
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Pediatr., 21 November 2023
Sec. Obstetric and Pediatric Pharmacology
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1288376
This article is part of the Research TopicSupporting the Pediatric Drug Development: From Basic Research to Clinical Studies and Technological AdvancementsView all 9 articles
 Marika A. de Hoop-Sommen1*
Marika A. de Hoop-Sommen1* Joyce E. M. van der Heijden1
Joyce E. M. van der Heijden1 Jolien J. M. Freriksen1
Jolien J. M. Freriksen1 Rick Greupink1
Rick Greupink1 Saskia N. de Wildt1,2,3
Saskia N. de Wildt1,2,3
Introduction: Modeling and simulation can support dosing recommendations for clinical practice, but a simple framework is missing. In this proof-of-concept study, we aimed to develop neonatal and infant gentamicin dosing guidelines, supported by a pragmatic physiologically-based pharmacokinetic (PBPK) modeling approach and a decision framework for implementation.
Methods: An already existing PBPK model was verified with data of 87 adults, 485 children and 912 neonates, based on visual predictive checks and predicted-to-observed pharmacokinetic (PK) parameter ratios. After acceptance of the model, dosages now recommended by the Dutch Pediatric Formulary (DPF) were simulated, along with several alternative dosing scenarios, aiming for recommended peak (i.e., 8–12 mg/L for neonates and 15–20 mg/L for infants) and trough (i.e., <1 mg/L) levels. We then used a decision framework to weigh benefits and risks for implementation.
Results: The PBPK model adequately described gentamicin PK. Simulations of current DPF dosages showed that the dosing interval for term neonates up to 6 weeks of age should be extended to 36–48 h to reach trough levels <1 mg/L. For infants, a 7.5 mg/kg/24 h dose will reach adequate peak levels. The benefits of these dose adaptations outweigh remaining uncertainties which can be minimized by routine drug monitoring.
Conclusion: We used a PBPK model to show that current DPF dosages for gentamicin in term neonates and infants needed to be optimized. In the context of potential uncertainties, the risk-benefit analysis proved positive; the model-informed dose is ready for clinical implementation.
Gentamicin is a widely used aminoglycoside antibiotic used for infections with gram-negative and gram-positive bacteria, such as in pneumonia, urinary tract infections, and sepsis. It is a hydrophilic drug that is excreted renally with a half-life of 2–3 h in adults. Initially, gentamicin was registered for multiple daily dosing, although its post antibiotic effect makes it suitable for once-daily dosing. Furthermore, a reduced risk of bacterial resistance and diminished accumulation in the renal tubules and inner ear, both target organs for toxicity, have been observed after once-daily dosing (1–4). Despite the current consensus on once-daily dosing, drug labels still differ in this regard (5, 6). The U.S. label still recommends the traditional thrice-daily dosing, while the European label prefers once-daily over twice-daily dosing (7–9). Also, there is no consensus with regard to the therapeutic targets, especially in special populations like pediatric, obese, and elderly patients (5, 10, 11).
Next to differences in dosing frequency and therapeutic targets, the differences in recommended dosages are also substantial among dosing guidelines and drug labels. This has been indicated specifically for neonates, but these differences also exist for infants, children and adolescents (Supplementary Table S1) (12, 13). In addition, the evidence used for the current doses in the Dutch Pediatric Formulary (DPF) in neonates and infants is limited; only 4 references were cited, 2 of which are very specific to children with febrile neutropenia, aiming at peak concentrations (Cmax) of at least 20 or 25 mg/L (14, 15). Another study aiming at similarly high Cmax values, proposed a dose of 9.5 mg/kg every 24 h (16). The fourth study, a population pharmacokinetic (popPK) study, suggested a dose of 7 mg/kg for infants to reach a Cmax of at least 10 mg/L, which is the currently recommended DPF dose (17). However, all 4 of these trials aimed at a different therapeutic window than suggested by therapeutic drug monitoring (TDM) guidelines, so the evidence base for this age category needed strengthening.
Many studies have been conducted to find “the” right dose (18, 19). Especially in preterm neonates, several popPK models have been developed, and also term neonates in their first week of life were extensively studied (19). Older neonates and young infants, however, are less studied, despite reported large dose ranges of 4.5–7.5 mg/kg every 24 h (19, 20). This variation in dosing partially reflects the wide variability in pharmacokinetics (PK), as body composition and organ maturation change tremendously during neonatal and infant periods (21). For example, gentamicin's volume of distribution (Vd) decreases from 0.48 L/kg in a preterm neonate to 0.35 L/kg in an infant, while clearance (CL) nearly triples from neonatal to infant age (19). Yet, this is a gradual process, where the dose certainly needs to be adjusted over time, but the question remains of exactly how and when to adjust it. Dose increases of 33%–52% (Supplementary Table S1) are partly explained by the change in therapeutic target (from 8–12 mg/L to 15–20 mg/L), but it is questionable if such a large, abrupt dose increase is indeed justified.
Pediatric physiologically-based pharmacokinetic (PBPK) models capture the developmental changes in PK related processes. Such models have been previously developed for gentamicin, but they did not aim to provide dosing guidelines or infants were not represented in these models (Supplementary Table S2) (22–25). At the same time abundant gentamicin PK data from neonates (i.e., 0–28 days postnatal age) and infants (i.e., 1–24 months of age) are widely available, enabling verification of dosing simulations. Previously, we have shown that PBPK modeling is feasible for the prediction of pediatric exposure of seven drugs and have also published a tutorial outlining a pragmatic approach to this methodology (26, 27). We now aim to expand this to establish pediatric doses for clinical implementation. Hence, the aim of our study was to pragmatically establish rational gentamicin doses for term neonates and young infants, while also assessing the risks and benefits of these model-informed doses for clinical implementation.
The PBPK platform Simcyp™ Simulator (version 21; Certara, Sheffield, UK) was used for our simulations. For our pragmatic approach, we compared the two recently published gentamicin PBPK models developed in Simcyp™ using exploratory simulations (23, 24). Gentamicin Cmax and trough concentration (Ctrough) were used as surrogate markers for efficacy and toxicity. The model that best predicted gentamicin PK, and more specifically Cmax and Ctrough, was the full distribution model reported by Abduljalil et al. (23). Next, the performance of this model was assessed according to the approach described by van der Heijden et al., using published adult and pediatric PK data (26).
To verify the model, we searched for published PK data that were not used in model development by Abduljalil et al. (Supplementary Tables S3a–c). In simulations, default Simcyp™ populations were used, for adults we used the “Sim-healthy volunteer” population or “Sim-NEurCaucasian”, in case patients above 65 years of age were included (28, 29). For children, both the “Sim-paediatric” and the “Sim-preterm” populations were used (23, 30, 31). Trial design (e.g., administered dose, age range, proportion of females, and trial duration) was matched to the corresponding clinical study, using ten trials of ten virtual subjects. For children younger than 1 year, the “redefining subjects over time” option was activated, to allow for virtual growth (physiologically and biochemically) of the virtual subjects during the simulation. Model performance was assessed by visual predictive checks (VPC) and by calculating predicted-to-observed (P/O) PK parameter ratios. Ratios within 2-fold were considered acceptable, the closer the ratio was to 1, the better the prediction. The ratios of surrogate markers Cmax and Ctrough were also compared to the bio-equivalence range (i.e., 1.25-fold).
Prior to the dose simulations, we had to define the therapeutic targets. For efficacy, the most commonly used therapeutic targets are the Cmax or the area under the curve (AUC), where the ratio to the minimal inhibitory concentration (MIC) of a bacterium should be within a certain range. For our target population, Cmax/MIC is the best therapeutic target, as a study in neonates found that AUC/MIC was not a determining factor for the efficacy of gentamicin (32). Moreover, most guidelines for gentamicin use Cmax and agree that a Cmax/MIC ratio of 8–10 should be aimed for (33–36). In neonatal infections, multiple studies have shown that most micro-organisms involved in these infections have an MIC of less than 1 mg/L (33, 36–40), which has been translated into a therapeutic window of 8–12 mg/L for Cmax in most guidelines. For all other age groups, i.e., infants through adults, a Cmax between 15 and 20 mg/L is recommended, as the infections treated with gentamicin are often caused by micro-organisms with MICs up to 2 mg/L (33, 35, 36, 41).
For toxicity, both AUC and Ctrough have been used (42–45). However, the upper threshold for daily or cumulative AUC for the occurrence of nephrotoxicity is currently based on multiple daily dosing (45), and no AUC based thresholds are known for once-daily dosing. Therefore, we assumed that Ctrough is a better predictor of toxicity in this study. A range of maximum trough levels of 0.5–2 mg/L has been used as target threshold (33–36, 46), though qualitative research is currently lacking whether a Ctrough of 2 mg/L results in more toxicity than 1 or 0.5 mg/L (42). Also drug labels differ in this regard; the U.S. label recommends a Ctrough ≤2 mg/L, while the EU label differentiates between a trough level for twice-daily administration (≤2 mg/L) and once-daily administration (≤1 mg/L) (7–9). As such, we used a Ctrough of 1 mg/L as a predictor of toxicity in this study.
To find the best model-informed dose, we simulated several dosing scenarios. For our age group of interest (term neonates—infants), these simulations were performed using the “Sim-Paediatric” population, using ten trials of ten virtual subjects. The current DPF dosing recommendations for term neonates (4 mg/kg every 24 h) and infants (7 mg/kg every 24 h) were simulated first, followed by simulations of several alternative dosing scenarios (Supplementary Table S4). Gentamicin was simulated as a single, intravenous infusion over 30 min for a 48-h period, as dosing thereafter is usually based on therapeutic drug monitoring (TDM) if longer therapy is needed. For each simulation, we assessed Cmax, Ctrough and the corresponding 5th and 95th percentiles (33, 34). In accordance with various TDM guidelines, also in our simulations Cmax was taken at 1 h after the start of infusion (i.e., 30 min after the end of the 30 min infusion period) and Ctrough 30 min before the next (theoretical) dose (33, 36). For neonates, we assessed Ctrough at 23.5, 35.5 and 47.5 h after the start of infusion in order to extensively evaluate trough levels, and assess the potential need for an extended dosing interval. For infants, Ctrough was assessed only at 23.5 h after the start of infusion as no elevated trough levels were expected based on literature. When the 5th to 95th percentile for simulated Cmax and/or Ctrough fell outside the therapeutic window for its specific ages [i.e., Cmax within 8–12 mg/L for neonates and within 15–20 mg/L for infants, Ctrough below 1 mg/L for both neonates and infants (33)], the dose and/or dose frequency was further adjusted in the simulations until desired levels were reached (Supplementary Table S4).
After completing model verification and conducting dose simulations of current DPF doses and alternative dosing regimens, we interpreted our simulation results in context of a previously published framework for implementing model-informed doses in a clinical setting (47). The following questions were addressed:
1. What is the level of certainty on the target concentrations?
2. What is the clinical risk of over- or underdosing?
3. What is the level of certainty of the model output?
4. Does the currently advised DPF dose result in adequate target exposure?
5. Which dose results in better target exposure, is this a significant improvement?
6. Is the proposed dose practical?
7. Is the population used to verify the PBPK model comparable to the intended population (e.g., with respect to demographics, severity of illness, underlying disease)? If not, will this impact the dosing requirements?
8. What is the overall conclusion for gentamicin?
Based on these questions, we derived model-informed dosing recommendations along with a recommendation for implementation in clinical practice.
To verify the gentamicin PBPK model in adults, pediatrics, and preterms, we used published PK data of 8 adult, 19 pediatric, and 3 preterm studies, representing 87, 485, and 912 patients, respectively (Supplementary Tables S3a–c). For adults, 82% of all calculated P/O PK parameter ratios were within 2-fold and for pediatric patients this percentage was as high as 91%. More specifically, for pediatric Cmax and Ctrough values, the P/O ratios were within 2-fold for 100% (n = 54) and 82% (n = 39), and within 1.25-fold for 65% and 29%, respectively (Figure 1 and Supplementary Table S5). Although in term neonates a slight overprediction of Cmax and Ctrough was visible, this fell within the prespecified acceptance ranges, as illustrated in Figures 1 and Supplementary Figures S1–S3.
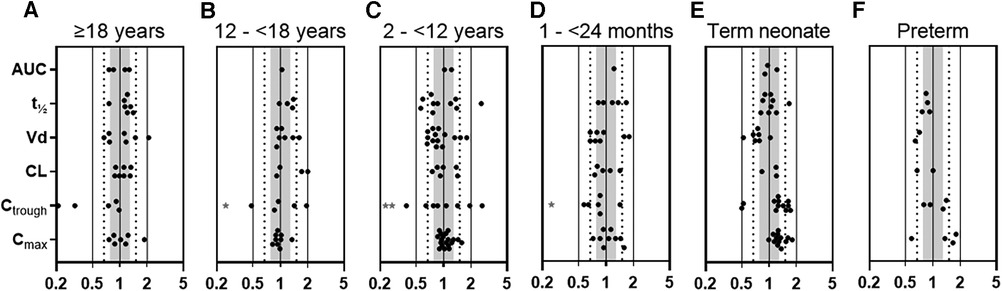
Figure 1. Predicted-to-observed PK parameter ratios for gentamicin, separated for each age group (i.e., (A) adults ≥18 years, (B) adolescents 12–<18 years, (C) children 2–<12 years, (D) infants 1–<24 months, (E) term neonates 0–28 days postnatal age, and (F) preterm neonates 0–28 days postnatal age). Solid lines, dotted lines, and shaded area indicate the 2-fold, 1.5-fold, and 1.25-fold range, respectively. * 1 or 2 (**) datapoints fell outside axis limits. AUC, area under the curve; t½, half-life; Vd, volume of distribution; CL, clearance, Ctrough, plasma trough concentration; Cmax, maximal plasma concentration.
Simulations of the current DPF dosing recommendations for term neonates showed that maximal concentrations fell within the therapeutic window with a median Cmax of 10.2 mg/L (Figure 2A, i.e., black squares). On the contrary, Ctrough only fell below 1 mg/L for neonates approximating 1 month of age (Figure 2B, i.e., black squares). For infants, almost all simulated Cmax levels were within the therapeutic window, although the 5th percentile for infants ≥21 months of age just dipped below the lower limit (Figure 2E, i.e., black diamonds). Similarly, the DPF dose led to adequate trough levels, except in one-month-old infants. In these children, the 95th percentile was above 1 mg/L (Figure 2F, i.e., black diamonds). Further simulations showed that from 6 weeks of age onwards, this 95th percentile would be below 1 mg/L again. Our simulations indicated that for infants 4–6 weeks of age an extended interval of 36 h was required to fulfil the target requirements (Figure 3).
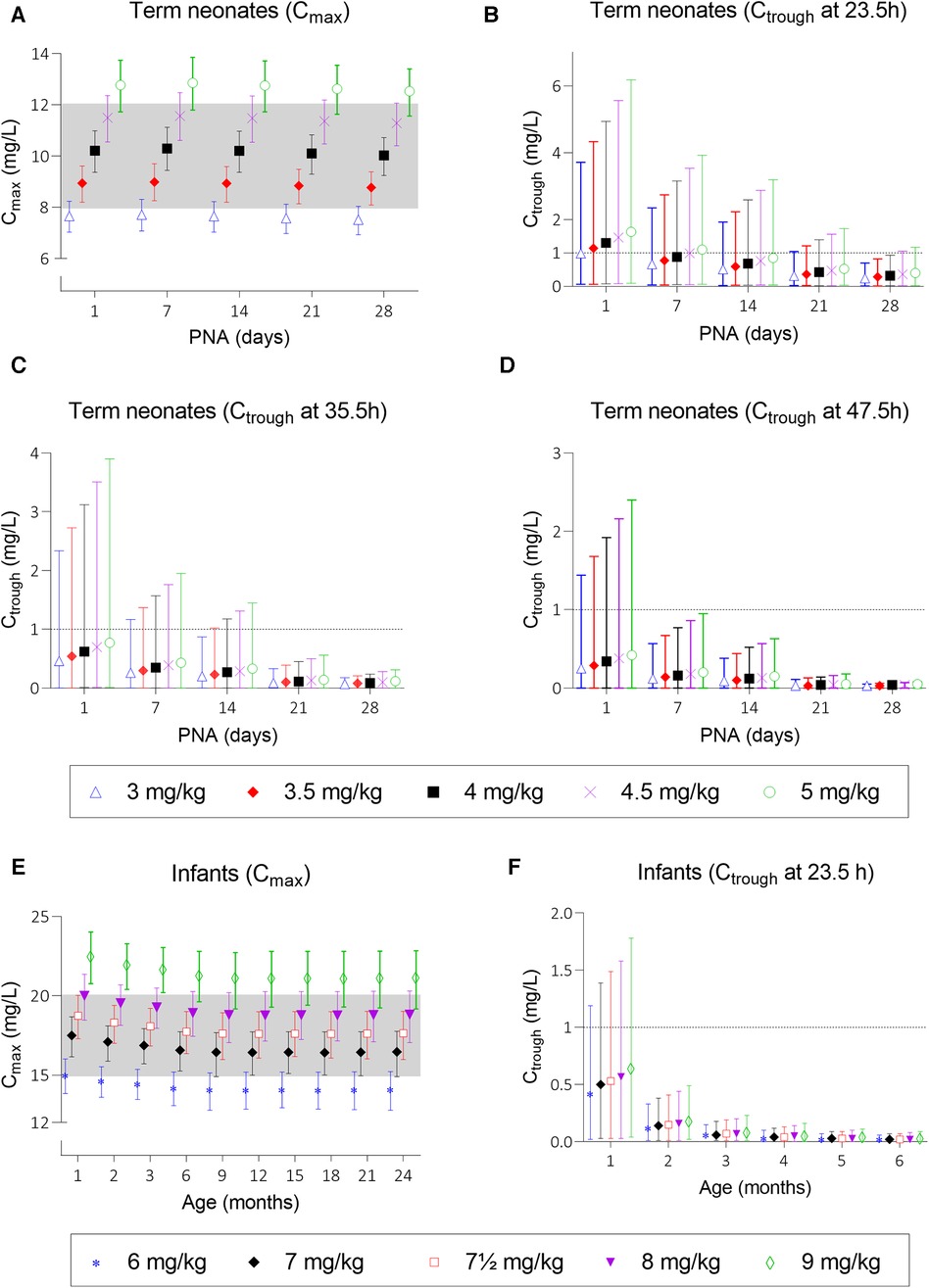
Figure 2. Mean predicted Cmax and Ctrough levels and their 5th and 95th percentiles at several dosing scenarios. Graph (A) shows Cmax levels after doses of 3, 3.5, 4, 4.5, and 5 mg/kg for neonates and graph (E) shows Cmax levels after doses of 6, 7, 7.5, 8, and 9 mg/kg for infants. Graph (B–D) show Ctrough levels at 23.5, 35.5, and 47.5 h after start of infusion for neonates and graph (F) shows Ctrough levels for infants until the age of 6 months; older infants all have trough levels below 1 mg/L and are therefore not depicted. Black squares (▪) in graphs (A–D) and black diamonds (◆) in graphs (E,F) represent the Dutch Pediatric Formulary dose. PNA, postnatal age.
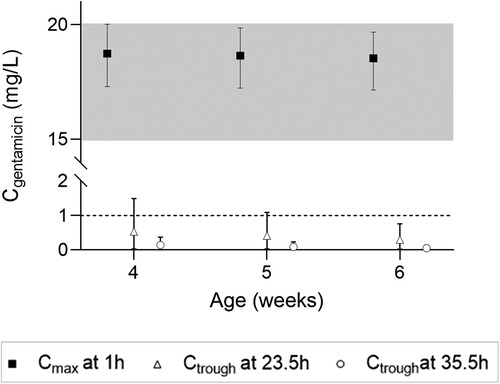
Figure 3. Cmax and Ctrough levels and their 5th and 95th percentiles in infants 4–6 weeks of age upon a 7.5 mg/kg dose.
Taking these findings as a starting point, we simulated a series of potential alternatives to the current DPF dosing scenarios in order to optimize gentamicin exposure (Supplementary Table S4). As for most neonates Ctrough levels at 23.5 h after dosing were too high, the dose interval was extended (36–48 h). Although neonatal Cmax was predicted in the middle of the therapeutic window, we simulated several other dosages to assess the effect on Ctrough. Figures 2A–D show the mean predicted Cmax and Ctrough levels with the 5th to 95th percentiles for each dosing regimen.
Based on these simulations, initial model-informed dosing recommendations could be derived. For example, the majority of the one-day-old neonates did not reach a Ctrough below 1 mg/L when a dose is administered every 24 h (Figure 2B). When administered every 48 h, at least 95% of the predicted population is expected to reach trough levels below 1 mg/L. Regarding Cmax, dose simulations showed that for a one-day-old child, doses of 3.5 and 4 mg/kg every 24 h resulted in adequate concentrations, as both the mean Cmax and the 5th and 95th percentiles were within the therapeutic range. In case several simulated doses seemed adequate, the highest dose was chosen for the sake of efficacy. For each simulated age, we made similar trade-offs (Supplementary Table S6) with the final model-informed dosing recommendations shown in Table 1.
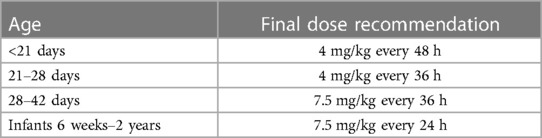
Table 1. Final model-informed dosing recommendations for neonates and infants.
Before proceeding to clinical application of these model-informed doses, we weighted uncertainties, by placing our simulation results in the context of the framework proposed by Hartman et al. (47). The following questions were addressed:
1. What is the level of certainty on the target concentrations? We assessed this as “high certainty”, based on a widely accepted Cmax/MIC ratio of 8–10 for infections with bacteria with an MIC of ≤1 mg/L for neonates and ≤2 mg/L for infants (33–36).
2. What is the clinical risk of over- or underdosing? Gentamicin's main concerns are its oto- and nephrotoxicity, which can be minimized by keeping through concentrations <1 mg/L. Based on our simulations we evaluated the risk as low. When only 1 dose is administered, this is not of concern, but when gentamicin is continued, measuring trough levels is part of routine TDM, at least in most clinical guidelines, and reduces the risk of dose-related toxicity due to increased trough levels. While underdosing may result in ineffective treatment, potentially worsening the disease state with an increased risk of death, our simulated dosages support a low risk of underdosing. Also, the risk of underdosing is reduced by routine TDM in case of continued use.
3. What is the level of certainty of the model output? High, as model performance could be verified against several available clinical data sets and model performance was robust, resulting in a high certainty of model outcomes.
4. Does the currently advised DPF dose result in adequate target exposure? No, simulations showed that the current neonatal DPF dose resulted in excessive trough levels, while the infant dose does not always result in adequate peak levels. This was supported by Hartman et al., who showed that 30% of term neonates had excessive trough levels and 87% of infants did not reach therapeutic levels (48).
5. Which dose results in better target exposure, is this a significant improvement? The extended dosing interval results in better trough levels for neonates. The model-supported increase in dose for infants prevents underdosing of Cmax when infected by a micro-organism with an MIC of ≤2 mg/L.
6. Is the proposed dose practical? Yes, it is acceptable, although there are now 4 instead of 2 dosing recommendations for neonates and infants.
7. Is the population used to verify the PBPK model comparable to the intended population (e.g., with respect to demographics, severity of illness, underlying disease)? If not, will this impact the dosing requirements? Yes, for model verification, we mostly used PK studies with patients of all ages, treated for various diseases (Supplementary Tables S3a–c). In addition, since the virtual populations in Simcyp™ are based on healthy subjects, a few PK studies in healthy volunteers were used for verification as well.
8. What is the overall conclusion for gentamicin? Our adjusted model-informed doses are likely to lead to more adequate Cmax and Ctrough levels. Remaining uncertainties, e.g., related to small changes in PK due to disease conditions and/or treatment [i.e., augmented CL in critically ill patients, decreased CL in congenital heart disease or hypothermia, increased Vd in extracorporeal membrane oxygenation (ECMO)] can be minimized by routine TDM, so that the optimized dose only contributes to better treatment of term neonates and infants.
We used an already existing PBPK model of gentamicin to optimize neonatal and infant gentamicin doses. We showed that the current neonatal DPF dose for term neonates up to 1 month of age (4 mg/kg every 24 h) likely results in adequate Cmax levels, but excessive, potentially toxic, Ctrough levels. Additionally, for the youngest infants (4–6 weeks of age) the current infant DPF dose (7 mg/kg every 24 h) likely results in Ctrough levels approximating the 1 mg/L limit. For older infants, a similar 7 mg/kg/24 h dose likely results in Cmax levels at the low end of the therapeutic range. To achieve both adequate Cmax as well as Ctrough levels, model-informed dosing recommendations were developed (Table 1). These allow for a more gradual adjustment in dosing for term neonates and infants compared to the currently proposed dosing scheme in the DPF, which advocates a sudden increase in dose when a child reaches 1 month of age.
Before implementing dosing guidelines resulting from simulations, we advocate the use of a decision framework to clearly identify the assumptions while weighing the benefits and risks of the new dosing guideline (47). We identified the following assumptions: (1) Cmax/MIC is the best measure of efficacy for our population (32–36, 49, 50), 2) a Cmax of 15–20 mg/L is required for infants and a Cmax of 8–12 mg/L for neonates (33–41), (3) Ctrough is the best predictor of toxicity (42–45), and (4) if a Ctrough <1 mg/L is achieved, toxicity is prevented (7, 8, 18, 33–36, 42, 46, 51, 52). Combined with the answers on the questions asked within the decision framework, we evaluated the benefit-risk as positive.
Our findings are supported by a retrospective study evaluating 1,288 trough levels of 353 children aged 1 month to 17 years after a dose of 7 mg/kg every 24 h. They found high trough levels in only 2.2%, which is comparable to the 1.3% of our simulations (53). Another retrospective study in critically ill neonates showed that after a dose of 4 mg/kg every 24 h, 10 out of 34 (29%) trough levels measured were above 1 mg/L, which corresponds to the 5%–37% found in our simulations using the same dose (48). This study, and also another prospective study, reported lower proportions of therapeutic concentrations than we simulated using the study doses, but because of incomplete reporting of the conditions under which these values were determined, these data cannot be compared with our simulations (54). Both studies are unclear with regard to the exact sampling times used to determine Cmax. Although TDM standards aim to determine Cmax 1 h after the start of infusion (i.e., 30 min after the end of infusion), these times may deviate in clinical practice. Sampling at later times will result in lower Cmax values.
Another approach to study PK and determine dosing recommendations, is popPK. PopPK modeling is a top-down approach, where the PK data itself is used to fit the model. PBPK modeling is a bottom-up approach, starting with human physiology and drug characteristics (e.g., molecular weight, pKa, etc.). The virtual populations within the PBPK software are very well validated, which enables us to specifically investigate the effect of age-related physiological changes on gentamicin PK. Still, many attempts have been done to provide dosing recommendations using popPK. We found 18 popPK studies providing dosing recommendations for term neonates and/or infants (17, 18, 55–70). Only 4 of these studied the same population and the same therapeutic window as we did (55, 58, 61, 69). For neonates, only Valitalo et al. differentiated on postnatal age, recommending a 4.5 mg/kg dose every 48, 36, or 24 h for neonates ≤5 days, 6–10 days, or ≥11 days, respectively (69). The dose proposed by Bijleveld et al. applies only to neonates less than 7 days old and appears to be based largely on data from preterm neonates (58). For infants, large age ranges were included (i.e., up to 12 years of age) and the dose recommendations were based on MIC (55, 61). The model of Ghoneim et al., for example, aiming for a Cmax of 20 mg/L, targeting an MIC of 2 mg/L, predicted that a dose of 6–7 mg/kg every 24 h according was required while the model of Alsultan et al. predicted that even a dose of 10 mg/kg would not be sufficient to achieve a Cmax of at least 16 mg/L. According to Ghoneim et al. these differences could be explained by their additional age stratification as applied by Ghoneim et al. (61), which highlights the importance of our extensive PBPK modeling work in this population.
Our model-informed dose for neonates and infants until the age of 6 weeks is quite different from the current DPF dose. For older infants, only a minor dose increase is recommended, which is still important given the relatively large interindividual variability. Figure 2E shows that this variability covers half of the therapeutic window and increasing age is associated with lower mean plasma concentrations. The 5th percentile of the 7 mg/kg dose for infants aged ≥21 months and the 95th percentile of the 8 mg/kg dose for all infants falls outside the therapeutic window. Therefore, a dose of 7.5 mg/kg is most likely to achieve a therapeutic level.
Our study has some limitations. First of all, choosing another therapeutic target or window would logically affect the dose. Secondly, our dosing recommendations do not address variation in renal clearance or volume of distribution due to, for example, acute kidney injury, renal replacement therapy, or extracorporeal membrane oxygenation. PBPK modeling could be used for these purposes as well and should be explored in future research. A third limitation is the broad acceptance range. Besides the fact that this 2-fold range is the most commonly applied criterion, the large observed interindividual variability also justifies this range. Lastly, the model predictions for Ctrough and Cmax might appear biased for term neonates. For Ctrough, the higher predicted than observed concentrations reduce the risk of toxicity. For Cmax, the question arises as to whether the reported Cmax is the “true Cmax”. A PBPK model can accurately predict Cmax, but sampling a few minutes too early or too late will always result in a lower observed Cmax.
In conclusion, our PBPK model was able to adequately capture pediatric PK of gentamicin. Our simulations indicate that until the age of 4 weeks postnatally, gentamicin should be administered at a dose of 4 mg/kg, but less frequently than currently recommended in the DPF. In order to achieve adequate Cmax levels in infants from 4 weeks of age onwards, the current dose needs to be increased from 7 to 7.5 mg/kg, although the dose interval should be extended to 36 h for infants 4–6 weeks of age. Since TDM is advised for gentamicin, the effects of altered PK due to special co-morbidities, critical illness or ECMO, will still be noticed and the dose can be further adjusted accordingly. Evaluation according to the framework proposed by Hartman et al. showed a positive risk-benefit analysis for clinical implementation of this model-informed dose. With this study, we have shown that a pragmatic approach to establish model-informed dosages is feasible and that a framework to assess the readiness of model-informed dosing for clinical implementation is useful. In the future, we plan to apply this approach to many other drug to provide dosing recommendations for neonates, infants, children, and adolescents.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
MH-S: Conceptualization, Formal Analysis, Methodology, Validation, Visualization, Writing – original draft. JH: Writing – review & editing. JF: Writing – review & editing. RG: Supervision, Writing – review & editing. SW: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This publication is based on research funded by the Bill & Melinda Gates Foundation (INV-001822). The findings and conclusions contained within are those of the authors and do not necessarily reflect positions or policies of the Bill & Melinda Gates Foundation.
We thank Nicolas Louca for his assistance with the modelling and Dr. Ping Zhao for useful discussions that improved the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1288376/full#supplementary-material
1. Contopoulos-Ioannidis DG, Giotis ND, Baliatsa DV, Ioannidis JP. Extended-interval aminoglycoside administration for children: a meta-analysis. Pediatrics. (2004) 114(1):e111–8. doi: 10.1542/peds.114.1.e111
2. Kashuba AD, Bertino JS Jr, Nafziger AN. Dosing of aminoglycosides to rapidly attain pharmacodynamic goals and hasten therapeutic response by using individualized pharmacokinetic monitoring of patients with pneumonia caused by gram-negative organisms. Antimicrob Agents Chemother. (1998) 42(7):1842–4. doi: 10.1128/AAC.42.7.1842
3. Moore RD, Lietman PS, Smith CR. Clinical response to aminoglycoside therapy: importance of the ratio of peak concentration to minimal inhibitory concentration. J Infect Dis. (1987) 155(1):93–9. doi: 10.1093/infdis/155.1.93
4. Zhanel GG, Craig WA. Pharmacokinetic contributions to postantibiotic effects. Focus on aminoglycosides. Clin Pharmacokinet. (1994) 27(5):377–92. doi: 10.2165/00003088-199427050-00005
5. Hodiamont CJ, van den Broek AK, de Vroom SL, Prins JM, Mathôt RAA, van Hest RM. Clinical pharmacokinetics of gentamicin in various patient populations and consequences for optimal dosing for gram-negative infections: an updated review. Clin Pharmacokinet. (2022) 61(8):1075–94. doi: 10.1007/s40262-022-01143-0
6. Rao SC, Srinivasjois R, Moon K. One dose per day compared to multiple doses per day of gentamicin for treatment of suspected or proven sepsis in neonates. Cochrane Database Syst Rev. (2016) 12(12):Cd005091. doi: 10.1002/14651858.CD005091.pub4
8. Centrafarm BV. Summary of Product Characteristics Gentamicine (RVG 57571/57572) (2021). (December 10).
9. Panpharma UK Ltd. Summary of Product Characteristics Gentamicin 40 mg/mL Solution for Injection/Infusion (2022). Available at: https://www.medicines.org.uk/emc
10. Roy C, Gray C, Ruda L, Bell A, High-Dose BJ. Extended-interval gentamicin and tobramycin for pediatric inpatients: a survey of Canadian hospital pharmacists. Can J Hosp Pharm. (2016) 69(5):367–75. doi: 10.4212/cjhp.v69i5.1591
11. Saddi V, Preddy J, Dalton S, Connors J, Patterson S. Variation in gentamicin dosing and monitoring in pediatric units across New South Wales. Pediatr Qual Saf. (2017) 2(2):e015. doi: 10.1097/pq9.0000000000000015
12. Liem TBY, Slob EMA, Termote JUM, Wolfs TFW, Egberts ACG, Rademaker CMA. Comparison of antibiotic dosing recommendations for neonatal sepsis from established reference sources. Int J Clin Pharm. (2018) 40(2):436–43. doi: 10.1007/s11096-018-0589-9
13. van Donge T, Pfister M, Bielicki J, Csajka C, Rodieux F, van den Anker J, et al. Quantitative analysis of gentamicin exposure in neonates and infants calls into question its current dosing recommendations. Antimicrob Agents Chemother. (2018) 62(4):1–12. doi: 10.1128/AAC.02004-17
14. Bialkowski S, Staatz CE, Clark J, Lawson R, Hennig S. Gentamicin pharmacokinetics and monitoring in pediatric patients with febrile neutropenia. Ther Drug Monit. (2016) 38(6):693–8. doi: 10.1097/FTD.0000000000000341.
15. Inparajah M, Wong C, Sibbald C, Boodhan S, Atenafu EG, Naqvi A, et al. Once-daily gentamicin dosing in children with febrile neutropenia resulting from antineoplastic therapy. Pharmacotherapy. (2010) 30(1):43–51. doi: 10.1592/phco.30.1.43
16. McDade EJ, Wagner JL, Moffett BS, Palazzi DL. Once-daily gentamicin dosing in pediatric patients without cystic fibrosis. Pharmacotherapy. (2010) 30(3):248–53. doi: 10.1592/phco.30.3.248
17. Medellín-Garibay SE, Rueda-Naharro A, Peña-Cabia S, García B, Romano-Moreno S, Barcia E. Population pharmacokinetics of gentamicin and dosing optimization for infants. Antimicrob Agents Chemother. (2015) 59(1):482–9. doi: 10.1128/AAC.03464-14
18. Llanos-Paez CC, Hennig S, Staatz CE. Population pharmacokinetic modelling, monte carlo simulation and semi-mechanistic pharmacodynamic modelling as tools to personalize gentamicin therapy. J Antimicrob Chemother. (2017) 72(3):639–67. doi: 10.1093/jac/dkw461
19. Crcek M, Zdovc J, Kerec Kos M. A review of population pharmacokinetic models of gentamicin in paediatric patients. J Clin Pharm Ther. (2019) 44(5):659–74. doi: 10.1111/jcpt.12850
20. Kato H, Hagihara M, Matsuda H, Iwamoto T. Gentamicin pharmacokinetics and optimal dosage in infant patients: a case report and literature review. Int J Environ Res Public Health. (2022) 19(22):1–7. doi: 10.3390/ijerph192215360
21. Smits A, Annaert P, Cavallaro G, De Cock P, de Wildt SN, Kindblom JM, et al. Current knowledge, challenges and innovations in developmental pharmacology: a combined conect4children expert group and European society for developmental, perinatal and paediatric pharmacology white paper. Br J Clin Pharmacol. (2022) 88(12):4965–84. doi: 10.1111/bcp.14958
22. Idkaidek N, Hamadi S, Bani-Domi R, Al-Adham I, Alsmadi M, Awaysheh F, et al. Saliva versus plasma therapeutic drug monitoring of gentamicin in Jordanian preterm infants. Development of a physiologically-based pharmacokinetic (PBPK) model and validation of class II drugs of salivary excretion classification system. Drug Res. (2020) 70(10):455–62. doi: 10.1055/a-1233-3582
23. Abduljalil K, Pan X, Pansari A, Jamei M, Johnson TN. Preterm physiologically based pharmacokinetic model. Part II: applications of the model to predict drug pharmacokinetics in the preterm population. Clin Pharmacokinet. (2020) 59(4):501–18. doi: 10.1007/s40262-019-00827-4
24. Neeli H, Hanna N, Abduljalil K, Cusumano J, Taft DR. Application of physiologically based pharmacokinetic-pharmacodynamic modeling in preterm neonates to guide gentamicin dosing decisions and predict antibacterial effect. J Clin Pharmacol. (2021) 61(10):1356–65. doi: 10.1002/jcph.1890
25. Zazo H, Lagarejos E, Prado-Velasco M, Sánchez-Herrero S, Serna J, Rueda-Ferreiro A, et al. Physiologically-based pharmacokinetic modelling and dosing evaluation of gentamicin in neonates using PhysPK. Front Pharmacol. (2022) 13:977372. doi: 10.3389/fphar.2022.977372
26. van der Heijden JEM, Freriksen JJM, de Hoop-Sommen MA, Greupink R, de Wildt SN. Physiologically-based pharmacokinetic modeling for drug dosing in pediatric patients: a tutorial for a pragmatic approach in clinical care. Clin Pharmacol Ther. (2023) 114(5):960–71. doi: 10.1002/cpt.3023
27. van der Heijden JEM, Freriksen JJM, de Hoop-Sommen MA, van Bussel LPM, Driessen SHP, Orlebeke AEM, et al. Feasibility of a pragmatic PBPK modeling approach: towards model-informed dosing in pediatric clinical care. Clin Pharmacokinet. (2022) 61(12):1705–17. doi: 10.1007/s40262-022-01181-8
28. Howgate EM, Rowland Yeo K, Proctor NJ, Tucker GT, Rostami-Hodjegan A. Prediction of in vivo drug clearance from in vitro data. I: impact of inter-individual variability. Xenobiotica. (2006) 36(6):473–97. doi: 10.1080/00498250600683197
29. Jamei M, Dickinson GL, Rostami-Hodjegan A. A framework for assessing inter-individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of 'bottom-up' vs 'top-down' recognition of covariates. Drug Metab Pharmacokinet. (2009) 24(1):53–75. doi: 10.2133/dmpk.24.53
30. Abduljalil K, Pan X, Pansari A, Jamei M, Johnson TN. A preterm physiologically based pharmacokinetic model. Part I: physiological parameters and model building. Clin Pharmacokinet. (2020) 59(4):485–500. doi: 10.1007/s40262-019-00825-6
31. Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. (2006) 45(9):931–56. doi: 10.2165/00003088-200645090-00005
32. Ibrahim NA, Manan MM. AUC/MIC ratio as a tool in determining effectiveness of garasent® for the prevention of early onset sepsis in hospitalized neonates. Int J Pharm Pharm Sci. (2014) 6(Suppl 3):24–8.
33. Coenradie S, Touw DJ, Holtkamp F, den Daas I. TDM Monografie Gentamicine (2018). Available at: https://tdm-monografie.org/monografieen/tdm-monografieen/
34. Update on good use of injectable aminoglycosides, gentamycin, tobramycin, netilmycin, amikacin. Pharmacological properties, indications, dosage, and mode of administration, treatment monitoring. Med Mal Infect. (2012) 42(7):301–8. doi: 10.1016/j.medmal.2011.07.007
35. ANMF consensus group. Australasian Neonatal Medicines Formulary—Gentamicin (2021). Available at: https://www.anmfonline.org/clinical-resources/
36. Wilson W. UNC Medical Center Guideline—Aminoglycoside Dosing & Monitoring: Neonatal & Pediatric Guideline (2022). Available at: https://www.med.unc.edu/pediatrics/cccp/wp-content/uploads/sites/1156/gravity_forms/1-c06e424ddddee8826f29e1bc5926a251/2022/04/Aminoglycoside-Dosing-and-Monitoring-Guideline-Pediatrics_2022_FINAL.pdf
37. Lim WH, Lien R, Huang YC, Chiang MC, Fu RH, Chu SM, et al. Prevalence and pathogen distribution of neonatal sepsis among very-low-birth-weight infants. Pediatr Neonatol. (2012) 53(4):228–34. doi: 10.1016/j.pedneo.2012.06.003
38. Muller-Pebody B, Johnson AP, Heath PT, Gilbert RE, Henderson KL, Sharland M. Empirical treatment of neonatal sepsis: are the current guidelines adequate? Arch Dis Child Fetal Neonatal Ed. (2011) 96(1):F4–8. doi: 10.1136/adc.2009.178483
39. Topcuoglu S, Demirhan S, Dincer E, Ozalkaya E, Karatekin G. Early-onset neonatal sepsis in Turkey: a single-center 7-year experience in etiology and antibiotic susceptibility. Children (Basel). (2022) 9(11):2–11. doi: 10.3390/children9111642
40. Wang J, Zhang H, Yan J, Zhang T. Literature review on the distribution characteristics and antimicrobial resistance of bacterial pathogens in neonatal sepsis. J Matern Fetal Neonatal Med. (2022) 35(5):861–70. doi: 10.1080/14767058.2020.1732342
41. European Committee on Antimicrobial Susceptibility Testing (EUCAST). Clinical breakpoints (v 13.1) (2023). Available at: https://www.eucast.org/clinical_breakpoints
42. Yamada T, Fujii S, Shigemi A, Takesue Y. A meta-analysis of the target trough concentration of gentamicin and amikacin for reducing the risk of nephrotoxicity. J Infect Chemother. (2021) 27(2):256–61. doi: 10.1016/j.jiac.2020.09.033
43. Murry KR, McKinnon PS, Mitrzyk B, Rybak MJ. Pharmacodynamic characterization of nephrotoxicity associated with once-daily aminoglycoside. Pharmacotherapy. (1999) 19(11):1252–60. doi: 10.1592/phco.19.16.1252.30876
44. Mingeot-Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother. (1999) 43(5):1003–12. doi: 10.1128/AAC.43.5.1003
45. Bland CM, Pai MP, Lodise TP. Reappraisal of contemporary pharmacokinetic and pharmacodynamic principles for informing aminoglycoside dosing. Pharmacotherapy. (2018) 38(12):1229–38. doi: 10.1002/phar.2193
46. SA Maternal NGCoP. South Australian Neonatal Medication Guidelines—gentamicin V4.0 (2017). Available at: https://www.sahealth.sa.gov.au/wps/wcm/connect/34c75d004cd7d772b93bb9a496684d9f/Gentamicin_Neo_v4_0.pdf?MOD=AJPERES&CACHEID=ROOTWORKSPACE-34c75d004cd7d772b93bb9a496684d9f-n5jf87U
47. Hartman SJF, Swaving JGE, van Beek SW, van Groen BD, de Hoop M, van der Zanden TM, et al. A new framework to implement model-informed dosing in clinical guidelines: piperacillin and amikacin as proof of concept. Front Pharmacol. (2020) 11:592204. doi: 10.3389/fphar.2020.592204
48. Hartman SJF, Orriëns LB, Zwaag SM, Poel T, de Hoop M, de Wildt SN. External validation of model-based dosing guidelines for vancomycin, gentamicin, and tobramycin in critically ill neonates and children: a pragmatic two-center study. Paediatr Drugs. (2020) 22(4):433–44. doi: 10.1007/s40272-020-00400-8
49. The European Committee on Antimicrobial Susceptibility Testing. Rationale Document Gentamicin v2.0 (2020). Available at: https://www.eucast.org/fleadmin/src/media/PDFs/EUCAST_fles/Rationale_documents/Gentamicin_rationale_1.2_0906.pdf
50. United States Committee on Antimicrobial Susceptibility Testing. USCAST report Aminoglycoside In Vitro Susceptibility Test Interpretation Criteria Evaluations v1.3 (2019). Available at: https://app.box.com/s/1hxc8inf8u3rranwmk3efx48upvwt0ww
51. Raveh D, Kopyt M, Hite Y, Rudensky B, Sonnenblick M, Yinnon AM. Risk factors for nephrotoxicity in elderly patients receiving once-daily aminoglycosides. QJM. (2002) 95(5):291–7. doi: 10.1093/qjmed/95.5.291
52. Setiabudy R, Suwento R, Rundjan L, Yasin FH, Louisa M, Dwijayanti A, et al. Lack of a relationship between the serum concentration of aminoglycosides and ototoxicity in neonates. Int J Clin Pharmacol Ther. (2013) 51(5):401–6. doi: 10.5414/CP201833
53. Grodås KTM, Døllner H, Thaulow CM, Knudsen PK, Tønnessen A, Skeibrok M, et al. Gentamicin serum concentration measurement in children. Tidsskr Nor Laegeforen. (2023) 143(1). doi: 10.4045/tidsskr.22.0238
54. El-Chaar GM, Supaswud-Franks T, Venugopalan L, Kohn N, Castro-Alcaraz S. Extended-interval gentamicin administration in neonates: a simplified approach. J Perinatol. (2016) 36(8):660–5. doi: 10.1038/jp.2016.37
55. Alsultan A, Abouelkheir M, Elsharawy Y, Alkoraishi A, Osman R, Neely MN, et al. Optimizing gentamicin dosing in pediatrics using monte carlo simulations. Pediatr Infect Dis J. (2019) 38(4):390–5. doi: 10.1097/INF.0000000000002120
56. Avedissian SN, Rohani R, Bradley J, Le J, Rhodes NJ. Optimizing aminoglycoside dosing regimens for critically ill pediatric patients with augmented renal clearance: a convergence of parametric and nonparametric population approaches. Antimicrob Agents Chemother. (2021) 65(4):1–14. doi: 10.1128/AAC.02629-20
57. Bijleveld YA, de Haan TR, van der Lee HJ, Groenendaal F, Dijk PH, van Heijst A, et al. Altered gentamicin pharmacokinetics in term neonates undergoing controlled hypothermia. Br J Clin Pharmacol. (2016) 81(6):1067–77. doi: 10.1111/bcp.12883
58. Bijleveld YA, van den Heuvel ME, Hodiamont CJ, Mathôt RA, de Haan TR. Population pharmacokinetics and dosing considerations for gentamicin in newborns with suspected or proven sepsis caused by gram-negative Bacteria. Antimicrob Agents Chemother. (2017) 61(1):1–11. doi: 10.1128/AAC.01304-16
59. Cies JJ, Habib T, Bains V, Young M, Menkiti OR. Population pharmacokinetics of gentamicin in neonates with hypoxemic-ischemic encephalopathy receiving controlled hypothermia. Pharmacotherapy. (2018) 38(11):1120–9. doi: 10.1002/phar.2186
60. Črček M, Grabnar I, Zdovc JA, Grosek Š, Kos MK. External validation of population pharmacokinetic models of gentamicin in paediatric population from preterm newborns to adolescents. Acta Pharm. (2023) 73(2):175–94. doi: 10.2478/acph-2023-0027
61. Ghoneim RH, Thabit AK, Lashkar MO, Ali AS. Optimizing gentamicin dosing in different pediatric age groups using population pharmacokinetics and monte carlo simulation. Ital J Pediatr. (2021) 47(1):167. doi: 10.1186/s13052-021-01114-4
62. Lanao JM, Calvo MV, Mesa JA, Martín-Suárez A, Carbajosa MT, Miguelez F, et al. Pharmacokinetic basis for the use of extended interval dosage regimens of gentamicin in neonates. J Antimicrob Chemother. (2004) 54(1):193–8. doi: 10.1093/jac/dkh261
63. Lares-Asseff I, Pérz-Guillé MG, Camacho Vieyra GA, Pérez AG, Peregrina NB, Lugo Goytia G. Population pharmacokinetics of gentamicin in Mexican children with severe malnutrition. Pediatr Infect Dis J. (2016) 35(8):872–8. doi: 10.1097/INF.0000000000001204
64. Llanos-Paez CC, Staatz C, Hennig S. Balancing antibacterial efficacy and reduction in renal function to optimise initial gentamicin dosing in paediatric oncology patients. AAPS J. (2017) 20(1):14. doi: 10.1208/s12248-017-0173-6
65. Llanos-Paez CC, Staatz CE, Lawson R, Hennig S. Differences in the pharmacokinetics of gentamicin between oncology and nononcology pediatric patients. Antimicrob Agents Chemother. (2020) 64(2):1–10. doi: 10.1128/AAC.01730-19
66. Lopez SA, Mulla H, Durward A, Tibby SM. Extended-interval gentamicin: population pharmacokinetics in pediatric critical illness. Pediatr Crit Care Med. (2010) 11(2):267–74. doi: 10.1097/PCC.0b013e3181b80693
67. Moffett BS, Morris J, Galati M, Munoz FM, Arikan AA. Population pharmacokinetic analysis of gentamicin in pediatric extracorporeal membrane oxygenation. Ther Drug Monit. (2018) 40(5):581–8. doi: 10.1097/FTD.0000000000000547
68. Nielsen EI, Sandström M, Honoré PH, Ewald U, Friberg LE. Developmental pharmacokinetics of gentamicin in preterm and term neonates: population modelling of a prospective study. Clin Pharmacokinet. (2009) 48(4):253–63. doi: 10.2165/00003088-200948040-00003
69. Valitalo PA, van den Anker JN, Allegaert K, de Cock RF, de Hoog M, Simons SH, et al. Novel model-based dosing guidelines for gentamicin and tobramycin in preterm and term neonates. J Antimicrob Chemother. (2015) 70(7):2074–7. doi: 10.1093/jac/dkv052
Keywords: term neonate, infant, PBPK, model-informed dose, gentamicin, pediatric pharmacology, clinical implementation
Citation: de Hoop-Sommen MA, van der Heijden JEM, Freriksen JJM, Greupink R and de Wildt SN (2023) Pragmatic physiologically-based pharmacokinetic modeling to support clinical implementation of optimized gentamicin dosing in term neonates and infants: proof-of-concept. Front. Pediatr. 11:1288376. doi: 10.3389/fped.2023.1288376
Received: 4 September 2023; Accepted: 2 November 2023;
Published: 21 November 2023.
Edited by:
Karel Allegaert, KU Leuven, BelgiumReviewed by:
Victoria Ziesenitz, University Children's Hospital Basel, Switzerland© 2023 de Hoop-Sommen, van der Heijden, Freriksen, Greupink and de Wildt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marika A. de Hoop-Sommen bWFyaWthLmRlaG9vcC1zb21tZW5AcmFkYm91ZHVtYy5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.