 Timmy Strauss1,2,3*†Julia Körholz1,2,3,†Hye Sun Kuehn4Agustin A. Gil Silva4Franziska Taube3,5Karolin Trautmann-Grill5Anna Stittrich6Leonora Pietzsch1,3Ralf Wiedemuth1,3Volker Wahn7Horst von Bernuth7,8,9Sergio D. Rosenzweig4Maria Fasshauer10Renate Krüger7Catharina Schuetz1,2,3*
Timmy Strauss1,2,3*†Julia Körholz1,2,3,†Hye Sun Kuehn4Agustin A. Gil Silva4Franziska Taube3,5Karolin Trautmann-Grill5Anna Stittrich6Leonora Pietzsch1,3Ralf Wiedemuth1,3Volker Wahn7Horst von Bernuth7,8,9Sergio D. Rosenzweig4Maria Fasshauer10Renate Krüger7Catharina Schuetz1,2,3*
- 1Department of Pediatrics, Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
- 2University Center for Rare Diseases, Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
- 3Faculty of Medicine and University Hospital Carl Gustav Carus, University Center for Chronic Immunodeficiencies (UCID), Technische Universität Dresden, Dresden, Germany
- 4Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, United States
- 5Department of Hematology and Oncology, Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
- 6Department of Human Genetics, Labor Berlin Charité-Vivantes GmbH, Berlin, Germany
- 7Department of Pediatric Respiratory Medicine, Immunology, and Critical Care Medicine, Charité–Universitätsmedizin Berlin, Freie Universität Berlin, Humboldt-Universität zu Berlin, Berlin Institute of Health, Berlin, Germany
- 8Berlin Institute of Health (BIH), Charité—Universitätsmedizin Berlin, Berlin, Germany
- 9Berlin-Brandenburg Center for Regenerative Therapies (BCRT), Charité—Universitätsmedizin Berlin, Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health (BIH), Berlin, Germany
- 10ImmunoDeficiencyCenter Leipzig (IDCL), Hospital St. Georg GGmbH Leipzig, Academic Teaching Hospital of the University of Leipzig, Leipzig, Germany
Heterozygous germline variants in human IKZF1 encoding for IKAROS define an inborn error of immunity with immunodeficiency, immune dysregulation and risk of malignancy with a broad phenotypic spectrum. Growing evidence of underlying pathophysiological genotype-phenotype correlations helps to improve our understanding of IKAROS-associated diseases. We describe 6 patients from 4 kindreds with two novel IKZF1 variants leading to haploinsufficiency from 3 centers in Germany. We also provide an overview of first symptoms to a final diagnosis including data from the literature.
1 Introduction
The protein IKAROS, encoded by IKZF1, is part of the zinc finger transcription factor family which plays a central role in lymphocyte, erythroid, myeloid and megakaryocyte differentiation and development (1–3). Germline heterozygous variants in human IKFZ1 result in inborn errors of immunity (IEI) with infection susceptibility, immune dysregulation and risk of malignancy. The broad clinical spectrum includes bacterial, viral or fungal infections, autoimmunity, atopy, lymphoproliferative disorders and hematologic malignancies (4–7). To date, 4 different mechanisms impairing the function of IKAROS proteins have been described: haploinsufficiency (HI), dimerization defect (DD), dominant negative (DN) and gain-of-function (GOF) (8). Interestingly, regardless of the affected domain (and its corresponding functional defect), patients with IKAROS-associated diseases present with a phenotype of increased infection susceptibility and signs of immune dysregulation. Most patients with IKAROS haploinsufficiency (HI) suffer from bacterial infections and symptoms of immune dysregulation (8). Around 5% of patients develop malignancies including B-cell acute leukemia (B-ALL) (9). The median age of symptom onset is 10 years but first symptoms may present up to the age of 60 (10, 11). Some carriers may be clinically asymptomatic (12–14). Immunological characterization of IKAROS-HI usually reveals an incomplete B-cell arrest with low B-cell numbers and serum immunoglobulin levels. Reported patients were treated with corticosteroids, immunoglobulin replacement therapy (IgRT), prophylactic and/or therapeutic antibiotics and hematopoietic stem cell transplantation (HSCT) depending on the severity of their clinical presentation (8).
Variants located in the dimerization domain or nonsense variants upstream of the dimerization domain so called “dimerization defect” (DD), are usually characterized by hypogammaglobulinemia and hematological manifestations, including autoimmune cytopenias, lymphoproliferative disorders and a variety of hematologic malignancies. In comparison to patients with HI variants, those with DD variants show moderate B-cell lymphopenia and hypogammaglobulinemia resulting in less frequent and severe bacterial infections (5). Most children manifest before the age of 10 and are treated with immunosuppressants and IgRT (10).
Patients with dominant negative (DN) IKZF1 variants present with severe and invasive infections [esp. Pneumocystis jirovecii pneumonia (PCP)], usually before 2 years of age. Immunologically they are characterized by a combined immunodeficiency (CID) with agammaglobulinemia, severe B-cell lymphopenia, absence of plasma cells and an abnormal T-cell compartment with increased naïve and reduced memory cells. Patients are often managed with antimicrobial therapy including PCP prophylaxis, IgRT, and, due to the severity of the disease, HSCT (4, 15).
While HI, DD and DN variants cause an IKAROS loss-of-function (LOF), gain-of-function variants in IKZF1 lead to an increased IKAROS DNA binding (16). The clinical phenotype is dominated by signs of immune dysregulation and atopy. Multiple autoimmune phenomena like gastrointestinal, endocrinological and hematologic manifestations have been reported. Individuals with GOF variants manifest with incomplete penetrance between 1 and 40 years of age and exhibit normal B-cell numbers and normal to slightly elevated immunoglobulin levels.
In summary, diagnosis and treatment of IKAROS-associated phenotypes can be challenging due to the broad spectrum of clinical signs and symptoms (Figure 1). We subsequently describe 6 affected patients from 4 kindreds with IKZF1 variants and functional IKAROS-HI, and provide a clinical overview from first symptoms to final diagnosis.

Figure 1. Phenotype-genotype correlation including overlapping features (review of the literature). PCP, pneumocystis jirovecii pneumonia.
2 Clinical vignettes and functional testing
2.1 Family A
Patient 1 is a 14-year-old boy presenting with osteomyelitis and streptococcal bacteremia at age 7. He has a history of recurrent otitis media, chronic purulent rhinitis and cough for 2 years as well as recurrent and long-lasting infections starting in childhood. His family reported frequent exhaustion upon physical activity. Between the age of 6 and 9 years, the child had intermittent diarrhea, without any causative pathogen ever detected. Intolerance for fructose or lactose was excluded, endoscopic investigation was not performed. Imaging at age 9 showed mild bronchiectasis and hepatomegaly. Further diagnostic workup revealed severe hypogammaglobulinemia and a lack of vaccination responses to tetanus, diphtheria and Haemophilus influenzae. Further immunologic workup showed severely reduced B cells with a predominance of naïve B cells, absent class-switch, a reduced CD4/CD8-T-cell ratio due to expanded CD8+ T cells, as well as severely reduced naïve T cells (summarized in Table 1, details in Supplementary Table S1). T-cell proliferation in vitro was reduced to specific antigens such as tetanus and cytomegalovirus (CMV) (positive tetanus vaccination status, unknown status of CMV exposure). A de novo IKZF1 missense variant [c.448T>C; p.(Cys150Arg)] was identified in whole exome sequencing (WES) (Figure 2). Considering the cumulative infectious burden with possible organ damage and risk for hematological malignancies led us to reconsider curative treatment options. HSCT from an HLA-identical sister was performed at 11.5 years of age following myeloablative conditioning with treosulfan, fludarabine and thiotepa. Three years post HSCT, the patient is well with full donor chimerism including a normal T- and B-cell compartment.
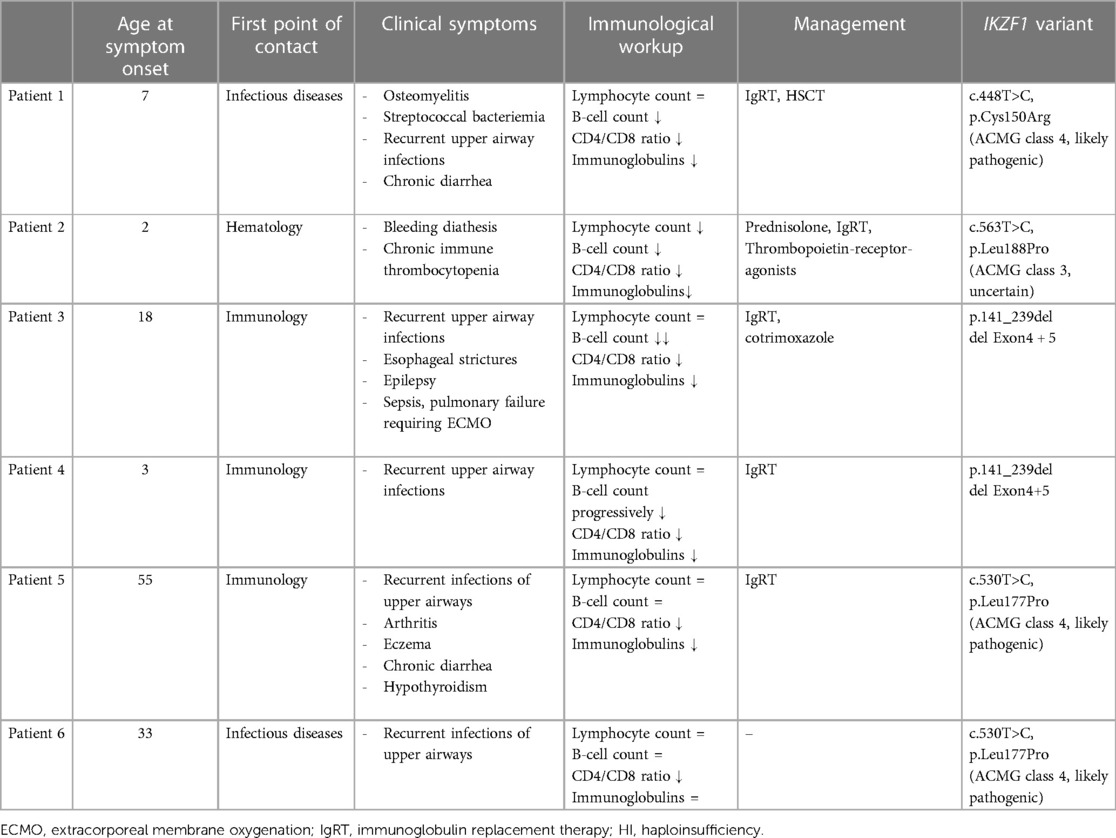
Table 1. Summary of clinical, immunological and genetic characteristics of IKZF1 HI and treatment of the 6 affected individuals.
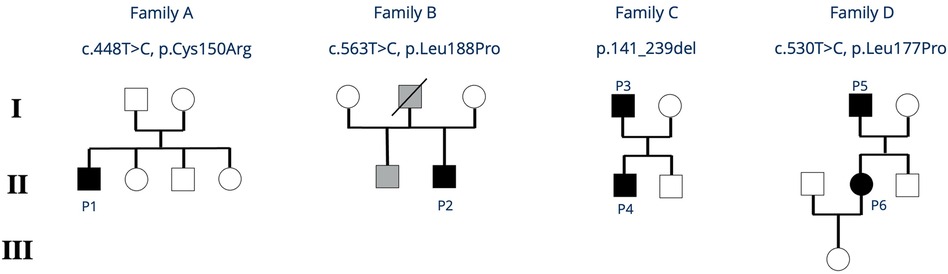
Figure 2. Family trees of all reported families with IKZF1 HI. Black symbols indicate clinically affected index patients, grey symbols indicate individuals who are clinically affected but not genetically tested. P, patient.
2.2 Family B
Patient 2 is a 19-year-old young adult, presenting at age 2 with epistaxis and hematomas. Persistently low platelet counts, and the presence of thrombocyte auto-antibodies (anti-GPIIb/IIIa) confirmed the diagnosis of immune thrombocytopenia (ITP) treated with prednisolone and intravenous immunoglobulins. In 2023, he was started on thrombopoietin-receptor-agonists (Revolade, NPlate) due to recurrent episodes of symptomatic thrombocytopenia. At that time, his laboratory workup showed mild hypogammaglobulinemia, reduced IgA and IgM, and the patient was started on subcutaneous immunoglobulin (SCIG) treatment. The patient's father died from thrombosis and pulmonary embolism. The patient's half-brother (same father) also suffers from chronic ITP (Figure 2). Further immunophenotyping of the patient revealed reduced B cells, a reduced CD4/CD8 ratio due to decreased CD4+ T cells, and impaired B- and T-cell maturation with increased naïve B cells, decreased class-switched B cells, as well as reduced naïve CD4+ T cells. Vaccine responses to a variety of vaccines were undetectable (Table 1 and Supplementary Table S1). A novel heterozygous missense variant IKZF1 [c.563T>C, p.(Leu188Pro)] was detected in WES. To date, the father and the brother have not been tested genetically.
2.3 Family C
Patient 3 is a 48-year-old male with a history of recurrent bacterial upper airway infections first presenting in adolescence. He reported recurrent and severe pneumonias, otitis and chronic pansinusitis (S. aureus, H. influenzae) requiring repeated antibiotic treatments. CT chest imaging showed bipulmonary micronodular lesions, but no bronchiectasis. At the age of 30 years, agammaglobulinemia was diagnosed. The patient was subsequently started on IgRT which improved the infection susceptibility. Of note, the patient requires 80 g IgG monthly to maintain IgG levels ≥10 g/L. Symptoms of sinusitis and productive cough further decreased when prophylaxis with cotrimoxazol was initiated. Immunophenotyping showed absent B cells, as well as a reduced CD4/CD8 ratio due to expanded CD8+ T-cells and borderline low naïve CD4+ T-cells. T cell proliferation to stimulation with mitogens and recall antigens was normal (Table 1 and Supplementary Table S2). At the age of 32 years, the patient was hospitalized with severe shingles [varicella zoster virus (VZV)]. At age 40, he started reporting recurrent episodes of diarrhea with weight loss. A previous episode of diarrhea could be explained by Salmonella infection, but the chronic intermittent course could not be attributed to any infectious agent. Histologically there were no signs of inflammation. To date the patient reports rare episodes of watery stools. In addition, he started complaining about swallowing difficulties. Endoscopy showed esophageal strictures, histology however did not confirm a suspected eosinophilic esophagitis. In his medical history, the patient reports of epilepsy with a first afebrile seizure at the age of 19, currently well-controlled with valproic acid and ethosuximide treatment. At present, the patient is obese and has both arterial hypertension and steatosis hepatis. At age 47 years, the patient was admitted to intensive care with pneumonia, pulmonary failure and fulminant sepsis. Following a very critical condition requiring extracorporeal membrane oxygenation, he has fortunately fully recovered and could be discharged from the hospital.
His now 10-year-old son (Patient 4, Figure 2) became symptomatic with recurrent upper airway infections (rhinitis, pharyngitis) starting from age 3. Due to the positive family history, clinical findings, and low IgG levels, he was started on IgRT at the age of 3 years. No major infections have occurred so far, but he also requires high doses of IgG (>0.9 g/kg body weight/month) to maintain IgG levels >6 g/L. His immunophenotyping shows progressive decline of B cells with reduced class-switch, but also a reduced CD4/CD8 T-cell ratio due to an expansion of CD8+ T-cells. Vaccination titers were low (tetanus) or undetectable (pneumococcus, measles) prior to IgRT despite respective vaccinations (summarized in Table 1, details in Supplementary Table S2).
Genetic testing of father and son revealed a deletion of exons 4 and 5 in the IKZF1 gene of father and son.
2.4 Family D
Patient 5 is a now 61-year-old male with a recent history of recurrent upper airway infections starting from the age of 55. He reports 4 hospital admissions due to recurrent and prolonged pneumonias revealing mild hypogammaglobulinemia with low IgG and absent IgA levels. Chest imaging showed bronchiectasis and pulmonary granuloma. He was started on IgRT which led to reduced infectious complications. Apart from infections, he also reports arthritis in his metacarpophalangeal and proximal interphalangeal joints, eczema, hypothyroidism and several episodes of non-infectious intermittent diarrhea for over one year's duration. At present he is being re-evaluated for exacerbation of non-bloody watery diarrhea, with negative results for stool pathogens including protozoa, and normal calprotectin. He is scheduled for colonoscopy. Other co-morbidities include metabolic syndrome with arterial hypertension, coronary sclerosis, Type 2 diabetes mellitus, hepatic steatosis and sleep apnea syndrome. Immunophenotyping in this patient shows normal B-cell counts with mildly reduced class-switched B-cells, and a reduced CD4/CD8 T-cell ratio due to expanded CD8+ T-cells. Vaccination titers to tetanus toxoid and diphtheria toxoid were low prior to IgRT (summarized in Table 1, details in Supplementary Table S3).
Patient 6 is the now 34-year-old daughter of patient 5 (Figure 2). She reports long lasting respiratory infections starting in childhood without increased need for antibiotic treatment and no need for hospitalization. Her medical history is almost unremarkable without the need for prophylaxes to prevent infections. Immunophenotyping revealed normal B-cell counts, mildly impaired B-cell class-switch, mildly reduced IgA levels, normal IgG and normal vaccination titers, and increased gamma-delta T-cells (summarized in Table 1, details in Supplementary Table S3). She was found to have elevated anti-nuclear antibody (ANA)-titers 1:640 (AC-21), antimitochondrial antibody (AMA) 1:1,280 (M2 15 U/ml) so far without clinical evidence of autoimmune disease. Abdominal ultrasound revealed mild hepatosplenomegaly. Genetic testing of patient 5 and 6 revealed a novel heterozygous, pathogenic missense variant in IKZF1 [c.530T>C, p.(Leu177Pro)].
2.5 Functional validation
Two of the herein reported four IKZF1 heterozygous germline variants are novel. To assess their functional consequences, the 3 missense variants were subjected to in-vitro functional testing by immunofluorescence and electrophoretic mobility shift assay (EMSA). Immunofluorescence data showed that while NIH3T3 cells expressing WT IKAROS protein exhibit a punctate staining pattern, characteristic of pericentromeric heterochromatin localization, all three mutant proteins display diffuse nuclear staining (Figure 3A top). However, when the mutant was co-expressed with WT IKAROS protein to mimic a heterozygous condition, the normal punctate staining pattern was again observed. These data suggest that while the three mutant IKZF1 variants lost their pericentromeric targeting, they did not exert dominant negative effects, as they did not abolish the pericentromeric localization of the WT protein under heterozygous conditions (Figure 3A bottom). Furthermore, EMSA data revealed that the ability of mutant IKAROS proteins to bind to corresponding DNA elements was completely abrogated. However, the mutants did not interfere with the binding of the WT protein under heterozygous conditions (Figure 3B). For these 3 missense variants, our data suggest that our patients with these IKAROS variants can be characterized as having IKAROS-HI. As for the patient with deletion of exons 4 and 5, the exact breaking points could not be detected and were presumably located outside of the sequenced regions. A similar deletion (chr7:50.435.843–50.452.713, 16.8 kb) was detected in a Norwegian patient with hypogammaglobulinemia and low B cells (12).
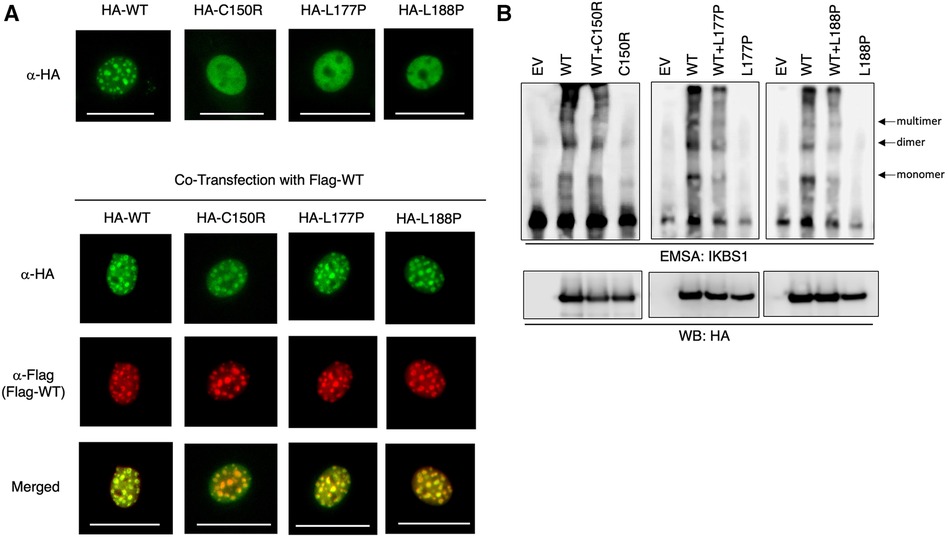
Figure 3. IKAROS mutants failed DNA binding and pericentromeric targeting. (A) NIH3T3 cells transfected with HA-tagged WT IKZF1 and mutant IKZF1 alone or together with Flag-tagged WT IKZF1. Pericentromeric heterochromatin localization of wild type and indicated mutant IKAROS proteins is revealed by fluorescence microscopy using tag specific antibodies. The scale bars indicate 25 μm. (B) Binding capacity of IKAROS mutants to corresponding DNA elements using an electrophoretic mobility shift assay (EMSA). Nuclear extracts of HEK293T cells transfected with HA-tagged WT IKZF1 and/or mutant IKZF1 were incubated with a IKBS1 DNA probe and separated by electrophoresis. Arrows indicate the monomer, dimer and multimer forms of IKAROS protein. Protein Expression of ectopic WT or mutant IKFZ1 protein was confirmed by western blot (WB) using a HA-specific antibody. EV, empty vector. Data shown are representative of three independent experiments.
3 Discussion
The herein described patients all carry germline IKZF1 variants with functional HI and have a history of recurrent bacterial or viral infections, immune thrombocytopenia and inflammatory signs like arthritis and eczema, but no history of malignant disease.
According to previous review articles, IKZF1 deficiency may present with a broad variety of clinical symptoms (Figures 1, 4) (5). In this IEI, a genotype-phenotype correlation has been described, i.e., signs and symptoms differ between IKZF1 genetic variants according to their functional impact (8).
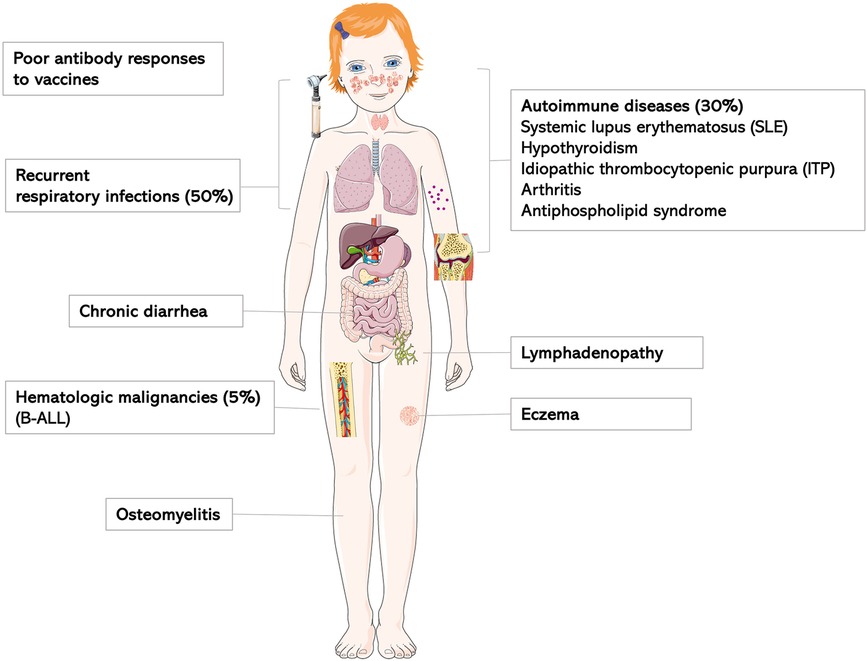
Figure 4. Clinical characteristics of IKZF1-haploinsufficiency patients as previously reviewed by PMID: 36433803.
In the cohort with LOF variants presented here, the age at symptom onset ranges from early childhood until late adulthood with incomplete clinical penetrance. In agreement with the literature, the clinical spectrum spans from severe invasive infections to clinically oligosymptomatic individuals (Figure 4). Three of the 6 patients of our cohort report a history of chronic or intermittent diarrhea of infectious or undefined origin; the latter with a self-limiting course. Previous reports describe IKAROS patients with common variable immunodeficiency (CVID)-like phenotypes, of whom a minority (3 of 29) individuals had chronic or recurrent diarrhea of infectious or unknown cause (12). As in any IEI, a history of persistent or chronic diarrhea requires histological work-up based on the severity and duration of symptoms, as infectious causes may be treatable, and e.g., celiac-like disease is not a rare occurence in other IEIs, and may impact quality of life.
The immunophenotype in most of our patients showed an incomplete B-cell maturation arrest leading to a decline of B-cell numbers and serum immunoglobulin levels. These findings confirm the importance of IKAROS function in B-cell development. The most severe reduction of B-cell counts is expected in patients with dominant negative mutations, followed by haploinsufficiency and dimerization defective variants. The decline of B-cell numbers can be progressive, and the severity of B-cell deficiency may vary within one kindred (8, 14). Especially in LOF missense variants, patients additionally have abnormalities in their T-cell compartment with T-cell lymphopenia, elevated CD8+ T-cells and a decreased CD4/CD8 ratio (8). This is in line with the immunophenotyping of our patients, including one family harboring a large genetic deletion. In contrast to former reports, we also identified reduced CD4+ and CD8+ naïve T-cell numbers in some of our IKAROS-HI patients regardless of the severity of their clinical phenotype. Of note, newborns with IKZF1 variants have been detected through severly reduced T-cell receptor excision circles (TREC) in newborn screening with severe combined immunodeficiency (SCID)-like phenotypes. Interestingly, some of these newborns had either functional T-cell abnormalities and/or partial immune-recovery on follow-up. The authors hypothesize an importance of fully functioning IKAROS during intrauterine lymphocyte development (17). Murine studies indeed confirm the importance of IKZF1 for T-cell development (18). The biology of abnormalities in T-cell differentiation needs more scientific attention. Lastly, abnormalities of the dendritic cell (DC) compartment have also been described for IKAROS-HI patients (11). Heterozygous variants in humans have been shown to reduce peripheral DCs and to expand conventional DC1 numbers. This may indicate a regulatory effect of IKZF1 in human DC development (19).
All variants described in our manuscript—including the two novel IKZF1 germline missense variants p.Leu188Pro (Family B) and p.Leu177Pro (Family D)—lead to the clinical phenotype of IKAROS-HI. The number of functionally proven variants is consistently growing (Figures 5A,B), with genetic heterogeneity (Figure 5A) affecting Zinc finger domains 2 and 3, which are essential for DNA binding. In our cohort, functional testing indeed confirmed the lack of DNA binding capacity and PC-HC targeting in our reported IKZF1 variants without having dominant negative effect (Figure 3). The clinical and immunological phenotypes mostly depend on the localization of the mutation within IKAROS protein. This has been elucidated by Kuehn et al., who have dissected protein impairment due to haploinsufficiency, dominant negative, dimerization vs. GOF (8). Our cohort who has functionally validated IKAROS-HI, however presented with a much broader range and severity of symptoms from mild upper airway infections to severe invasive infectious complications. Therefore with the here presented cohort, we contribute to the already reported clinical spectrum of IKAROS-HI with respect to genetic variants, immunophenotype, age at manifestation, the variety of symptoms, severity of clinical presentation and therapeutic management. Nevertheless, the here reported patients show symptom overlaps with other disease phenotypes associated with IKZF1 genetic variants such as IKAROS-DD (e.g., immune cytopenia), which is illustrated in Figure 1. This challenges a strict genotype-phenotype correlation and illustrates the importance of the interplay between genetics, clinical symptoms, functional immunological workup as well as interprofessional care to make the correct diagnosis and patient management.
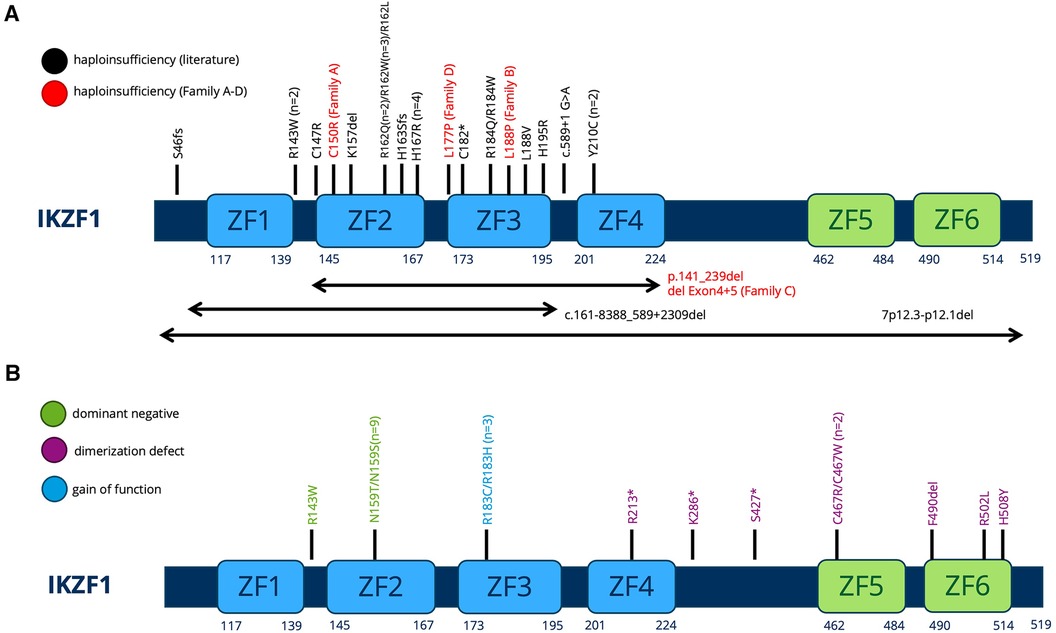
Figure 5. (A,B) Schematic of the IKAROS protein with indicated locations of the mutations of all patients previously described. The protein consists of 6 major Zinc finger (ZF) domains important for DNA binding and dimerization. (A) Variants leading to HI. Previously described variants are printed in black (PMID: 36433803). The families described in our manuscript are printed in red. (B) Dominant negative, Dimerization defect, Gain-of-function variants as previously described in PMID: 36433803.
Most patients reported in this manuscript have a sustained clinical response to IgRT. The patients with ITP were treated with corticosteroids and thrombopoietin-receptor-agonists. This is in in line with the successful conservative management of most IKAROS-HI patients in the literature: IgRT and prophylactic antibiotics for infection susceptibility, corticosteroids, high dose IgRT, rituximab for ITP and corticosteroids, immunosuppressants and anticoagulation for SLE and antiphospholipid syndrome (8, 12, 20). One of our patients received HSCT from his HLA-identical sister and shows persistent donor chimerism with cure from his IEI. This adds to the two previously reported patients with IKAROS-HI who underwent HSCT (8, 12, 20). As IKZF1 is expressed in hematologic cells only, HSCT is a potentially curative treatment option. As in other IEI, HSCT outcomes depend on patient age and pre-transplant organ damage (21). Hence, the decision for or against HSCT should be discussed on a case-by-case basis, and be a shared decision between patients, families and treating physicians.
Alongside with the lifelong risk of severe infectious complications, one of the most decisive arguments in favour of early HSCT might be the risk of developing hematologic malignancies. IKZF1 somatic variants are part of most oncological gene panels and a risk factor for poor outcome in hematological malignancies, mostly B-ALL (including pediatric B-cell precursor ALL) and entail more intense treatment protocols. Identification of somatic variants is relevant as these are of relevance in current protocols to guide tailoring of treatment intensity (22–24). IKZF1 is located on chromosome 7p12.2, and comprises 8 exons. The most frequent somatic variations in B-ALL are deletions of one or more exons (25), whereas missense variants are rather rare (26, 27). Additionally, IKZF1 copy-number variations have also been described especially in BCR-ABL1-positive ALL (25, 28).
Associations with hematologic malignancies are not restricted to somatic variants and have been reported in 2009 for germline IKZF1 variants and ALL (29). The association of these variants with hematologic malignancies was revealed through identification of IKAROS-deficient patients with CVID who also developed leukemia: the here reported variants were heterozygous, and the suspected mechanism of dominance was haploinsufficiency (12). An infant diagnosed with CID and a germline IKZF1 variant (attributed to DN IKAROS deficiency) later developed T cell leukaemia at the age of 13. In her T-ALL blasts, an additional NOTCH1 variant could be detected (30). This further illustrates hematological malignancies on the basis of germline IKZF1 variants, and sometimes additional somatic variants identified in hematological progenitors. Of note, hematological malignancies were reported in patients carrying variants leading to either HI, DN or DD (Figure 1).
Genetic screening of pediatric ALL cohorts, has revealed an accumulation of IKZF1 germline variants (missense, nonsense, frameshift) (31). This raises the question if germline genetic testing should be performed in all leukemia patients, especially when they report a history of severe infections, hypogammaglobulinemia or other IKAROS-related symptoms, or in case of a positive family history (32, 33).
Pediatricians of any specialization should be vigilant when looking after patients with recurrent infections and signs of autoinflammation or autoimmunity, and/or malignancies. Taking a careful family history and initiating basic immunological workup is at the reach of most professional caretakers around the globe. In case of clinical manifestations, a combination of warning signs and/or abnormal immunophenotyping, suggestive of possible IEI genetic testing should be initiated without delay, in order not to miss variants in genes such as IKZF1. This may influence prophylactic and potentially curative treatment decisions like HSCT. Additionally, the risk to develop malignant disease has an impact on medical management. With this report we have provided further insight into the genotype-phenotype-correlation of IKAROS-HI and its astonishing clinical variability and lifethreatening complications, as in one individual of our cohort. The growing number of variants reported will help to broaden our understanding of gene dosage effects and understand additional mechanisms impairing IKAROS function.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical approval was not required for the study involving human samples in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
TS: Conceptualization, Data curation, Visualization, Writing – original draft, Writing – review & editing, Investigation. JK: Conceptualization, Data curation, Visualization, Writing – original draft, Writing – review & editing, Investigation. HK: Data curation, Investigation, Methodology, Writing – review & editing. AG: Data curation, Writing – review & editing. FT: Writing – review & editing. KT-G: Writing – review & editing. AS: Methodology, Writing – review & editing. LP: Visualization, Writing – review & editing. RW: Writing – review & editing, Methodology. VW: Writing – review & editing. HB: Writing – review & editing. SR: Methodology, Writing – review & editing. MF: Writing – original draft, Writing – review & editing, Investigation. RK: Writing – original draft, Writing – review & editing, Investigation. CS: Conceptualization, Data curation, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by Else-Kröner-Fresenius-Stiftung (EKFK) to TS and JK. Rosemarie-Germscheid Stiftung (Foundation) to CS. HK and SR are supported by the Intramural Research Program, NIH Clinical Center and the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH), USA. The publication fee was financed by SLUB Dresden.
Acknowledgments
We are grateful to the patients, their families and referring physicians who made this study possible. We thank Sebastian Thieme for helping with figure formatting and scientific advice.
Conflict of interest
Author AS and HB were employed by the company Labor Berlin Charité-Vivantes GmbH and author MF was employed by the company Hospital St. Georg GGmbH Leipzig.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2024.1345730/full#supplementary-material
References
1. Georgopoulos K, Bigby M, Wang J-H, Molnar A, Wu P, Winandy S, et al. The IKAROS gene is required for the development of all lymphoid lineages. Cell. (1994) 79:143–56. doi: 10.1016/0092-8674(94)90407-3
2. Wang J-H, Nichogiannopoulou A, Wu L, Sun L, Sharpe AH, Bigby M, et al. Selective defects in the development of the fetal and adult lymphoid system in mice with an IKAROS null mutation. Immunity. (1996) 5:537–49. doi: 10.1016/S1074-7613(00)80269-1
3. Winandy S, Wu P, Georgopoulos K. A dominant mutation in the IKAROS gene leads to rapid development of leukemia and lymphoma. Cell. (1995) 83:289–99. doi: 10.1016/0092-8674(95)90170-1
4. Boutboul D, Kuehn HS, Van De Wyngaert Z, Niemela JE, Callebaut I, Stoddard J, et al. Dominant-negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest. (2018) 128:3071–87. doi: 10.1172/JCI98164
5. Kuehn HS, Nunes-Santos CJ, Rosenzweig SD. Germline IKZF1 mutations and their impact on immunity: IKAROS-associated diseases and pathophysiology. Expert Rev Clin Immunol. (2021) 17:407–16. doi: 10.1080/1744666X.2021.1901582
6. Eskandarian Z, Fliegauf M, Bulashevska A, Proietti M, Hague R, Smulski CR, et al. Assessing the functional relevance of variants in the IKAROS family zinc finger protein 1 (IKZF1) in a cohort of patients with primary immunodeficiency. Front Immunol. (2019) 10:568. doi: 10.3389/fimmu.2019.00568
7. Banday AZ, Jindal AK, Kaur A, Saka R, Parwaiz A, Sachdeva MUS, Rawat A. Cutaneous IgA vasculitis-presenting manifestation of a novel mutation in the IKZF1 gene. Rheumatology (Oxford). (2021) 60:e101–3. doi: 10.1093/rheumatology/keaa492
8. Kuehn HS, Boast B, Rosenzweig SD. Inborn errors of human IKAROS: lOF and GOF variants associated with primary immunodeficiency. Clin Exp Immunol. (2023) 212:129–36. doi: 10.1093/cei/uxac109
9. Chen Q, Wang X, Wang W, Zhou Q, Liu D, Wang Y. B-cell deficiency: a de novo IKZF1 patient and review of the literature. J Investig Allergol Clin Immunol. (2018) 28:53–6. doi: 10.18176/jiaci.0207
10. Kuehn HS, Nunes-Santos CJ, Rosenzweig SD. IKAROS-associated diseases in 2020: genotypes, phenotypes, and outcomes in primary immune deficiency/inborn errors of immunity. J Clin Immunol. (2021) 41:1–10. doi: 10.1007/s10875-020-00936-x
11. Nunes-Santos CJ, Kuehn HS, Rosenzweig SD. IKAROS family zinc finger 1–associated diseases in primary immunodeficiency patients. Immunol Allergy Clin North Am. (2020) 40:461–70. doi: 10.1016/j.iac.2020.04.004
12. Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, et al. Loss of B cells in patients with heterozygous mutations in IKAROS. N Engl J Med. (2016) 374:1032–43. doi: 10.1056/NEJMoa1512234
13. Hoshino A, Okada S, Yoshida K, Nishida N, Okuno Y, Ueno H, et al. Abnormal hematopoiesis and autoimmunity in human subjects with germline IKZF1 mutations. J Allergy Clin Immunol. (2017) 140:223–31. doi: 10.1016/j.jaci.2016.09.029
14. Bogaert DJ, Kuehn HS, Bonroy C, Calvo KR, Dehoorne J, Vanlander AV, et al. A novel IKAROS haploinsufficiency kindred with unexpectedly late and variable B-cell maturation defects. J Allergy Clin Immunol. (2018) 141:432–435.e7. doi: 10.1016/j.jaci.2017.08.019
15. Kellner ES, Krupski C, Kuehn HS, Rosenzweig SD, Yoshida N, Kojima S, et al. Allogeneic hematopoietic stem cell transplant outcomes for patients with dominant negative IKZF1/IKAROS mutations. J Allergy Clin Immunol. (2019) 144:339–42. doi: 10.1016/j.jaci.2019.03.025
16. Hoshino A, Boutboul D, Zhang Y, Kuehn HS, Hadjadj J, Özdemir N, et al. Gain-of-function IKZF1 variants in humans cause immune dysregulation associated with abnormal T/B cell late differentiation. Sci Immunol. (2022) 7:eabi7160. doi: 10.1126/sciimmunol.abi7160
17. Kuehn HS, Gloude NJ, Dimmock D, Tokita M, Wright M, Rosenzweig SD, et al. Abnormal SCID newborn screening and spontaneous recovery associated with a novel haploinsufficiency IKZF1 mutation. J Clin Immunol. (2021) 41:1241–9. doi: 10.1007/s10875-021-01035-1
18. Boast B, Miosge LA, Kuehn HS, Cho V, Athanasopoulos V, McNamara HA, et al. A point mutation in IKAROS ZF1 causes a B cell deficiency in mice. J Immunol. (2021) 206:1505–14. doi: 10.4049/jimmunol.1901464
19. Cytlak U, Resteu A, Bogaert D, Kuehn HS, Altmann T, Gennery A, et al. IKAROS family zinc finger 1 regulates dendritic cell development and function in humans. Nat Commun. (2018) 9:1239. doi: 10.1038/s41467-018-02977-8
20. Goldman FD, Gurel Z, Al-Zubeidi D, Fried AJ, Icardi M, Song C, et al. Congenital pancytopenia and absence of B lymphocytes in a neonate with a mutation in the IKAROS gene. Pediatr Blood Cancer. (2012) 58:591–7. doi: 10.1002/pbc.23160
21. Albert MH, Sirait T, Eikema D-J, Bakunina K, Wehr C, Suarez F, et al. Hematopoietic stem cell transplantation for adolescents and adults with inborn errors of immunity: an EBMT IEWP study. Blood. (2022) 140:1635–49. doi: 10.1182/blood.2022015506
22. Stanulla M, Dagdan E, Zaliova M, Möricke A, Palmi C, Cazzaniga G, et al. IKZF1plus defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric B-cell precursor acute lymphoblastic leukemia. J Clin Oncol. (2018) 36:1240–9. doi: 10.1200/JCO.2017.74.3617
23. Olsson L, Ivanov Öfverholm I, Norén-Nyström U, Zachariadis V, Nordlund J, Sjögren H, et al. The clinical impact of IKZF1 deletions in paediatric B-cell precursor acute lymphoblastic leukaemia is independent of minimal residual disease stratification in nordic society for paediatric haematology and oncology treatment protocols used between 1992 and 2013. Br J Haematol. (2015) 170:847–58. doi: 10.1111/bjh.13514
24. Boer JM, van der Veer A, Rizopoulos D, Fiocco M, Sonneveld E, de Groot-Kruseman HA, et al. Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia. (2016) 30:32–8. doi: 10.1038/leu.2015.199
25. Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of IKAROS. Nature. (2008) 453:110–4. doi: 10.1038/nature06866
26. Mullighan CG, Su X, Zhang J, Radtke I, Phillips LAA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. (2009) 360:470–80. doi: 10.1056/NEJMoa0808253
27. Vairy S, Tran TH. IKZF1 alterations in acute lymphoblastic leukemia: the good, the bad and the ugly. Blood Rev. (2020) 44:100677. doi: 10.1016/j.blre.2020.100677
28. van der Veer A, Zaliova M, Mottadelli F, De Lorenzo P, Te Kronnie G, Harrison CJ, et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood. (2014) 123:1691–8. doi: 10.1182/blood-2013-06-509794
29. Treviño LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. (2009) 41:1001–5. doi: 10.1038/ng.432
30. Yoshida N, Sakaguchi H, Muramatsu H, Okuno Y, Song C, Dovat S, et al. Germline IKAROS mutation associated with primary immunodeficiency that progressed to T-cell acute lymphoblastic leukemia. Leukemia. (2017) 31:1221–3. doi: 10.1038/leu.2017.25
31. Churchman ML, Qian M, Te Kronnie G, Zhang R, Yang W, Zhang H, et al. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell. (2018) 33:937–48.e8. doi: 10.1016/j.ccell.2018.03.021
32. van der Werff Ten Bosch J, van den Akker M. Genetic predisposition and hematopoietic malignancies in children: primary immunodeficiency. Eur J Med Genet. (2016) 59:647–53. doi: 10.1016/j.ejmg.2016.03.001
Keywords: IKZF1, IKAROS, transcription factor, haploinsufficiency, immunodeficiency
Citation: Strauss T, Körholz J, Kuehn HS, Gil Silva AA, Taube F, Trautmann-Grill K, Stittrich A, Pietzsch L, Wiedemuth R, Wahn V, von Bernuth H, Rosenzweig SD, Fasshauer M, Krüger R and Schuetz C (2024) IKAROS—how many feathers have you lost: mild and severe phenotypes in IKZF1 deficiency. Front. Pediatr. 12:1345730. doi: 10.3389/fped.2024.1345730
Received: 28 November 2023; Accepted: 15 April 2024;
Published: 9 May 2024.
Edited by:
Guzide Aksu, Ege University, TürkiyeReviewed by:
Ankur Kumar Jindal, Post Graduate Institute of Medical Education and Research (PGIMER), IndiaSinisa Dovat, Penn State Hershey Children's Hospital, United States
© 2024 Strauss, Körholz, Kuehn, Gil Silva, Taube, Trautmann-Grill, Stittrich, Pietzsch, Wiedemuth, Wahn, von Bernuth, Rosenzweig, Fasshauer, Krüger and Schuetz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timmy Strauss, timmy.strauss@ukdd.de
Catharina Schuetz, catharina.schuetz@ukdd.de
†These authors share first authorship