 Xin-Wei Shi
Xin-Wei Shi Jiang-Hong Deng†
Jiang-Hong Deng†- Department of Rheumatology, National Center for Children’s Health, Beijing Children’s Hospital, Capital Medical University, Beijing, China
Degos disease also known as malignant atrophic papulosis (MAP), is an autoinflammatory disease that mainly affects small- to medium-sized arteries. Gastrointestinal and nervous system are most commonly affected systems. Herein, we reported a case of Degos disease with disease onset during infantile and had severe neurological involvement.
Introduction
Degos disease, also known as malignant atrophic papulosis (MAP), is an autoinflammatory disease that mainly affects small- to medium-sized arteries (1). Initially described in 1941, it was later reclassified as malignant atrophic papulosis due to its involvement in multiple organs. While Degos disease commonly manifests in the fourth decade of adulthood, there have been reported cases in infants (2). The first infantile Degos disease was reported by Henkind and Clark, who had disease onset at three weeks of life and died due to neurological complications. Subsequent cases with neurological involvement have been reported, alluding to an overall unfavorable prognosis. Until now, more than 200 cases of Degos disease have been reported, and approximately 30 cases of childhood Degos disease have been published. Despite its rarity, there are no reported cases of infantile Degos disease in China. This report details a patient exhibiting severe neurological manifestations with an overall grim prognosis.
Case presentation
A 6-year-old Chinese girl presented with three months of numbness and two months of weakness in her extremities. Three months before admission, the patient developed right foot numbness and subsequently recovered. Two months before admission, weakness in the right lower limb developed, which manifested as claudication. Later, her symptoms worsened to flaccid paralysis of the bilateral limbs, and the patient was referred to the neurological department of the local hospital. Physical examination revealed right peripheral facial paralysis, right eye ptosis and flaccid paralysis of bilateral limbs. Laboratory examinations, such as blood cell levels, were unremarkable, and the patient was presented with normal liver and kidney function. The erythrocyte sedimentation rate (ESR) was 33 mm/h. Cerebrospinal fluid (CSF) protein concentration was 311 mg/dl. CSF revealed the presence of pleocytosis (25 × 106/l, normal range 0–15 ×106/l), with lymphocyte predominance. CSF IgM was 0.638 mg/ml. Cultivation of CSF and autoimmune encephalitis-associated antibodies was negative. Brain Magnetic Resonance Imaging (MRI) showed subdural effusion. The patient received two doses of methylprednisolone pulse and intravenous immunoglobulin (IVIG) therapy, but the disease progressed to incontinence of the stool and urine and involuntary movement of both lower extremities. The patient was subsequently transferred to the neurological department of our hospital to determine the etiology of the disease. On examination, right facial paralysis was noted. Decreased muscle strength and involuntary movement of both lower limbs were observed. The sensation of both lower limbs was lost to pin, touch, temperature and position. Cerebellar signs and cranial nerve examination were negative. Deep tendon reflexes showed hyporeflexia. Skin examination revealed dark-red papules throughout the body with a white atrophic center. After a detailed inquiry of her medical history, the patient had round to oval dark-red papules with white calcification centers on her left forefinger and dorsal wrist at the age of 20 days. These papules progressed throughout the body, subsided within two weeks and subsequently recurred. Additional laboratory results were as follows: routine blood and urine tests were unremarkable, and the patients presented with normal liver and kidney function. Repeated ESR was 10 mm/h. Cerebrospinal fluid (CSF) protein concentration was 2,215 mg/dl which is higher than before. CSF revealed the normal range of granulocyte. Her plasma D-dimer level was 0.76 mg/l (normal range 0–0.243 mg/l). The serum folate, lactate and vitamin B12 levels were normal. Antinuclear antibodies and anti-double stranded DNA antibodies were negative. The patient was positive for an antineutrophil cytoplasmic antibody. Vascular ultrasound was normal. Brain Magnetic Resonance Imaging (MRI) showed left hemisphere atrophy, subdural effusion, widespread calcification foci, hemorrhagic foci and local vasculitis (Figure 1A). Due to the possible diagnosis of autoimmune-associated vasculitis, the patient was then transferred to our rheumatic department. No specific genetic mutations were found after whole-genome sequencing was completed. Skin biopsy of papules on the left lower limb and right upper limb revealed dermal necrosis, thickening of the vascular wall, local hyperkeratosis and hypokeratosis, as well as perivascular lymphocytic infiltration in the upper dermis without mucin disposition (Figure 2). The diagnosis of Degos disease was made based on characteristic papules, disease course and histopathological findings. A methylprednisolone pulse (20 mg/kg/d), oral prednisone (2 mg/kg), intravenous immunoglobulin (IVIG) (2 g/kg), intravenous cyclophosphamide (0.8 g/m2) and aspirin were prescribed. The skin lesions recovered, but there was no significant improvement in neurological manifestations after 1 month of combined treatment. Repeated brain MRI showed lacunar infarction, and the size of the ischemic area increased (Figure 1B). Second doses of methylprednisolone pulse, IVIG, and intravenous cyclophosphamide were prescribed. Approximately half a month later, involuntary movement of both lower limbs was more frequent, and repeated brain MRI showed an increased amount of subdural effusion (Figure 1C). Later, the patient underwent surgery involving the right blurred hole on the right scalp with external drainage. The specimen collected during surgery showed inflammatory cell infiltration (Figure 3). Oral hydroxychloroquine (5 mg/kg), thalidomide (3 mg/kg), and methotrexate (10 mg/m2) were added as prednisolone-sparing agents. However, the neurological symptoms of the patient deteriorated to lower limb paralysis and areflexia. Her parent asked for discharge and was not followed up later. Unfortunately, the patient died 10 months after the first initiation.
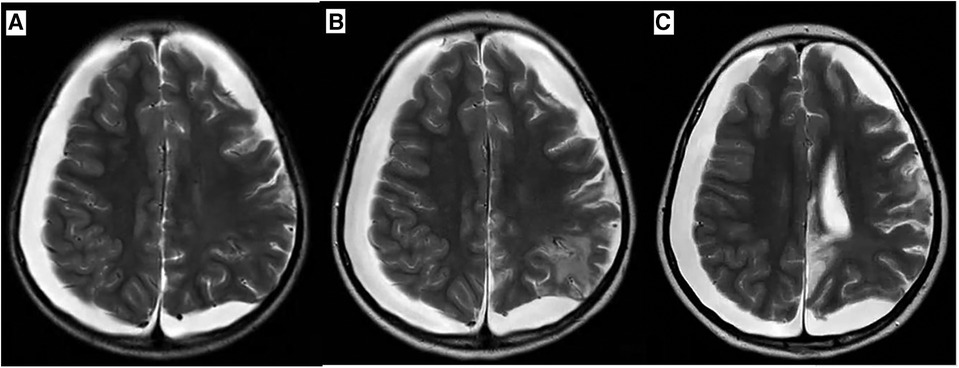
Figure 1 Axial T2 weighted Brain MR image during disease progression. (A) Brain MRI at first visit revealed atrophy of left hemisphere, subdural effusion, calcification foci, hemorrhagic region, ischemic region and vasculitis. (B) Brain MRI after one month of treatment which revealed the size of ischemic region increased. (C) Brain MRI after one and a half months of treatment which revealed medium amount of subdural effusion and size of ischemic region increased.
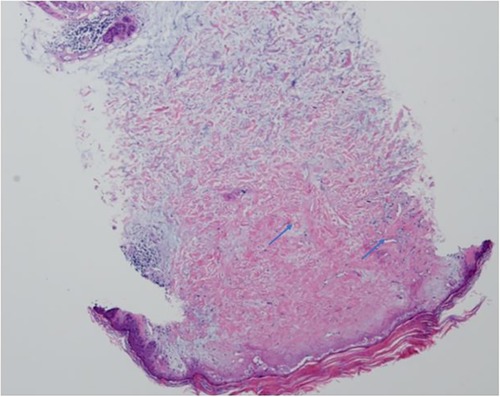
Figure 2 Histopathological examination of skin lesions showed dermis necrosis , thickness of vascular wall and perivascular lymphocytic infiltration (arrow) (H&E,20X).
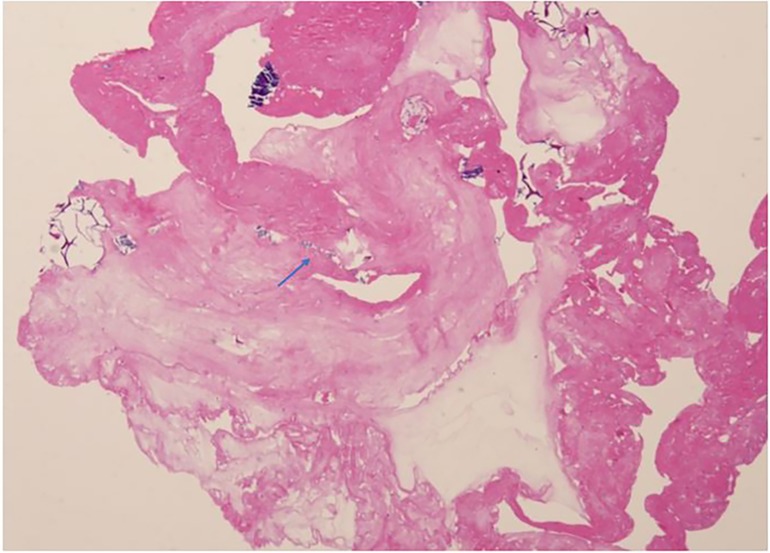
Figure 3 Histopathological examination of subdural effusion inflammation cells infiltration (arrow) (H&E,20X).
Literature review
A literature search for malignant atrophic papulosis, Degos disease and pediatric was carried out using PubMed database until April 2024. The search yielded 34 cases and the general information was summarized in Table 1 (2–28). According to these studies, 15 were female and 19 were male. Median age of disease onset was 7.52 ± 1.14 years.
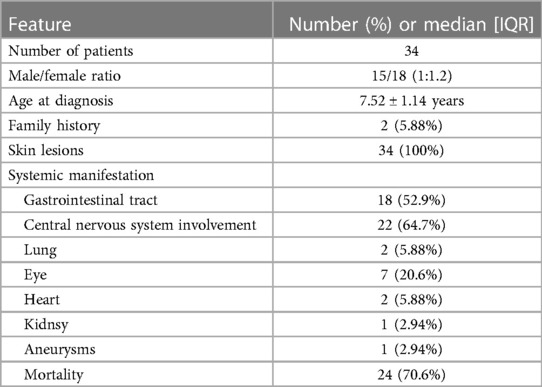
Table 1 Data of pediatric malignant atrophic paplosis cases from previous studies.
In two patients, disease onset soon after birth. Skin lesions were the most frequent clinical manifestation, with a frequency of 100%. Central nervous system and gastrointestinal tract involvements were common. Other system like eye, heart and kidney could also be affected. The mortality rate of Degos disease was high which is almost 70%.
Even treated with eculizumab, neurological symptoms exacerbated and patient died. Most patients died due to neurological involvement.
Discussion
Degos disease, also known as malignant atrophic papulosis, typically presents with erythematous papules that evolve into porcelain-white atrophic papules surrounded by erythematous rims. These lesions are asymptomatic and subside. The papules range in size from 0.5 to 1 cm in diameter, with the trunk and extremities being the most commonly affected areas (29). The most commonly affected systems are the gastrointestinal system (73%) and nervous system (20%–64%) (29). In pediatrics, there is a preference for nervous system involvement (30). Other systems, such as the optic, cardiovascular, pulmonary and hepatorenal systems, could also be affected.
The first case of neurological involvement in Degos disease was reported in 1960 by Nomland and Layton (31). The clinical manifestations of Neuro-MAP can be divided into central nervous system (CNS) presentations and peripheral nervous system (PNS) presentations. CNS involvements are more common (14). CNS presentations can be divided into parenchymal forms accompanied by meningoencephalitis and meningomyelitis and neurovascular forms accompanied by hemorrhage, infarction and thrombosis. PNS manifestations may include neuropathy, myopathy, or polyradiculopathy (32). The patient in our study had both CNS (intracranial hemorrhage, infarction and thrombosis) and PNS (facial paralysis) involvement.
Histopathological examination revealed hyperkeratosis, atrophy of the epidermis, dermo-epidermal separation and edema (33). Cerebrospinal fluid analysis revealed pleocytosis with elevated protein levels, while CSF glucose levels were within normal range, and CSF culture yielded negative results. Ischemic foci can be observed via brain MRI, regarded as the gold standard (32). Given the typical papules and severe neurological involvement, along with skin biopsy findings indicative of vasculitis, the patient was diagnosed with Degos disease.
Degos disease poses a therapeutic challenge, lacking uniform treatment options. There are few cases in which combined therapy comprising IVIG, prednisolone, and cyclophosphamide can induce remission; therefore, a high dose of IVIG and steroids followed by cyclophosphamide was initiated in our patient. Patients with Degos disease may present with elevated fibrinogen levels in plasma, aggravated platelet aggregation and reduced fibrinolytic activity, and previous studies have reported that antiplatelet agents may be useful in resolving skin lesions (29); therefore, antiplatelet agents (aspirin) were added to the patient's regimen. Unfortunately, despite the combined therapy, the patient's symptoms progressed, leading to a fatal outcome.
Recent studies have shown that Eculizumab which blocks terminal complement, has effects on skin and gastrointestinal lesions, and treprostinil, which is a prostacyclin analog, is effective for treating gastrointestinal (GI) and central nervous system manifestations (34). More advanced therapeutic therapies are needed.
For patients with neurological involvement, the average time from papulosis occurrence to death is 2 years (ranging from 1 to 12 years) (29). Unfortunately, in the case of the patient in our study, recommended treatment and follow-up were not adhered to, and the exact cause of death remained unclear. The patient succumbed to neurological complications associated with Degos disease.
Our study is unique because Degos disease is rarer in children than in infants. There are few cases of infantile Degos disease reported worldwide, and this is the first case reported in China. Degos disease is rare and urgently diagnosed, and new treatment options, such as eculizumab and treprostinil, are imperative. Our case also highlighted that neurologists should consider autoinflammatory diseases when neurological and dermatological manifestations coexist.
Conclusions
Degos disease is a rare and challenging condition to diagnose. Rapid and accurate diagnosis is crucial, and there is a pressing need for additional treatment options and novel medications to improve prognosis in affected individuals.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Beijing Children's Hospital ([2022]-E-155-R). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
X-WS: Writing – original draft. J-HD: Writing – original draft. C-FL: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank the patient and her family for consent to participate this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
MAP, malignant atrophic papulosis; ESR, erythrocyte sedimentation rate; MRI, Magnetic Resonance Imaging; CSF, cerebrospinal fluid; IVIG, intravenous immunoglobulin; CNS, central nervous system; PNS, peripheral nervous system.
References
1. Degos R. Malignant atrophic papulosis. Br J Dermatol. (1979) 100(1):21–35. doi: 10.1111/j.1365-2133.1979.tb03566.x
2. Torrelo A, Sevilla J, Mediero IG, Candelas D, Zambrano A. Malignant atrophic papulosis in an infant. Br J Dermatol. (2002) 146(5):916–8. doi: 10.1046/j.1365-2133.2002.04677.x
3. Basset A, Guilaine A, Monfort J. Papulose atrophiante maligne de degos. Bull Soc Fr Dermatol Syphiligr. (1956) 63(4):358–60. (French).13396476
4. Henkind P, Clark WE. Ocular pathology in malignant atrophic papulosis. Degos’ disease. Am J Ophthalmol. (1968) 65(2):164–9. doi: 10.1016/0002-9394(68)93580-0
5. Hall-Smith P. Malignant atrophic papulosis (Degos’ disease) two cases occurring in the same family. Br J Dermatol. (1969) 81(11):817–22. doi: 10.1111/j.1365-2133.1969.tb15949.x
6. Enjolras O, Debray H, Bedouelle J, Hewitt J. Un cas juvenile de maladie de Degos. Bull Soc Fr Dermatol Syphiligr. (1974) 81.
7. Horner FA, Myers GJ, Stumpf DA, Oseroff BJ, Choi BH. Malignant atrophic papulosis (Kohlmeier Degos disease) in childhood. Neurology. (1976) 26(4):317–21. doi: 10.1212/WNL.26.4.317
8. Sotrel A, Lacson AG, Huff KR. Childhood Köhlmeier-Degos disease with atypical skin lesions. Neurology. (1983) 81(2):205–7. doi: 10.1212/wnl.33.9.1146
9. Su WP, Schroeter AL, Lee DA, Hsu T, Muller SA. Clinical and histologic findings in Degos’ syndrome (malignant atrophic papulosis). Cutis. (1985) 35(2):131–8.3884281
10. Schneider A, Tschumi A, Egloff B, Ott F, Fanconi A. Maligne atrophisierende papulose (Degos-syndrom) bei einem saugling. Helv Paediatr Acta. (1986) 41(5):447–54. (German).3818332
11. Schade FB, Monnens L, Hendriks JHCL, Rosenbusch G. Renovascular hypertension in a child with Degos-Köhlmeier disease. Pediatr Radiol. (1987) 17(3):260–1. doi: 10.1007/BF02388181
12. Rosemberg S, Lopes MBS, Sotto MN, Graudenz MS. Childhood Degos disease with prominent neurological symptoms: report of a clinicopathological case. J Child Neurol. (1988) 3(1):43–6. doi: 10.1177/088307388800300110
13. Barabino A, Pesce F, Gatti R, Colotto P, Nobili F, Colacino R, et al. An atypical paediatric case of malignant atrophic papulosis (Köhlmeier-Degos disease). Eur J Pediatr. (1990) 149(7):457–8. doi: 10.1007/BF01959394
14. Subbiah P, Wijdicks E, Muenter M, Carter J, Connolly S. Skin lesion with a fatal neurologic outcome (Degos’ disease). Neurology. (1996) 46(3):636–40. doi: 10.1212/WNL.46.3.636
15. Yoshikawa H, Maruta T, Yokoji H, Takamori M, Yachie A, Torii Y. Degos’ disease: radiological and immunological aspects. Acta Neurol Scand. (1996) 94(5):353–6. doi: 10.1111/j.1600-0404.1996.tb07079.x
16. Lankisch MR, Johst P, Scolapio JS, Fleming CR. Acute abdominal pain as a leading symptom for Degos’ disease (malignant atrophic papulosis). Am J Gastroenterol. (1999) 94(4):1098–9. doi: 10.1111/j.1572-0241.1999.1022_a.x
17. Caviness VS Jr, Sagar P, Israel EJ, Mackool BT, Grabowski EF, Frosch MP. Case records of the Massachusetts General Hospital. Case 38-2006. A 5-year-old boy with headache and abdominal pain. N Engl J Med. (2006) 355(24):2575–84. doi: 10.1056/NEJMcpc069029
18. Jalil J, Shafique M, Rashid Dar N. Dermatological clue to diagnosis of Degos disease in a 2-year-old with obscure chronic abdominal pain. Clin Pediatr (Phila). (2008) 47(2):180–2. doi: 10.1177/0009922807306776
19. Gutiérrez-Pascual M, Hernández-Martín A, Colmenero I, García-Peñas JJ, López-Pino MA, Torrelo A. Malignant atrophic papulosis: a case report with severe visual and neurological impairment. Pediatr Dermatol. (2011) 28(3):302–5. doi: 10.1111/j.1525-1470.2010.01287.x
20. Guo YF, Pan WH, Cheng RH, Yu H, Liao WQ, Yao ZR. Successful treatment of neurological malignant atrophic papulosis in child by corticosteroid combined with intravenous immunoglobulin. CNS Neurosci Ther. (2014) 20(1):88–91. doi: 10.1111/cns.12196
21. Gmuca S, Boos MD, Treece A, Narula S, Billinghurst L, Bhatti T, et al. Degos disease mimicking primary vasculitis of the CNS. Neurol Neuroimmunol Neuroinflamm. (2016) 3(2):e206. doi: 10.1212/NXI.0000000000000206
22. Huang YC, Wang JD, Lee FY, Fu LS. Pediatric malignant atrophic papulosis. Pediatrics. (2018) 141(Suppl 5):S481–S484. doi: 10.1542/peds.2016-4206
23. Moran EJ, Lapin WB, Calame D, Bray M, Wright LN, Desai NK, et al. Degos disease: a radiological-pathological correlation of the neuroradiological aspects of the disease. Ann Diagn Pathol. (2020) 47:151545. doi: 10.1016/j.anndiagpath.2020.151545
24. Wang HQ, Guan Y, Gong XP, Chen YT, Ji C. Case report: pediatric malignant atrophic papulosis with small bowel perforation and positivity of anticardiolipin antibody. Front Pediatr. (2021) 9:764797. doi: 10.3389/fped.2021.764797
25. Schaefer LS, Wampler Muskardin T, Tillema JM, Wieland C, Tollefson MM. A fatal case of malignant atrophic papulosis in a pediatric patient. Pediatr Dermatol. (2022) 39(1):112–114. doi: 10.1111/pde.14878
26. Cabre J, S N-D, J B-C. Papulose atrophiante maligne de degos chez unnourisson. Bull Soc Fr Dermatol Syphiligr. (1974):652–3.
27. Warot P, Caron JC, Lehembre P, Houcke M. Maladie de Degos à forme cérébrale. Rev Neurol (Paris). (1977) 133:353–8.897442
28. Paudel A, Bhurtel MR, Gautam A, Gautam A, Bista M, Singh P. Malignant atrophic papulosis presenting with intestinal perforation: a case report. JNMA J Nepal Med Assoc. (2023) 61(262):549–551. doi: 10.31729/jnma.8192
29. Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease)—a review. Orphanet J Rare Dis. (2013) 8:10. doi: 10.1186/1750-1172-8-10
30. Wilson J, Walling HW, Stone MS. Benign cutaneous Degos disease in a 16-year-old girl. Pediatr Dermatol. (2007) 24(1):18–24. doi: 10.1111/j.1525-1470.2007.00326.x
31. Nomland R, Layton JM. Malignant papulosis with atrophy (Degos): fatal cutaneointestinal syndrome. AMA Arch Dermatol. (1960) 81:181–8. doi: 10.1001/archderm.1960.03730020017003
32. Yousefi S, Borhani-Haghighi A, Safari A, Shapiro L. Neurological involvement in malignant atrophic papulosis: a comprehensive review of literature. Neurol India. (2022) 70(1):5–10. doi: 10.4103/0028-3886.338719
33. Slater D. Histologic diagnosis of inflammatory skin diseases. An algorithmic method based on pattern analysis, 3rd edn. Histopathology. (2005) 46(6):707–8. doi: 10.1111/j.1365-2559.2005.02174.x
Keywords: Degos disease, malignant atrophic papulosis, infantile onset, neurological involvement, prognosis
Citation: Shi X-W, Deng J-H and Li C-F (2024) Case Report: Infant-onset Degos disease with nervous system involvement and a literature review. Front. Pediatr. 12:1374150. doi: 10.3389/fped.2024.1374150
Received: 4 February 2024; Accepted: 9 May 2024;
Published: 5 July 2024.
Edited by:
Erkan Demirkaya, Western University, CanadaReviewed by:
David Piskin, Lawson Health Research Institute, CanadaVincenza Gragnaniello, University Hospital of Padua, Italy
© 2024 Shi, Deng and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cai-Feng Li, Y2FpZmVuZ19saUB5ZWFoLm5ldA==
†These authors share co-authorship