 Ziad T. Basuni1
Ziad T. Basuni1 Dania A. Monagel
Dania A. Monagel Amany Ahmed
Amany Ahmed- 1Department of Oncology, Ministry of the National Guard- Health Affairs, Jeddah, Saudi Arabia
- 2King Abdullah International Medical Research Center, Jeddah, Saudi Arabia
- 3College of Medicine, King Saud bin Abdul-Aziz University for Health Sciences, Jeddah, Saudi Arabia
Introduction: Sickle cell disease (SCD) is a common inherited blood disorder characterized by the production of abnormal sickle-shaped red blood cells. SCD can lead to various complications including neurological issues. Early detection and treatment are crucial for preventing these complications. This study aimed to describe the neurological manifestations, radiological findings, and neurological diagnosis related to SCD in Saudi children with the aim of contributing to the formulation of population-based guidelines for screening and treating SCD-related neurological complications.
Methods: This descriptive retrospective study included pediatric patients aged < 14 years diagnosed with SCD who were regularly followed up at the hematology clinic in KAMC, Jeddah, Saudi Arabia, from January 2008 to January 2022. Demographic and clinical data were collected from the clinical charts of 101 participants.
Results: This study included 101 patients with SCD with a mean age of 23 months at diagnosis. Among these, 59% had SCD and high fetal hemoglobin (HbF) levels. Neurological sequelae, including seizures, stroke, and other abnormalities, were observed in 26.7% of patients. There were no significant differences in the onset of neurological issues between the patients with SCD-high HbF and those with other SCD phenotypes.
Discussion: This study highlights the increased risk of brain injury and neurocognitive deficits in children with SCD. The occurrence of neurological sequelae in many patients emphasizes the need for early detection and intervention. Some patients experience neurological complications despite having high HbF levels, suggesting that further interventions are needed. This study has some limitations, including its small sample size and retrospective nature.
Conclusion: Early detection and intervention are crucial for neurological complications in patients with SCD. This study emphasizes the need for further research and effective treatment strategies considering the presence of neurological complications despite the presence of high HbF levels. Large-scale studies and population-specific guidelines are warranted for better understanding and management of SCD-related neurological complications in the Saudi population.
Background
Sickle cell disease (SCD) is one of the most common single-gene mutations worldwide (1). Mutation in the beta-globin gene leads to the production of sickle hemoglobin, which has a reduced solubility compared to regular fetal (HbF) or adult hemoglobin (HbA). This contributes to hemoglobin polymerization and abnormal red blood cell shapes, resulting in vessel blockage and ischemia-reperfusion injury (2). Individuals with either a homozygous disease state or a compound heterozygous state involving sickle hemoglobin (HbS) are generally affected by SCD (2).
Molecular analysis of patients with sickle cell anemia SCD revealed the existence of significant haplotypes, including African and Arab-Indian (AI) haplotypes, also known as the Saudi haplotype. The AI haplotype is commonly found in the eastern region of Saudi Arabia and other Arab Gulf countries. Although the genetic abnormality responsible for the sickle cell gene is the same in all haplotypes, there is considerable variability in the clinical presentation and severity of the disease (3). Previous studies indicated that African haplotypes exhibit more severe and clinically significant manifestations of SCD, whereas individuals with the AI haplotype typically experience milder clinical symptoms (4, 5).
In Saudi Arabia, two prevalent haplotypes have been observed: the AI and Benin (African) haplotypes (6). Studies have indicated that individuals with the AI haplotype typically exhibit higher HbF levels and tend to experience milder symptoms of SCD, although the disease is not asymptomatic in these cases (7).
SCD is a complex disorder that affects multiple body systems and leads to high morbidity and mortality rates. Neurological complications are common and severe in SCD. This disease can cause neurological problems including stroke, seizures, transient ischemic attack (TIA), posterior reversible encephalopathy (PRES), moyamoya syndrome, silent cerebral infarcts, cognitive impairment, and neuropathic pain (8).
A retrospective cohort study of 189 newborns screened and diagnosed with SCD found that silent cerebral infarction was the most common cause of permanent neurological injury in children and adults with sickle hemoglobin (HbSS) and hemoglobin beta thalassemia (HbSb0 thalassemia). By the age of 18 years, approximately 39% of the children had experienced a silent cerebral infarct (9). Both stroke and silent cerebral infarcts are associated with significant cognitive impairment, resulting in an average 5-point decrement in full-scale IQ scores and negative impacts on educational attainment, employment status, and overall quality of life (8, 10). Children with SCD are at a higher risk of stroke, especially if they have abnormal transcranial doppler ultrasound (TCD) test results. Therefore, early detection and treatment are critical for preventing neurological complications of SCD (11).
To the best of our knowledge, there are no Saudi national data regarding the neurological outcomes in pediatric patients with SCD. In this retrospective study, we reviewed the neurological manifestations, radiological findings, and neurological complications related to SCD in children. We hypothesized that Saudi children with SCD would have higher HbF concentrations. We performed a subgroup analysis in patients with SCD associated with high HbF to determine if they have a lower rate of neurological sequelae due to the effect of circulating HbF. In future, our results could be combined with other available evidence to formulate population-based guidelines for the screening and treatment of SCD-related neurological complications.
Methods
This descriptive retrospective study included all patients who were diagnosed with (HbSS, HbSS with high HbF, HbS-β thalassemia, and HbSC) and were younger than 14 years of age (as this is the upper age limit for the pediatric age group in our hospital). The group is regularly followed up at the hematology clinic at King Abdulaziz Medical City in Jeddah (KAMC-J) from January 2008 to January 2022.
A chart review was conducted between January 2021 and January 2022. Patients older than 14 years old, with sickle cell traits (HbAS), or lost to follow-up at KAMC-J were excluded. The research team reviewed the clinical charts of all the participants (N = 101) to collect demographic and clinical data using a Case Report Form (CRF) and the best care system. This study was approved by the King Abdullah International Medical Research Center (KAIMRC).
The Data collected included the patient's demographics, SCD diagnosis-related information (age at diagnosis since universal newborn screening for SCD is not available; SCD phenotype; genetics if available; and high HbF at initial diagnosis), and SCD management as hydroxyurea and transfusion history. Details of SCD complications, particularly neurological issues, were identified. Radiological images, including TCD and Magnetic resonance image (MRI), were reviewed. All patients with SCD in our clinic, regardless of genotype, underwent baseline TCD and MRI (can be performed without general anesthesia).
Our study included patients with SCD and high HbF levels as part of our subgroup analysis. Although there is no universally standardized definition of high HbF levels, Steinbergin et al. extensively reviewed the topic and found that HbF levels in SCD depend on several factors. However, they concluded that an HbF level of 20.7 ± 8.2% can be considered high (12). At our institution, a high HbF level was defined as 20% or higher.
Statistical analysis
The analysis was performed using SPSS Statistics version 29 for Macintosh. Mean, median, range, and standard deviation (SD) were used for continuous variables, and frequency distributions were used to assess categorical data. The independent sample t-test, one-way ANOVA, and χ2 test were used to compare groups, and a p-value <0.05 was considered significant.
Results
Patient demographics
Between January 2021 and January 2022, data were collected from 101 patients who met the inclusion criteria. Of these, 58 were male and 43 were female. The mean age at diagnosis was 23 months (SD = 20) and that at data collection was 105 months (SD = 44). Based on Hb electrophoresis analysis before the initiation of hydroxyurea (HU) treatment 59 (58%) patients had HbSS with high HbF, 23 (22.8%) had HbSS disease, 18 (17.8%) had sickle beta thalassemia, and 1 (1%) had HbSC disease.
Clinical characteristics
Five patients underwent whole exome sequencing (WES) in addition to SCD diagnosis, which revealed the following mutations: G6PD (n = 2), SPTA1 (n = 1), alpha thalassemia (n = 1), a compound heterozygous mutation causing sickle beta thalassemia (n = 1), and a FANCA variant of unknown significance (n = 1). WES is not a standard of care, but was performed in cases with other clinical indications.
Of the 101 patients included in the study, 81 (80%) underwent Hb electrophoresis at the time of diagnosis before starting HU; mean HbF: 26% (SD = 11.3) and HbS: 69% (SD = 10.3). Only 12 (11.8%) patients had no reported crises in the study center, with a mean number of admissions per patient of 6.4 (SD = 7.7) and an average of 0.8 admissions per patient per year (SD = 0.9). Additionally, 21% of patients had at least one admission to the pediatric intensive care unit. Figure 1 summarizes the most common SCD-related crises (other than neurological issues) in this cohort.
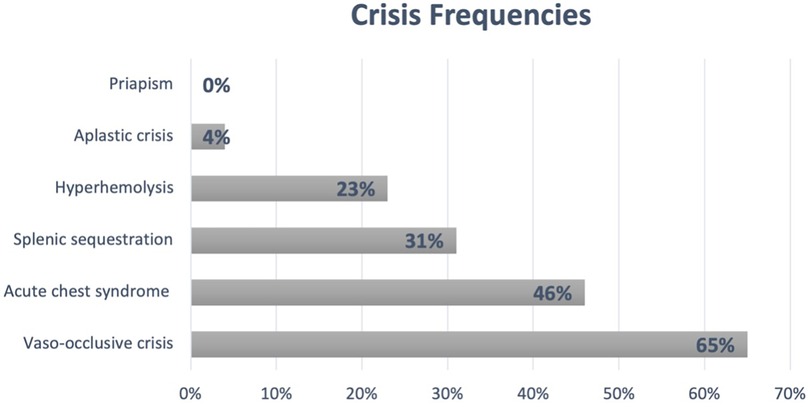
Figure 1. SCD crisis frequencies. Patients who experienced at least one episode.
The mean Hb, white blood cell (WBC), neutrophil, and platelet counts prior to HU initiation were 8.9 g/dl (SD = 1), 12 × 109/L (SD = 4.3), 4.3 × 109/L (SD = 4.2), and 421 × 109/L (SD = 192) respectively. Eighty-five patients underwent baseline TCD and 75 underwent at least one MRI/MRA.
The study found that 98% of participants (n = 99) were started on HU as a disease-modifying therapy, with a mean age at HU initiation of 46 months (SD = 32) and a mean maximum tolerated dose of HU of 21 mg/kg/day (SD = 9.3). Thirty percent of the patients required one or more exchange transfusions for different indications, including stroke, abnormal TCD, high hemoglobin level at the time of crisis, or preoperative preparation for any procedure, whereas 61% (n = 62) required simple blood transfusions, with 47% of the transfused group requiring five or more red blood cells (RBC) transfusions.
Neurological sequelae
Of the 101 patients in the study, 27 (26.7%) had neurological sequelae (clinical or radiological), of whom 15 were male and 12 were female (Table 1). The mean age at sickle cell anemia diagnosis was 21 months (SD = 20) and 55% (n = 15) had sickle cell anemia with high HbF levels, 26% (n = 7) had HbSS disease, 15% (n = 4) had sickle beta thalassemia, and 3% (n = 1) had HbSC based on hemoglobin electrophoresis prior to starting HU.The mean Hb, WBC, neutrophil, and platelet counts before starting HU were 8.7 g/dl (SD = 1), 11.5 × 109/L (SD = 3.3), 5.5 × 109/L (SD = 6.8) and 471 × 109/L (SD = 193), respectively. The mean age at the onset of neurological issues was 83 months (SD = 41), with 19% of affected children (n = 5) being ≤ 36 months old at the onset of the neurological abnormality. Eight patients had seizures, four had overt stroke, one had moyamoya syndrome, 13 had only radiological changes, one had sensory neuronal hearing loss, and one reported deterioration in school performance based on IQ assessment. The mean Hb, WBC, and platelet counts, and creatinine and urea levels at time of neurological insult were 8.7 g/dl (SD = 1.04), 11.7 × 109/L (SD = 4.7), and 460 × 109/L (SD = 207), and 36 µmol/L (SD = 8.2) and 4.4 mmol/L (SD = 9.3), respectively.
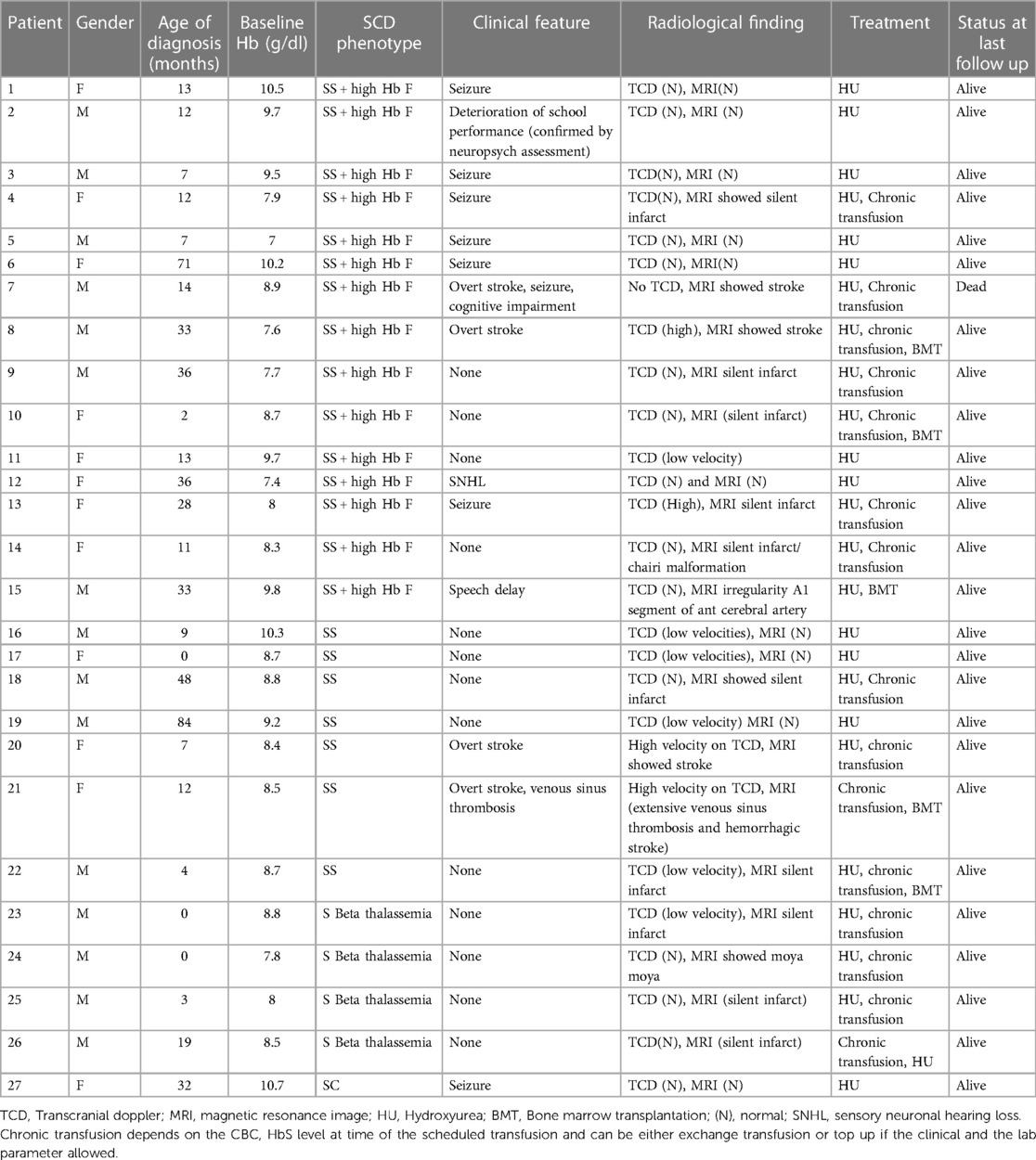
Table 1. Patients with neurological sequalae characteristics.
All patients except one were receiving HU, with a mean age of 54 months (SD = 36 months) at initiation. The mean maximum tolerated dose of HU was 21 mg/kg/day (SD = 9). All patients with stroke, silent infarct, or abnormal TCD were placed in a chronic transfusion program, and four patients underwent bone marrow transplantation.
Although a significant proportion of patients with neurological injury had sickle cell-high HbF (56%), there was no statistically significant difference between patients with sickle cell-high HbF and those with other SCD phenotypes (p = 0.7). The mean age of onset of neurological diagnosis in children with sickle cell-high HbF ( months, SD = 42) and those with other SCD phenotypes (, SD = 37) was not significantly different [t(25) = −1.4, p = 0.9]. Table 2 summarizes the clinical and hematological profiles of patients with and without neurological complications.
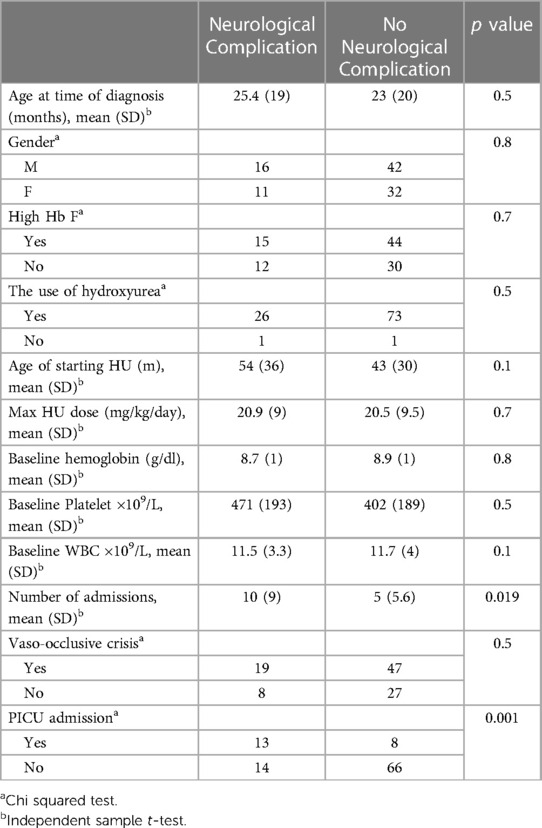
Table 2. A comparison of the medical profile of children with SCD with and without neurological complication.
Discussion
SCD predisposes children to an increased risk of developing brain injury (13). Patients with neurological insults develop a further decline in academic function and neurocognitive deficits, typically manifesting as executive dysfunction (14). The findings of this study provide a broad overview of the clinical and hematological profiles of Saudi children with SCD and highlight the occurrence of neurological sequelae in a significant proportion of patients.
SCD causes a broad range of neurological complications. Stroke is a significant contributor to morbidity and mortality in SCD, and The Cooperative Study of Sickle Cell Disease, which monitored approximately 4,082 adults and children over five years, showed a stroke prevalence of 3.75% with two peaks at 2–5 and >30 years. Interestingly, asymptomatic silent cerebral infarcts develop in 27% of patients before the age of 6 years and in 39% by 18 years of age. Moyamoya syndrome is a vasculopathy that develops in 30%–40% of patients and manifests at a young age. In addition, individuals with SCD have a higher risk of epilepsy. Nigeran et al. showed that seizures were 2–10 times more frequent in patients with SCD than in the general population. Other complications include PRES, paraplegia due to cord infarction, and venous sinus thrombosis (15, 16). In our cohorts, the most common manifestations of SCD were picked by routine surveillance (TCD followed by MRI) in asymptomatic participants, and in patients with seizures and stroke. Other documented complications include poor school performance, cerebral venous thrombosis, speech delay, and hearing loss. Most of our patients with neurological injuries received optimal supportive care measures, including HU, and there was only one death related to stroke and its complications. The study further highlights the role of HU, exchange transfusion, and simple blood transfusion in preventing further neurological damage in patients with SCD and neurological sequelae, as confirmed previously in international trials (11, 17). The study also identified various accompanying genetic mutations other than the sickle cell gene; although not routinely performed, it is important to consider genetic testing to identify other genes that may play a role in disease modification.
There is controversial evidence regarding the protective effects of HbF; by 5–10 years of age, children with SCD usually achieve stable HbF levels. The gene cluster in SCD has four main haplotypes: Bantu, Senegal, Benin, and AI. The highest HbF levels were found in the AI and Senegal haplotypes; these individuals usually have milder disease, although they can still be anemic and symptomatic. The modulatory effect of high HbF levels is induced by the inhibition of deoxy-HbS polymerization, which is one of the many functions of HbF. The HbF concentration in the blood does not represent HbF/F cells (F cells = number of cells with detectable HbF), meaning that HbF is not evenly distributed among F cells. Therefore, some cells have inadequate concentrations of HbF to protect against HbS polymerization, which explains why even patients with high HbF levels can still develop severe disease (18). In a cross-sectional study by Steinberg et al., high HbF in SCD was correlated with fewer pain crises and leg ulcers and lower occurrence rates of avascular necrosis and acute chest syndrome. Nonetheless, there was no apparent link to SCD nephropathy, blood pressure issues, priapism, stroke, or silent cerebral infarcts (19).
Furthermore, the majority of Saudi patients with SCD (AI haplotype) had high HbF levels and were likely to have milder disease. However, our data showed that even in this group, neurological complications (clinical or radiological) were observed in a significant number of patients. There were no statistically significant differences when comparing this finding between patients with high HbF levels and those with other SCD phenotypes. This finding suggests that the proposed protective effect of high HbF levels may not be sufficient to prevent neurological complications in patients with SCD and that the course of illness may not always be as benign as expected.
Clinical observations in Saudi patients with SCD by Sultan et al. demonstrated that the disease has a milder phenotype in children with HbF concentrations close to 30%, but symptoms become more prevalent in adults as the HbF concentration falls to 15%–20% (6). In contrast, we found that the mean age at SCD diagnosis was 21 months, which underscores the early onset of signs and symptoms of the disease, regardless of a high HbF status. Additionally, nearly one-fifth of the affected children were ≤ 36 months old at the onset of neurological abnormalities. This observation emphasizes that a high HbF level is one of our population's most potent genetic modifiers; however, other modifiers may counteract the effect of HbF and therefore further population-based studies are needed.
Neuropsychological assessments and sequelae in pediatric patients with SCD have not been sufficiently studied in our Saudi Patients with SCD. Gruntorad et al. had performed neurocognitive evaluations for 20 children with SCD and demonstrated significant neurocognitive deficits even in cases without major clinical or radiological changes. They highlighted that patients with SCD at increased risk not only for cognitive deterioration but also can suffer from other mental and psychological disorders such as attention-deficit hyperactivity disorder, adjustment disorder, depressed mood, impulse control, and conduct disorders (20).
Neurocognitive decline is usually linked to important illness-related factors, such as sickle cell genotype, neuroimaging findings, and the presence or absence of overt/silent infarcts. These children usually suffer from IQ deficits, attention/memory issues, and disorders of language/math/spelling/reading, with poor academic and career achievements overall. The availability of clinical neuropsychologists is extremely important in SCD programs. Their roles usually guide the selection of the most appropriate tools and answer pertinent questions while providing good insight into developmental considerations. Unfortunately, the lack such services in most of our centers affects multiple aspects of children's lives, including the proper transition into adulthood (21).
Overall, the findings of this study have important implications for the management and treatment of patients with SCD in Saudi Arabia and emphasize the need for continued research into effective treatment strategies to prevent or minimize the occurrence of neurological complications. In conclusion, this study provides valuable insights into the clinical and hematological profiles of patients with SCD and highlights the significant occurrence of neurological sequelae in affected patients. Despite the protective effects of high HbF levels, many patients still experience neurological complications, emphasizing the need for early detection and intervention. Future research should focus on a larger-scale multicenter national prospective study to better characterize the nature of neurological complications and develop a well-accepted national guideline for the Saudi population.
Limitations
Our study had some limitations. First, it had a relatively small sample size (101 patients), which may limit the generalizability of the findings. Second, the study was conducted at a single center, which may limit the representativeness of the patient population. However, it is a tertiary care center that offers well-defined supportive care and bone marrow transplantation. Third, the lack of formal neurocognitive assessment is a significant limitation in assessing the population with SCD; in particular, it has a major impact on future performance. Unfortunately, our clinic as well is not based on multidisciplinary approach and the involvement of the neurologist is usually based on the case upon discretion of treating physician. Finally, this study relied on a retrospective review of medical records, which may have introduced bias or incomplete data.
Data availability statement
The datasets presented in this article are not readily available to maintain the confidentiality of our subjects. Requests to access the datasets should be directed toZGFuaWFfbW9uYWdlbEBob3RtYWlsLmNvbQ==.
Ethics statement
The studies involving humans were approved by King Abdullah International Medical Research Center. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
ZB: Conceptualization, Data curation, Formal Analysis, Investigation, Writing – original draft. DM: Conceptualization, Formal Analysis, Investigation, Methodology, Resources, Software, Supervision, Validation, Writing – original draft, Writing – review & editing. AT: Conceptualization, Data curation, Methodology, Writing – original draft. NA: Conceptualization, Data curation, Methodology, Resources, Writing – original draft. AA: Conceptualization, Data curation, Methodology, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We would like to thank Editage (http://www.editage.com) for editing and reviewing this manuscript for English language.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet (London, England). (2010) 376(9757):2018–31. doi: 10.1016/S0140-6736(10)61029-X
2. Brewin J, Kaya B, Chakravorty S. How i manage sickle cell patients with high transcranial Doppler results. Br J Haematol. (2017) 179(3):377–88. doi: 10.1111/bjh.14850
3. Jastaniah W. Epidemiology of sickle cell disease in Saudi Arabia. Ann Saudi Med. (2011) 31(3):289–93. doi: 10.4103/0256-4947.81540
4. Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore). (2005) 84(6):363–76. doi: 10.1097/01.md.0000189089.45003.52
5. Powars DR, Schroeder WA, Weiss JN, Chan LS, Azen SP. Lack of influence of fetal hemoglobin levels or erythrocyte indices on the severity of sickle cell anemia. J Clin Invest. (1980) 65(3):732–40. doi: 10.1172/JCI109720
6. Alsultan A, Alabdulaali MK, Griffin PJ, Alsuliman AM, Ghabbour HA, Sebastiani P, et al. Sickle cell disease in Saudi Arabia: the phenotype in adults with the arab-Indian haplotype is not benign. Br J Haematol. (2014) 164(4):597–604. doi: 10.1111/bjh.12650
7. Akinsheye I, Alsultan A, Solovieff N, Ngo D, Baldwin CT, Sebastiani P, et al. Fetal hemoglobin in sickle cell anemia. Blood. (2011) 118(1):19–27. doi: 10.1182/blood-2011-03-325258
8. DeBaun MR, Jordan LC, King AA, Schatz J, Vichinsky E, Fox CK, et al. American society of hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv. (2020) 4(8):1554–88. doi: 10.1182/bloodadvances.2019001142
9. Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Vasile M, Kasbi F, et al. Chronic and acute anemia and extracranial internal carotid stenosis are risk factors for silent cerebral infarcts in sickle cell anemia. Blood. (2015) 125(10):1653–61. doi: 10.1182/blood-2014-09-599852
10. Kassim AA, Galadanci NA, Pruthi S, DeBaun MR. How i treat and manage strokes in sickle cell disease. Blood. (2015) 125(22):3401–10. doi: 10.1182/blood-2014-09-551564
11. Lee MT, Piomelli S, Granger S, Miller ST, Harkness S, Brambilla DJ, et al. Stroke prevention trial in sickle cell anemia (STOP): extended follow-up and final results. Blood. (2006) 108(3):847–52. doi: 10.1182/blood-2005-10-009506
12. Steinberg MH. Fetal hemoglobin in sickle hemoglobinopathies: high HbF genotypes and phenotypes. J Clin Med. (2020) 9(11):3782. PMID: 33238542; PMCID: PMC7700170. doi: 10.3390/jcm9113782
13. Sarnaik SA, Lusher JM. Neurological complications of sickle cell anemia. Am J Pediatr Hematol Oncol. (1982) 4(4):386–94. doi: 10.1097/00043426-198224000-00006
14. Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet (London, England). (2013) 381(9861):142–51. doi: 10.1016/S0140-6736(12)61229-X
15. Farooq S, Testai FD. Neurologic complications of sickle cell disease. Curr Neurol Neurosci Rep. (2019) 19(4):17. doi: 10.1007/s11910-019-0932-0
16. Lagunju IA, Brown BJ. Adverse neurological outcomes in Nigerian children with sickle cell disease. Int J Hematol. (2012) 96(6):710–8. doi: 10.1007/s12185-012-1204-9
17. Ware RE, Davis BR, Schultz WH, Brown RC, Aygun B, Sarnaik S, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial Doppler flow velocities in children with sickle cell anaemia-TCD with transfusions changing to hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial. Lancet (London, England). (2016) 387(10019):661–70. doi: 10.1016/S0140-6736(15)01041-7
18. Steinberg MH, Chui DH, Dover GJ, Sebastiani P, Alsultan A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. (2014) 123(4):481–5. doi: 10.1182/blood-2013-09-528067
19. Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. Am J Hematol. (2012) 87(8):795–803. doi: 10.1002/ajh.23232
20. Gruntorad S, Searcy J, Peddinti R. Neurocognitive testing in pediatric sickle cell disease: full scale IQ assessment may not be the full story. Blood. (2019) 134(Supplement_1):4833. doi: 10.1182/blood-2019-129178
Keywords: sickle cell disease, pediatric, neurological complications, stroke, seizure, transient ischemic attack, high hemoglobin F
Citation: Basuni ZT, Monagel DA, Taha A, Ahmed N and Ahmed A (2024) Neurological abnormalities among pediatric patients with sickle cell disease in Saudi Arabia: a single-center retrospective study. Front. Pediatr. 11:1290314. doi: 10.3389/fped.2023.1290314
Received: 7 September 2023; Accepted: 26 December 2023;
Published: 10 January 2024.
Edited by:
Carsten Heilmann, Rigshospitalet, DenmarkReviewed by:
Grace Onimoe, The MetroHealth System, United StatesMarianne Hutchings Hoffmann, University of Copenhagen, Denmark
© 2024 Basuni, Monagel, Taha, Ahmed and Ahmed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dania A. Monagel ZGFuaWFfbW9uYWdlbEBob3RtYWlsLmNvbQ==
Abbreviations HbS, sickle hemoglobin; HbF, fetal hemoglobin; HbA, adult hemoglobin; SCD, sickle cell disease; QoL, quality of Life; KAIMRC, king abdullah international medical research center; NGHA, national guard health affairs; SPSS, statistical package for the social sciences.