. Mondal S, Vignesh P, Loganathan SK, Arora K, Das J, Rawat A, et al. Case report: chronic granulomatous disease presenting with early-onset inflammatory bowel disease and normal oxidative burst testing. Front Pediatr. (2022) 10:964025. doi: 10.3389/fped.2022.964025
91. Suematsu M, Suzuki M, Miura S, Nagata H, Oshio C, Asakura H, et al. Sulfasalazine and its metabolites attenuate respiratory burst of leukocytes: a possible mechanism of anti-inflammatory effects. J Clin Lab Immunol. (1987) 23:31–3.2886669
92. Costa D, Marques AP, Reis RL, Lima JLFC, Fernandes E. Inhibition of human neutrophil oxidative burst by pyrazolone derivatives. Free Radic Biol Med. (2006) 40:632–40. doi: 10.1016/j.freeradbiomed.2005.09.017
93. Almutairi A, Zaman F, Day-Lewis M, Tsitsikov E, Reiter A, Xue K, et al. Acetaminophen inhibits the neutrophil oxidative burst: implications for diagnostic testing. J Allergy Clin Immunol Pract. (2020) 8:3543–8. doi: 10.1016/j.jaip.2020.07.012
94. Milligan KL, Mann D, Rump A, Anderson VL, Hsu AP, Kuhns DB, et al. Complete myeloperoxidase deficiency: beware the “false-positive” dihydrorhodamine oxidation. J Pediatr. (2016) 176:204–6. doi: 10.1016/j.jpeds.2016.05.047
95. Margolis DM, Melnick DA, Alling DW, Gallin JI. Trimethoprim-sulfamethoxazole prophylaxis in the management of chronic granulomatous disease. J Infect Dis. (1990) 162:723–6. doi: 10.1093/infdis/162.3.723
96. Hauck F, Koletzko S, Walz C, von Bernuth H, Klenk A, Schmid I, et al. Diagnostic and treatment options for severe IBD in female X-CGD carriers with non-random X-inactivation. J Crohns Colitis. (2016) 10:112–5. doi: 10.1093/ecco-jcc/jjv186
97. Wu C-Y, Chen Y-C, Lee W-I, Huang J-L, Chen L-C, Ou L-S, et al. Clinical features of female Taiwanese carriers with X-linked chronic granulomatous disease from 2004 to 2019. J Clin Immunol. (2021) 41:1303–14. doi: 10.1007/s10875-021-01055-x
98. Zhang Y, Shu Z, Li Y, Piao Y, Sun F, Han T, et al. X-linked chronic granulomatous disease secondary to skewed X-chromosome inactivation in female patients. Clin Exp Immunol. (2024) 215:261–7. doi: 10.1093/cei/uxad129
99. The International Chronic Granulomatous Disease Cooperative Study Group. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N Engl J Med. (1991) 324:509–16. doi: 10.1056/NEJM199102213240801
100. Condino-Neto A, Newburger PE. Interferon-gamma improves splicing efficiency of CYBB gene transcripts in an interferon-responsive variant of chronic granulomatous disease due to a splice site consensus region mutation. Blood. (2000) 95:3548–54. doi: 10.1182/blood.V95.11.3548
101. Lugo Reyes SO, González Garay A, González Bobadilla NY, Rivera Lizárraga DA, Madrigal Paz AC, Medina-Torres EA, et al. Efficacy and safety of interferon-gamma in chronic granulomatous disease: a systematic review and meta-analysis. J Clin Immunol. (2023) 43:578–84. doi: 10.1007/s10875-022-01391-6
102. Roesler J, Koch A, Pörksen G, von Bernuth H, Brenner S, Hahn G, et al. Benefit assessment of preventive medical check-ups in patients suffering from chronic granulomatous disease (CGD). J Eval Clin Pract. (2005) 11:513–21. doi: 10.1111/j.1365-2753.2005.00584.x
103. Hemsworth S, Pizer B. Pet ownership in immunocompromised children: a review of the literature and survey of existing guidelines. Eur J Oncol Nurs. (2006) 10:117–27. doi: 10.1016/j.ejon.2005.08.001
104. Magnani A, Mahlaoui N. Managing inflammatory manifestations in patients with chronic granulomatous disease. Paediatr Drugs. (2016) 18:335–45. doi: 10.1007/s40272-016-0182-4
105. BMJ Best Practice. Chronic granulomatous disease: symptoms, diagnosis and treatment. (2022). Available online at: https://bestpractice.bmj.com/topics/en-gb/703 (accessed May 8, 2024).
106. Uzel G, Orange JS, Poliak N, Marciano BE, Heller T, Holland SM. Complications of tumor necrosis factor-α blockade in chronic granulomatous disease-related colitis. Clin Infect Dis. (2010) 51:1429–34. doi: 10.1086/657308
107. Conrad A, Neven B, Mahlaoui N, Suarez F, Sokol H, Ruemmele FM, et al. Infections in patients with chronic granulomatous disease treated with tumor necrosis factor alpha blockers for inflammatory complications. J Clin Immunol. (2021) 41:185–93. doi: 10.1007/s10875-020-00901-8
108. Hahn KJ, Ho N, Yockey L, Kreuzberg S, Daub J, Rump A, et al. Treatment with Anakinra, a recombinant IL-1 receptor antagonist, unlikely to induce lasting remission in patients with CGD colitis. Am J Gastroenterol. (2015) 110:938–9. doi: 10.1038/ajg.2015.135
109. Bhattacharya S, Marciano BE, Malech HL, Quezado M, Holland SM, De Ravin SS, et al. Safety and efficacy of ustekinumab in the inflammatory bowel disease of chronic granulomatous disease. Clin Gastroenterol Hepatol. (2022) 20:461–4.e2. doi: 10.1016/j.cgh.2021.03.039
110. Cole T, McKendrick F, Titman P, Cant AJ, Pearce MS, Cale CM, et al. Health related quality of life and emotional health in children with chronic granulomatous disease: a comparison of those managed conservatively with those that have undergone haematopoietic stem cell transplant. J Clin Immunol. (2013) 33:8–13. doi: 10.1007/s10875-012-9758-0
111. Dunogué B, Pilmis B, Mahlaoui N, Elie C, Coignard-Biehler H, Amazzough K, et al. Chronic granulomatous disease in patients reaching adulthood: a nationwide study in France. Clin Infect Dis. (2017) 64:767–75. doi: 10.1093/cid/ciw837
112. Horwitz ME, Barrett AJ, Brown MR, Carter CS, Childs R, Gallin JI, et al. Treatment of chronic granulomatous disease with nonmyeloablative conditioning and a T-cell-depleted hematopoietic allograft. N Engl J Med. (2001) 344:881–8. doi: 10.1056/NEJM200103223441203
113. Seger RA, Gungor T, Belohradsky BH, Blanche S, Bordigoni P, Di Bartolomeo P, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985–2000. Blood. (2002) 100:4344–50. doi: 10.1182/blood-2002-02-0583
114. Åhlin A, Fugeläng J, de Boer M, Ringden O, Fasth A, Winiarski J. Chronic granulomatous disease-haematopoietic stem cell transplantation versus conventional treatment. Acta Paediatr. (2013) 102:1087–94. doi: 10.1111/apa.12384
115. Yonkof JR, Gupta A, Fu P, Garabedian E, Dalal J, the United States Immunodeficiency Network Consortium. Role of allogeneic hematopoietic stem cell transplant for chronic granulomatous disease (CGD): a report of the United States immunodeficiency network. J Clin Immunol (2019) 39:448–58. doi: 10.1007/s10875-019-00635-2
116. Güngör T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. (2014) 383:436–48. doi: 10.1016/S0140-6736(13)62069-3
117. Morillo-Gutierrez B, Beier R, Rao K, Burroughs L, Schulz A, Ewins A-M, et al. Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience. Blood. (2016) 128:440–8. doi: 10.1182/blood-2016-03-704015
118. Parta M, Kelly C, Kwatemaa N, Theobald N, Hilligoss D, Qin J, et al. Allogeneic reduced-intensity hematopoietic stem cell transplantation for chronic granulomatous disease: a single-center prospective trial. J Clin Immunol. (2017) 37:548–58. doi: 10.1007/s10875-017-0422-6
119. Lum SH, Flood T, Hambleton S, McNaughton P, Watson H, Abinun M, et al. Two decades of excellent transplant survival for chronic granulomatous disease: a supraregional immunology transplant center report. Blood. (2019) 133:2546–9. doi: 10.1182/blood.2019000021
120. Chiesa R, Wang J, Blok H-J, Hazelaar S, Neven B, Moshous D, et al. Hematopoietic cell transplantation in chronic granulomatous disease: a study of 712 children and adults. Blood. (2020) 136:1201–11. doi: 10.1182/blood.2020005590
121. Tsilifis C, Torppa T, Williams EJ, Albert MH, Hauck F, Soncini E, et al. Allogeneic HSCT for symptomatic female X-linked chronic granulomatous disease carriers. J Clin Immunol. (2023) 43:1964–73. doi: 10.1007/s10875-023-01570-z
122. Scheiermann J, Künkele A, von Stackelberg A, Eggert A, Lang P, Zirngibl F, et al. Case report: HLA-haploidentical HSCT rescued with donor lymphocytes infusions in a patient with X-linked chronic granulomatous disease. Front Immunol. (2023) 14:1042650. doi: 10.3389/fimmu.2023.1042650
123. Keller MD, Notarangelo LD, Malech HL. Future of care for patients with chronic granulomatous disease: gene therapy and targeted molecular medicine. J Pediatr Infect Dis Soc. (2018) 7:S40. doi: 10.1093/jpids/piy011
124. Kohn DB, Booth C, Kang EM, Pai S-Y, Shaw KL, Santilli G, et al. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat Med. (2020) 26:200–6. doi: 10.1038/s41591-019-0735-5
125. Hoenig M, Niehues T, Siepermann K, Jacobsen E-M, Schütz C, Furlan I, et al. Successful HLA haploidentical hematopoietic SCT in chronic granulomatous disease. Bone Marrow Transplant. (2014) 49:1337–8. doi: 10.1038/bmt.2014.125
126. Parta M, Hilligoss D, Kelly C, Kwatemaa N, Theobald N, Malech H, et al. Haploidentical hematopoietic cell transplantation with post-transplant cyclophosphamide in a patient with chronic granulomatous disease and active infection: a first report. J Clin Immunol. (2015) 35:675–80. doi: 10.1007/s10875-015-0204-y
127. Zhou L, Dong L-J, Gao Z-Y, Yu X-J, Lu D-P. Haploidentical hematopoietic stem cell transplantation for a case with X-linked chronic granulomatous disease. Pediatr Transplant. (2017) 21:e12861. doi: 10.1111/petr.12861
128. Leiding JW, Arnold DE, Parikh S, Logan B, Marsh RA, Griffith LM, et al. Genotype, oxidase status, and preceding infection or autoinflammation do not affect allogeneic HCT outcomes for CGD. Blood. (2023) 142:2105–18. doi: 10.1182/blood.2022019586
129. Güngör T, Chiesa R. Cellular therapies in chronic granulomatous disease. Front Pediatr. (2020) 8:327. doi: 10.3389/fped.2020.00327
Keywords: chronic granolumatous disease, diagnosis, clinical presentation, HSCT, hematopoietic stem cell transplantation, therapy
Citation: Staudacher O and von Bernuth H (2024) Clinical presentation, diagnosis, and treatment of chronic granulomatous disease. Front. Pediatr. 12:1384550. doi: 10.3389/fped.2024.1384550
Received: 9 February 2024; Accepted: 14 June 2024;
Published: 28 June 2024.
Edited by:
Catharina Schuetz, University Hospital Carl Gustav Carus, GermanyReviewed by:
Christa Zerbe, Clinical Center (NIH), United States© 2024 Staudacher and von Bernuth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Horst von Bernuth, aG9yc3Qudm9uLWJlcm51dGhAY2hhcml0ZS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.