 Amal Y. Kentab1,2*
Amal Y. Kentab1,2* Yara Alsalloum2
Yara Alsalloum2 Mai Labani3
Mai Labani3 Abrar Hudairi2
Abrar Hudairi2 Muddathir H. Hamad2
Muddathir H. Hamad2 Dima Z. Jamjoom4
Dima Z. Jamjoom4 Ali H. Alwadei2,5
Ali H. Alwadei2,5 Reem M. Alhammad6
Reem M. Alhammad6 Fahad A. Bashiri1,2
Fahad A. Bashiri1,2
- 1Division of Pediatric Neurology, Department of Pediatrics, College of Medicine, King Saud University, Riyadh, Saudi Arabia
- 2Department of Pediatrics, King Saud University Medical City, Riyadh, Saudi Arabia
- 3Pediatric Intensive Care Unit, Department of Pediatrics, King Khalid University Hospital, King Saud University Medical City, Riyadh, Saudi Arabia
- 4Department of Radiology and Medical Imaging, College of Medicine, King Saud University, Riyadh, Saudi Arabia
- 5Pediatric Neurology Department, National Neuroscience Institute, King Fahd Medical City, Riyadh, Saudi Arabia
- 6Department of Internal Medicine, College of Medicine, King Saud University, Riyadh, Saudi Arabia
Background: Brown-Vialetto-Van Laere (BVVL) syndrome is an extremely rare autosomal recessive progressive motoneuron disease that is caused by a defect in the riboflavin transporter genes SLC52A2 and SLC52A3. BVVL syndrome has a variable age of presentation, and it is characterized by progressive auditory neuropathy, bulbar palsy, stridor, muscle weakness, and respiratory compromise secondary to diaphragmatic and vocal cord paralysis. BVVL syndrome has a poor prognosis in the absence of treatment, including morbidity with quadriparesis and sensorineural hearing loss, with mortality in the younger age group. Early administration of riboflavin is associated with prolonged survival, low morbidity, and reversal of some clinical manifestations.
Case presentation: We describe an 18-month-old male infant with progressive pontobulbar palsy, loss of developmental milestones, and a clinical picture suggestive of chronic inflammatory demyelinating neuropathy. A nerve conduction study revealed axonal neuropathy, while molecular analysis revealed a homozygous mutation in one of the riboflavin transporter genes, SLC52A3, confirming BVVL syndrome. The patient needed long-term respiratory support and a gastrostomy tube to support feeding. With high-dose riboflavin supplementation, he experienced moderate recovery of motor function.
Conclusion: This report highlights the importance of considering BVVL syndrome in any patient who presents with the clinical phenotype of pontobulbar palsy and peripheral axonal neuropathy, as early riboflavin treatment may improve or halt disease progression, thus reducing the associated mortality and morbidity.
Introduction
Brown-Vialetto-Van Laere (BVVL) syndrome is a rare progressive motor neuron disorder with an autosomal recessive inheritance that results from a defect in riboflavin transporter genes; SLC52A2, SLC52A3 under the umbrella of recently called Riboflavin Transporter Deficiency (RTD) (1). It was first reported by Dr. Charles Henry Brown (1894) (2) as a form of familial infantile amyotrophic lateral sclerosis. Later on, further familial cases with an autosomal recessive pattern of inheritance and female predominance were reported in the literature (3, 4). Classically characterized by sensorineural hearing loss (SNHL), progressive pontobulbar palsy involving the motor components of the seventh and ninth to twelfth cranial nerves, progressive muscle weakness causing respiratory compromise, limb weakness, and upper motor signs. Disease onset usually occurs in the second decade of life, but earlier and later ages of onset have been reported (3). Symptoms, severity, and disease duration are variable. BVVL syndrome exhibits genetic heterogeneity; BVVL1 (OMIM # 211530) is caused by homozygous or compound heterozygous mutations in the SLC52A3 gene (OMIM #613350) on chromosome 20p13 (5), whereas BVVL2 (OMIM #614707) is caused by a mutation in the SLC52A2 gene (OMIM #607882) on chromosome 8q. Fazio Londe syndrome (OMIM #211500) is another RTD syndrome with a similar clinical phenotype to BVVL syndrome but without sensorineural deafness (3, 4, 6).
BVVL syndrome can mimic chronic inflammatory demyelinating neuropathy and other neuroimmune disorders, as it may present with positive cerebrospinal fluid autoantibody titers and may transiently respond to intravenous immunoglobulin (7–9). An excellent response to treatment with high doses of riboflavin was reported in the literature, with the arrest of the disease in the majority of patients and the reversal of symptom progression in some patients (5, 6, 10–12).
Herein, we report a case of BVVL1 (RTD3) with a clinical phenotype masquerading as chronic inflammatory demyelinating polyneuropathy (CIDP) at presentation, highlighting the importance of early recognition, genetic diagnosis, and riboflavin introduction to halt progression, accelerate recovery, and reduce associated morbidity and mortality.
Case presentation
Patient information
The patient was an 18-month-old boy who was a product of a first-degree consanguineous marriage with an uneventful pregnancy and an uncomplicated elective cesarean section at full term. He had a normal neonatal period. His developmental milestones, mainly motor and speech, were mildly delayed; he sat at age 7 months, crawled at 10 months, and stood with support at 1 year. Currently, he can stand with support and can vocalize only two words. There was no family history of spontaneous abortion or neurogenetic disorders. His father had multiple sclerosis.
He presented with insidious-onset progressive stridor (obvious during crying and feeding) and difficulty swallowing for 2 months, preceded by fever and upper respiratory tract infection (URTI) for 5 days. Afterward, he was noted to have impaired sucking, drooling of saliva, frequent choking, changes in his facial expressions, drooping of the right eyelid and less interaction with episodes of retrocollis and weight loss. He was inattentive most of the time, with a gradual decrease in verbal output and the use of nonverbal communication to indicate needs. No alteration of sensorium was observed.
At 16 months, he was evaluated by an ear, nose and throat (ENT) specialist who observed evidence of tracheal tag, subcostal retraction and biphasic stridor; however, nasopharyngeal scope, direct laryngoscopy and bronchoscopy (DLB), were unremarkable, ruling out the possibility of foreign body inhalation. Esophagoscopy was also normal.
Clinical findings
Examination revealed an active, alert, and thriving child with normal vital signs. Intermittent soft stridor with no grunting was observed at rest. The ENT exam results were normal with no lymphadenopathy. Cranial nerve examination showed no ptosis of the left eye with normal pupils, fundus, and extraocular movements. He had bilateral lower motor neuron (LMN) facial (VII) nerve palsy, which was more pronounced on the right side; weak palatal and gag reflexes with excessive drooling; no uvular deviation; and normal tongue movements. He had a reduced response to loud sounds by simple distraction test. He had decreased strength and tone in both the upper and lower limbs and depressed deep tendon reflexes (DTRs) in the lower limbs. The plantar reflex was bilaterally flexor. His gait was unsteady with obvious hand tremors. No paradoxical abdominal movements when breathing was noted. The remainder of his systemic examination results were unremarkable (Table 1).
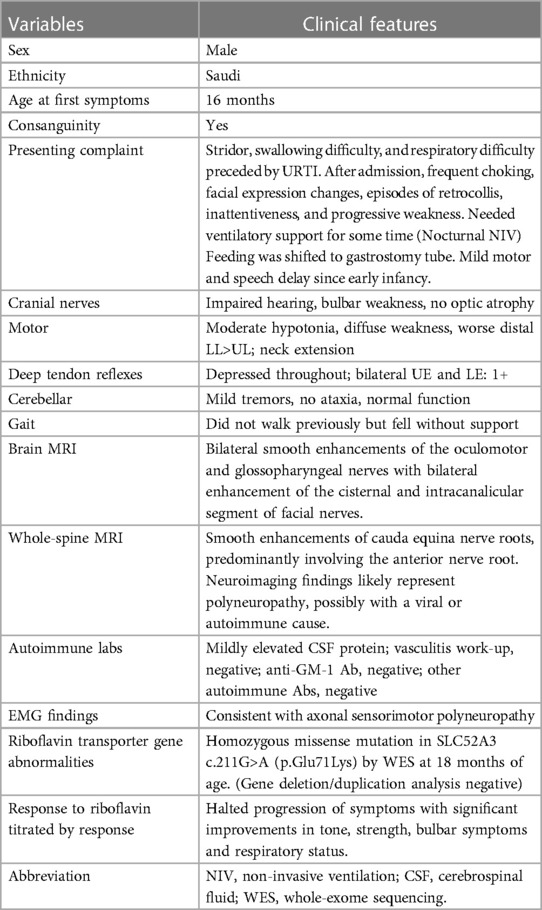
Table 1. Clinical characteristics and laboratory profile of a patient with Brown-Vialetto-Van Laere syndrome caused by a mutation in SLC52A3.
Diagnostic assessment
Based on the findings of subacute-onset multiple cranial neuropathies, specifically pontobulbar palsy and impaired hearing preceded by viral illness, the diagnoses listed in (Table 2) were considered after ruling out the possibility of posterior fossa/cerebellopontine angle mass lesion.
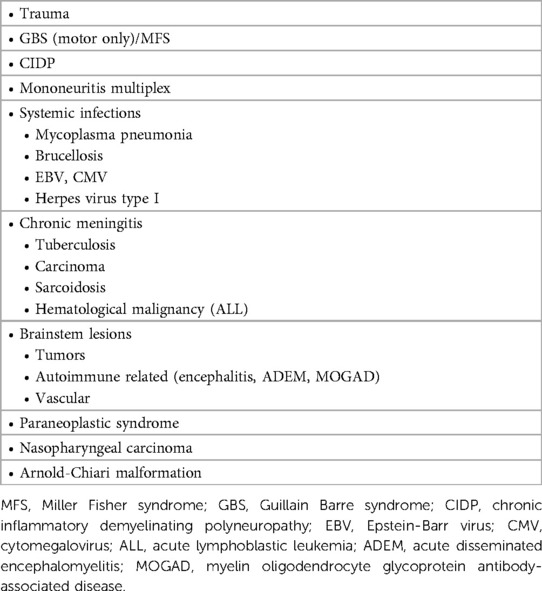
Table 2. Etiology of multiple cranial nerve lesions/enhancement.
The patient's basic hemogram, peripheral smear, and biochemistry lab results were normal. His throat swab and nasopharyngeal aspirate for common viruses were negative. Magnetic resonance imaging (MRI) with contrast revealed bilateral smooth enhancement of the oculomotor and glossopharyngeal nerves, with a possible bilateral enhancement of the cisternal and intracanalicular segments of the facial nerves (Figure 1). A whole-spine MRI with contrast showed cauda equina enhancement suggestive of chronic inflammatory demyelinating disorder (Figure 2). Cerebrospinal fluid (CSF) analysis was unremarkable with a slightly elevated protein level. The metabolic panel, including venous blood gas (VBG), serum ammonia, lactate, tandem amino acids, and urinary organic acids, was unremarkable. His thyroid panel and vitamin B12 level were normal. Anti-streptolysin O titer (ASO), mycoplasma, brucella titers, and vasculitis workup were negative. Bone marrow aspiration results were unremarkable. The findings of the electromyogram (EMG) and nerve conduction study (NCS) of the right upper and lower extremities were suggestive of a non-length dependent, predominantly axonal, sensorimotor polyneuropathy with features of ongoing denervation in the distal right upper extremity muscles (Tables 1, 3). Brain Auditory Evoked Potential (BAEP) later showed bilateral sensorineural hearing loss, while ophthalmological evaluation and visual evoked potential (VEP) were unremarkable.
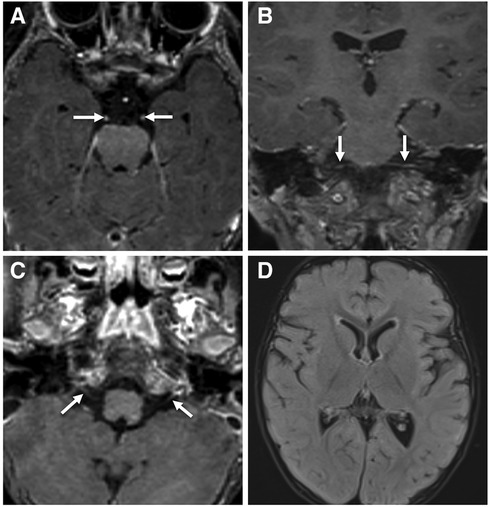
Figure 1. Contrast-enhanced brain MRI images acquired at 18-months of age demonstrate smooth bilateral enhancement of multiple cranial nerves, including the oculomotor nerves, cisternal and intracanalicular segments of the facial nerves and the glossopharyngeal nerves (white arrows in A–C). Axial FLAIR image (D) shows no signal abnormality in the brain parenchyma.

Figure 2. Contrast enhanced MRI of the lumbar spine obtained at the same time of the brain MRI shows smooth enhancement of the cauda equina nerve roots, particularly the anterior nerve fibres (white arrowheads in A–C).
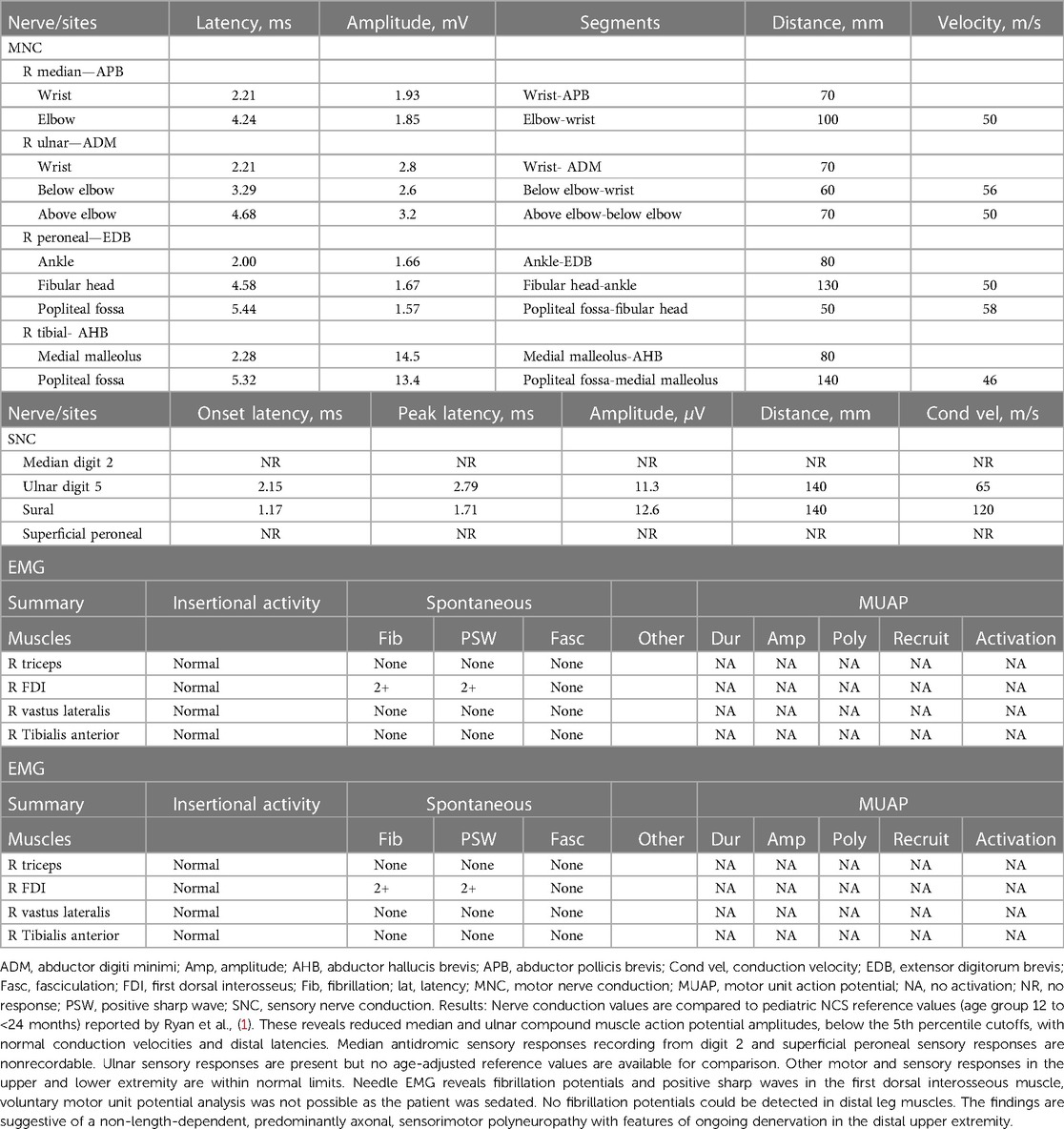
Table 3. Summary of electromyographic (EMG) and nerve conduction study (NCS) findings in this patient at time of admission).
Therapeutic intervention
The patient was started on intravenous immunoglobulin (IVIG) 2 g/kg over two consecutive days with no obvious improvement, and empirical mega-multivitamins were added, including riboflavin 200 mg/day (20 mg/kg). During his admission, he suffered arrest twice secondary to recurrent lung aspiration, received different modalities of ventilatory support and was kept in the intensive care unit (ICU) for close observation. He had a complex clinical course in the ICU for more than two months with fluctuations in his respiratory status and a need for oxygen before finalizing his diagnosis. A trial of atropine drops was administered for excessive drooling, and botulinum toxin was injected into the salivary glands. Paradoxical breathing and tachypnea were observed in the ICU. x-ray of the chest showed an eventration of the left diaphragm, and fluoroscopy confirmed diaphragmatic paralysis, probably secondary to phrenic nerve involvement, which was managed conservatively. Finally, a gastrostomy tube was inserted to minimize choking episodes. His carnitine profile showed normal levels of total carnitine, very low levels of free carnitine (7.88 μmol/L; normal range, 24.7–66.6 μmol/L) and a normal acylcarnitine profile. The possibility of motor neuron disease was considered, and the diagnosis of BVVL1 was confirmed by rapid clinical whole-exome sequencing (WES), which identified a previously reported homozygous missense mutation, NM_033409.3: c.211G>A: p. Glu71Lys, in the SLC52A3 gene (4), which resulted in the amino acid substitution of glutamic acid with lysine. No mutations were found in the SLC52A1 and SLC52A2 genes. The patient's parents were heterozygous carriers and were asymptomatic. The genetic result was published previously (13) as a part of the mass screen by Rapid WES for critically sick patients admitted to the intensive care unit. Accordingly, coenzyme Q10 supplementation was initiated, and riboflavin was increased to 400 mg (80 mg/kg/d) in two divided doses with obvious clinical improvement in facial weakness and palatal movement.
Follow-up and outcomes
After 3 months of follow-up on riboflavin, the child had a normal gag reflex, accepted sips of water orally, and had mild facial weakness. He has started vocalizing, and subjectively, there is some response to high-frequency sounds.
A follow-up brain MRI at 22 months showed no further enhancement of the cranial nerves or cauda equina and no evidence of acute hypoxic-ischemic insult secondary to repeated arrest, only mild cerebral atrophy (Figure 3). Clinically, he showed impressive improvement in interaction with and awareness of surroundings, facial expression, eye closure during the night, response to painful stimuli, and retention of a spontaneous cough and gag reflex. He was planned for a cochlear implant after further assessment by an ENT specialist. Figure 4 represents the timeline course for this patient.
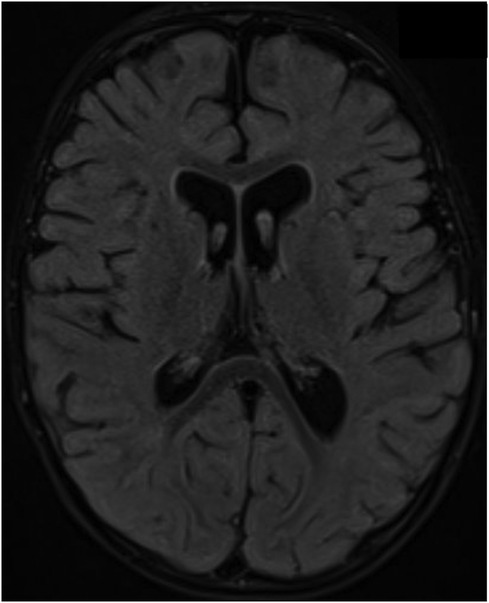
Figure 3. Brain MRI acquired at 19- months of age after an episode of cardiac arrest showed no evidence of hypoxic ischemic insult on the axial FLAIR image; however, there is evidence of mild brain volume loss with enlarged ventricles and prominent subarachnoid spaces.
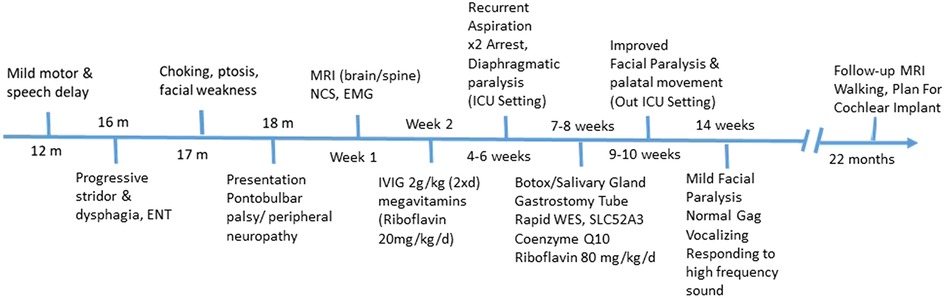
Figure 4. Diagnostic and patient management timeline. RTD, riboflavin transporter deficiency; CSF, cerebrospinal fluid; MRI, magnetic resonance imaging; EMG, electromyogram; NCS, nerve conduction study; WES, whole exome sequencing.
Discussion
Three human riboflavin transporters (RFVT) homologues have been identified: RFVT1-3 encoded by genes SLC52A1-3, respectively. SLC52A1 is mainly expressed in the placenta and intestine. SLC52A2 is ubiquitously expressed but is particularly abundant in nervous tissues. SLC52A3 is most highly expressed in testis but also intestine and prostate. Pathogenic mutations in SLC52A1 result in transient riboflavin deficiency with onset in the newborn children of mothers harboring one heterozygous SLC52A1 mutation (OMIM 615026), where the clinical symptoms are not typical of Riboflavin transporter deficiency (RTD) phenotype, and it usually subsides by 2 years of age (14). While, pathogenic mutations in the SLC52A2 and SLC52A3 (previously C20orf54) genes that encode human riboflavin transporters RFVT2 and RFVT3, respectively result in an RTD, which is a rare autosomal recessive motor neuron neurological disorder. More than one hundred RTD patients have been described to date (6).
Both Brown-Vialetto-Van Laere (BVVL) and Fazio-Londe (FL) syndromes are phenotypically continuous syndromes that lie under the umbrella of RTD caused by biallelic mutations in the human riboflavin transporter genes SLC52A2, and SLC52A3, and re-named RTD2 and RTD3, respectively (15, 16).
RTD runs a neurodegenerative course of inherited motor neuron disease with variable age of presentation, symptoms severity, and disease duration. It is characterized by both motor cranial neuropathy and sensorimotor peripheral neuropathy. Major manifestations include bulbar palsy (dysphagia, dysphonia, and tongue atrophy), facial palsy, neck and shoulder weakness, vision loss, deafness, progressive axial and distal muscle weakness, sensory ataxia, and respiratory compromise due to combined muscle weakness, and diaphragm paralysis. It is associated with high mortality in the very young age group if left untreated (10, 15, 17). Sensorineural deafness is present in BVVL only, and may not be obvious initially in twenty percent of patients (3). Other cranial nerve involvement (III, V, and VI) is less common. Less frequent manifestations include ocular abnormalities, autonomic dysfunction, epilepsy, and mental retardation (18).
Our patient had a previously reported SLC52A3 homozygous missense mutation causing clinical characteristics of RTD3, consistent with BVVL syndrome (4). Although both RTD2 and RTD3 are present early in life only RTD3 has a late onset (as late as the third decade) (19). Hearing loss, muscle weakness, and respiratory dysfunction are among the most common presenting symptoms at onset for both, but abnormal gait and/or ataxia is rarely a presenting feature of RTD3. RTD3, as in our patient commonly presents with early onset bulbar symptoms, and facial weakness whereas it is observed either later in the disease course or rarely in RTD2. Optic nerve atrophy causing nystagmus, blindness, SNHL, weakness of neck extension and upper limbs, early-onset ataxia, and sensory abnormalities are more prevalent features of RTD2 (6, 10, 18). Our patient had normal vision, but bilateral SNHL was clinically suspected based on poor attentiveness and decreased interest in the surroundings, as well as an inadequate response to various stimuli by his mother. Furthermore, he had abnormal BAEP results that showed bilateral SNHL. Based on the latest published literature, the most common clinical features of RTD3 were muscle weakness (RTD2 83%, RTD3 84%), hearing loss (RTD2 90%, RTD3 84%), respiratory distress (RTD2 53%, RTD3 73%), and bulbar palsy (RTD2 50%, RTD3 61%), usually presenting before 3 years of age (6).
In RTD neuropathy, patients predominantly present with motor neuropathy with loss of DTRs and SNHL (1), but can also present with predominantly sensory symptoms (20). The exact pathophysiology of RTD-related neuropathy is not known, but a mitochondrial dysfunction has been postulated as riboflavin is a precursor for several metabolites that are involved in the electron transport chain (5). A preceding viral illness or an infection with fever has been reported to precipitate the disease onset in some genetically predisposed patients (7, 21). Based on this observation, infectious or autoimmune factors have been postulated to play a role in the presentation of RTD-related neuropathy. Motor and sensory nerve conduction studies (NCS), and electromyogram (EMG) in most RTD cases will reveal an axonal rather than demyelinating neuropathic phenotype, with signs of anterior horn dysfunction, and chronic denervation. RTD3 cases may have normal NCS, and EMG results initially (6). Our patient had similar findings of sensorimotor abnormalities and predominantly axonal polyneuropathy with features of ongoing denervation.
MRI is usually normal in RTD (16), but RTD3 patients specifically may have hyperintense T2-weighted signals within cerebellar peduncles (22), cortical, subcortical (basal ganglia and internal capsule), and brainstem (vestibular nuclei and central tegmental tract) regions (22–24). In contrast, RTD2 may have mild atrophy of the cerebellar vermis (25), optic nerve abnormalities (26), and thinning/shortening of the corpus callosum (26). In general, Spinal MRI in RTD may show abnormal T2-weighted intensities in ventral nerve roots and dorsal regions of the spinal cord (22, 23, 27, 28). MRI in our patient showed multiple cranial nerves (3rd, 7th, and 9th) and cauda equina enhancement, not consistent with the expected changes seen on the brain MRI of RTD3 patients.
Acute inflammatory demyelinating polyradiculoneuropathies (AIDPs), autoimmune axonal motor neuropathy, juvenile-onset amyotrophic lateral sclerosis, myasthenia gravis, and mitochondrial myopathy should be considered in the differential diagnosis of RTD, especially in the presence of bulbar weakness, and absence of SNHL. Several patients received intravenous immunoglobulin (IVIG) and steroids prior to receiving autoantibody testing results (7, 29, 30), with some responding at least transiently to immunotherapies (7, 21). A unique response to riboflavin supplementation with reversal of some symptoms is a key difference distinguishing RTD from other disorders.
Based on the constellation of our patient's symptoms, and signs, a prodrome of viral illness, duration of illness, initial MRI findings suggesting acquired inflammatory autoimmune polyneuropathy and elevated CSF protein, Chronic inflammatory demyelinating polyneuropathy (CIDP) was suspected, and he was treated with IVIG accordingly. No improvement was observed, and as his autoimmune, vascular, and paraneoplastic panel results were negative, a trial of megavitamins including riboflavin was initiated at 20 mg/kg/day, but he responded to higher doses of 50 mg/kg/day, which was titrated further to 80 mg/kg/day after genetic confirmation of BVVL1, and initial stabilization of his rapidly progressive weakness, bulbar palsy, and the respiratory compromise was achieved on this dose.
Our patient is unique in his presentation compared with the three patients reported previously by Allison T, et al. (8), who had progressive weakness secondary to CIDP. The main manifestation in our patient was pontobulbar palsy with respiratory compromise rather than hypotonia or progressive peripheral weakness. One of the previously reported patients, a 3-year-old child, had a positive anti-GM1 antibody test (1:100), consistent with autoimmune-mediated motor neuropathy, which may have made it more difficult to associate hearing loss with the disease entity causing muscle weakness (8).These findings had been reported previously in this syndrome and led to an initial diagnosis of multifocal motor neuropathy (9) with a transient response to steroid and/or intravenous immunoglobulin treatment, which falsely confirmed the suspicions of autoimmune disorders (7, 9).
Multiple factors can potentially result in missing a treatable RTD, including positive serology for autoimmune antibodies (anti-LGl1, anti-GM1), clinical response to intravenous immunoglobulin, and lack of any history of hearing loss at presentation, which only occurs in approximately two-thirds of patients with RTD with onset prior to age 4.
Our patient had an initial low free carnitine level, while the plasma acylcarnitine profile was normal. Urine organic acid analysis was suggestive of ethylmalonic acidemia. These were consistent with the previously reported metabolic abnormalities in BVVL that occur secondary to impaired riboflavin absorption, which includes multiple acyl-CoA dehydrogenation defects (MADD) like- picture with elevated plasma acylcarnitine secondary to an impairment in the metabolism of fatty acids by mitochondrial β-oxidation, and urine organic acid analysis abnormalities in nearly half of the RTD cases. Ethylmalonic aciduria suggestive of impairments in fatty acid, methionine, and/or isoleucine oxidation is the most common abnormality noted (RTD2 4/10, RTD3 5/12) (6).
RTD is a treatable genetic disorder, where maintaining a high index of suspicion, and early institution of riboflavin is crucial to stop disease progression and reverse some clinical manifestations until another diagnosis has been confirmed or genetic testing for RTDs has returned negative results. Early high-dose oral riboflavin supplementation is effective in preventing mortality, and the need for longer ventilatory support as well as reducing morbidity related to deficits in cranial nerves, muscle strength, motor abilities, and respiratory function in over 70% of patients (10). Effective doses vary between 10 and 80 mg/kg /kg body weight per day, and titration of the dose is recommended based on the best patient`s response (6, 11, 12). A small dose of riboflavin 20 mg/kg/day was not effective with our patient as reported previously (1), but a high dose of 80 mg/kg/day was effective in reversing multiple deficits and maintaining a steady improvement over months. He started to walk while supported, and his speech as well as his ability to talk and swallow also improved. There was a subjective improvement only in hearing, and eventually, he needed cochlear implantation. The response and persistence of hearing loss posttreatment have not been well-studied in patients with BVVL syndrome, and the results of cochlear implants have not been encouraging (31). Interestingly, post-therapy the follow-up MRI showed the disappearance of previous findings, and the presence of mild generalized atrophy probably not related to hypoxia but similar to previously reported findings of the brainstem, and cerebellum atrophy detected in some patients with BVVL syndrome (32).
The disease has a variable course and prognosis, which is also influenced by the timing of initiation of riboflavin treatment. High-dose riboflavin was reported to produce clinical improvement in BVVL syndrome (30), and prolongation of survival (1). Without riboflavin treatment, a more severe disease course, irreversible morbidity, mainly quadriparesis and hearing loss, and early death were reported in those with younger age at presentation; the mean time to death was reported to be approximately 10 months in those presenting before 3 years of age, while the mean time to death was approximately 13 years in children presenting from 3 to 18 years of age even in patients with similar genetic mutations, probably due to unknown altered pathophysiological mechanisms.
In conclusion, although BVVL syndrome (riboflavin transporter disorders) is a rare inherited disorder, it should be suspected in the presence of pontobulbar palsy, progressive weakness, respiratory compromise, and motor cranial neuropathy even in the absence of hearing loss. Although riboflavin trials can help in early clinical diagnosis, mutational molecular analysis can confirm the genetic diagnosis and facilitate early lifesaving riboflavin treatment, genetic counseling, and future family planning.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to Amal Y. Kentab,YWtlbnRhYkBrc3UuZWR1LnNh.
Ethics statement
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
AK: Conceptualization, Data curation, Investigation, Methodology, Project administration, Supervision, Validation, Writing – review & editing. YS: Data curation, Formal Analysis, Methodology, Writing – original draft, Writing – review & editing. ML: Data curation, Formal Analysis, Methodology, Writing – original draft, Writing – review & editing. AH: Conceptualization, Data curation, Methodology, Writing – review & editing. MH: Conceptualization, Data curation, Methodology, Writing – review & editing. DJ: Data curation, Investigation, Writing – review & editing. AA: Conceptualization, Data curation, Methodology, Writing – review & editing. RH: Data curation, Investigation, Writing – review & editing. FB: Conceptualization, Data curation, Methodology, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bosch AM, Stroek K, Abeling NG, Waterham HR, Ijlst L, Wanders RJ. The Brown-Vialetto-Van Laere and fazio londe syndrome revisited: natural history, genetics, treatment and future perspectives. Orphanet J Rare Dis. (2012) 7:83. doi: 10.1186/1750-1172-7-83
2. Brown CH. Infantile amyotrophic lateral sclerosis of the family type. J Nerv Ment Dis. (1894) 19(11):707–16. doi: 10.1097/00005053-189411000-00003
3. Green P, Wiseman M, Crow YJ, Houlden H, Riphagen S, Lin JP, et al. Brown-Vialetto-Van Laere syndrome, a ponto-bulbar palsy with deafness, is caused by mutations in c20orf54. Am J Hum Genet. (2010) 86(3):485–9. doi: 10.1016/j.ajhg.2010.02.006
4. Johnson JO, Gibbs JR, Van Maldergem L, Houlden H, Singleton AB. Exome sequencing in Brown-Vialetto-Van Laere syndrome. Am J Hum Genet. (2010) 87(4):567–9. author reply 9-70, doi: 10.1016/j.ajhg.2010.05.021
5. Manole A, Jaunmuktane Z, Hargreaves I, Ludtmann MHR, Salpietro V, Bello OD, et al. Clinical, pathological and functional characterization of riboflavin-responsive neuropathy. Brain. (2017) 140(11):2820–37. doi: 10.1093/brain/awx231
6. O'Callaghan B, Bosch AM, Houlden H. An update on the genetics, clinical presentation, and pathomechanisms of human riboflavin transporter deficiency. J Inherit Metab Dis. (2019) 42(4):598–607. doi: 10.1002/jimd.12053
7. Ciccolella M, Catteruccia M, Benedetti S, Moroni I, Uziel G, Pantaleoni C, et al. Brown-Vialetto-Van Laere and fazio-londe overlap syndromes: a clinical, biochemical and genetic study. Neuromuscul Disord. (2012) 22(12):1075–82. doi: 10.1016/j.nmd.2012.05.007
8. Allison T, Roncero I, Forsyth R, Coffman K, Pichon JL. Brown-Vialetto-Van Laere syndrome as a mimic of neuroimmune disorders: 3 cases from the clinic and review of the literature. J Child Neurol. (2017) 32(6):528–32. doi: 10.1177/0883073816689517
9. Sztajzel R, Kohler A, Reichart M, Djientcheu VP, Chofflon M, Magistris MR. Brown-Vialetto-Van Laere syndrome: a case with anti-ganglioside GM1 antibodies and literature review. Rev Neurol (Paris). (1998) 154(1):51–4. 9773026.9773026
10. Jaeger B, Bosch AM. Clinical presentation and outcome of riboflavin transporter deficiency: mini review after five years of experience. J Inherit Metab Dis. (2016) 39(4):559–64. doi: 10.1007/s10545-016-9924-2
11. Chaya S, Zampoli M, Gray D, Booth J, Riordan G, Ndondo A, et al. The first case of riboflavin transporter deficiency in Sub-Saharan Africa. Semin Pediatr Neurol. (2018) 26:10–4. doi: 10.1016/j.spen.2017.03.002
12. Forman EB, Foley AR, King MD. Dramatic improvement of a rare syndrome with high dose riboflavin treatment. Pediatr Neurol. (2018) 86:77–8. doi: 10.1016/j.pediatrneurol.2018.05.005
13. Monies D, Goljan E, Assoum M, Albreacan M, Binhumaid F, Subhani S, et al. The clinical utility of rapid exome sequencing in a consanguineous population. Genome Med. (2023) 15(1):44. doi: 10.1186/s13073-023-01192-5
14. Ho G, Yonezawa A, Masuda S, Inui K, Sim KG, Carpenter K, et al. Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum Mutat. (2011) 32(1):E1976–84. doi: 10.1002/humu.21399
15. Manole A, Houlden H. Riboflavin Transporter Deficiency Neuronopathy. Seattle, Washington: University of Washington (2015).
16. Foley AR, Menezes MP, Pandraud A, Gonzalez MA, Al-Odaib A, Abrams AJ, et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain. (2014) 137(Pt 1):44–56. doi: 10.1093/brain/awt315
17. Horvath R. Update on clinical aspects and treatment of selected vitamin-responsive disorders II (riboflavin and CoQ 10). J Inherit Metab Dis. (2012) 35(4):679–87. doi: 10.1007/s10545-011-9434-1
18. Sathasivam S. Brown-Vialetto-Van Laere syndrome. Orphanet J Rare Dis. (2008) 3:9. doi: 10.1186/1750-1172-3-9
19. Camargos S, Guerreiro R, Bras J, Mageste LS. Late-onset and acute presentation of Brown-Vialetto-Van Laere syndrome in a Brazilian family. Neurol Genet. (2018) 4(1):e215. doi: 10.1212/nxg.0000000000000215
20. Srour M, Putorti ML, Schwartzentruber J, Bolduc V, Shevell MI, Poulin C, et al. Mutations in riboflavin transporter present with severe sensory loss and deafness in childhood. Muscle Nerve. (2014) 50(5):775–9. doi: 10.1002/mus.24224
21. Bandettini Di Poggio M, Monti Bragadin M, Reni L, Doria-Lamba L, Cereda C, Pardini M, et al. Brown-Vialetto-Van Laere syndrome: clinical and neuroradiological findings of a genetically proven patient. Amyotroph Lateral Scler Frontotemporal Degener. (2014) 15(1-2):141–4. doi: 10.3109/21678421.2013.837931
22. Spagnoli C, Pitt MC, Rahman S, de Sousa C. Brown-Vialetto-Van Laere syndrome: a riboflavin responsive neuronopathy of infancy with singular features. Eur J Paediatr Neurol. (2014) 18(2):231–4. doi: 10.1016/j.ejpn.2013.09.006
23. Koy A, Pillekamp F, Hoehn T, Waterham H, Klee D, Mayatepek E, et al. Brown-Vialetto-Van Laere syndrome: a riboflavin-unresponsive patient with a novel mutation in the C20orf54 gene. Pediatr Neurol. (2012) 46(6):407–9. doi: 10.1016/j.pediatrneurol.2012.03.008
24. Nimmo GAM, Ejaz R, Cordeiro D, Kannu P, Mercimek-Andrews S. Riboflavin transporter deficiency mimicking mitochondrial myopathy caused by complex II deficiency. Am J Med Genet A. (2018) 176(2):399–403. doi: 10.1002/ajmg.a.38530
25. Petrovski S, Shashi V, Petrou S, Schoch K, McSweeney KM, Dhindsa RS, et al. Exome sequencing results in successful riboflavin treatment of a rapidly progressive neurological condition. Cold Spring Harb Mol Case Stud. (2015) 1(1):a000257. doi: 10.1101/mcs.a000257
26. Set KK, Weber ARB, Serajee FJ, Huq AM. Clinical reasoning: siblings with progressive weakness, hypotonia, nystagmus, and hearing loss. Neurology. (2018) 90(7):e625–e31. doi: 10.1212/wnl.0000000000004973
27. Woodcock IR, Menezes MP, Coleman L, Yaplito-Lee J, Peters H, White SM, et al. Genetic, radiologic, and clinical variability in Brown-Vialetto-Van Laere syndrome. Semin Pediatr Neurol. (2018) 26:2–9. doi: 10.1016/j.spen.2017.03.001
28. Khadilkar SV, Faldu HD, Udani V, Patil SB, Malvadkar S. Reversible posterior column dysfunction in Brown-Vialetto-Von Laere syndrome. Muscle Nerve. (2017) 56(4):E28–e31. doi: 10.1002/mus.25694
29. Cosgrove J, Datta S, Busby M. Adult onset Brown-Vialetto-Van Laere syndrome with opsoclonus and a novel heterozygous mutation: a case report. Clin Neurol Neurosurg. (2015) 128:1–3. doi: 10.1016/j.clineuro.2014.10.016
30. Anand G, Hasan N, Jayapal S, Huma Z, Ali T, Hull J, et al. Early use of high-dose riboflavin in a case of Brown-Vialetto-Van Laere syndrome. Dev Med Child Neurol. (2012) 54(2):187–9. doi: 10.1111/j.1469-8749.2011.04142.x
31. Sinnathuray AR, Watson DR, Fruhstorfer B, Olarte JR, Toner JG. Cochlear implantation in Brown-Vialetto-Van-Laere syndrome. J Laryngol Otol. (2011) 125(3):314–7. doi: 10.1017/s0022215110001982
Keywords: pontobulbar palsy, riboflavin transporter deficiency, Brown-Vialetto-Van Laere syndrome, MRI, chronic inflammatory demyelinating polyneuropathy
Citation: Kentab AY, Alsalloum Y, Labani M, Hudairi A, Hamad MH, Jamjoom DZ, Alwadei AH, Alhammad RM and Bashiri FA (2024) Case Report: A rare treatable metabolic syndrome (Brown-Vialetto-Van Laere syndrome) masquerading as chronic inflammatory demyelinating polyneuropathy from Saudi Arabia. Front. Pediatr. 12:1377515. doi: 10.3389/fped.2024.1377515
Received: 31 January 2024; Accepted: 12 April 2024;
Published: 30 April 2024.
Edited by:
Cesare Indiveri, University of Calabria, ItalyReviewed by:
Maria Tolomeo, University of Calabria, ItalyZachary McPherson, Children’s Hospital at Westmead, Australia
© 2024 Kentab, Alsalloum, Labani, Hudairi, Hamad, Jamjoom, Alwadei, Alhammad and Bashiri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amal Y. Kentab YWtlbnRhYkBrc3UuZWR1LnNh