
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 10 November 2023
Sec. Pediatric Nephrology
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1283325
This article is part of the Research TopicCystic Kidney Diseases in Children and Adults: From Diagnosis to Etiology and BackView all 8 articles
 Margareta Fistrek Prlic1
Margareta Fistrek Prlic1 Sanda Huljev Frkovic2,3
Sanda Huljev Frkovic2,3 Bodo Beck4Ivana Tonkovic Durisevic5Stela Bulimbasic3,6
Bodo Beck4Ivana Tonkovic Durisevic5Stela Bulimbasic3,6 Marijana Coric3,6
Marijana Coric3,6 Lovro Lamot3,7
Lovro Lamot3,7 Ema Ivandic1
Ema Ivandic1 Ivana Vukovic Brinar1,3*
Ivana Vukovic Brinar1,3*
Introduction: Genetic kidney diseases are underdiagnosed; namely, from 7% to 40% of patients suffering from chronic kidney disease (CKD) can carry a pathogenic variant, depending on population characteristics. Hereditary tubulointerstitial kidney diseases, including autosomal dominant tubulointerstitial kidney diseases (ADTKD), are even more challenging to diagnose. ADTKD is a rare form of genetic kidney disease resulting from pathogenic variants in the MUC1, UMOD, HNF1B, REN, SEC61A1, and DNAJB11 genes. There is no typical clinical or histopathological sign of ADTKD, it is characterized by progressive CKD, an autosomal dominant inheritance pattern, and tubular atrophy with interstitial fibrosis on kidney biopsy. There is no significant proteinuria, and the urinary sediment is bland. The patients usually do not have severe arterial hypertension. There can be a history of early gout, especially when compared to the UMOD gene variants. Children can have enuresis due to a loss of renal concentration. On ultrasound, the kidneys can appear normal or small in size. Renal cysts are not pathognomonic for any of the named diseases. End-stage renal disease (ESRD) develops at the average age of 45, but this can be very variable. Family history that suggests autosomal dominant inheritance and CKD fulfilling the aforementioned characteristics of tubulointerstitial kidney disease should raise suspicion of ADTKD. In the setting of a negative family history for CKD, clinical suspicion should be raised based on clinical characteristics, including early onset of hyperuricemia or gout and compatible histology on the kidney biopsy. Contrary to the aforementioned characteristics of ADTKD, in the case of HNF1B-related disease, there is a more complex clinical presentation with extrarenal manifestations of the disease (diabetes mellitus, hypomagnesemia, neurologic and psychiatric disturbances, etc.). The diagnosis of ADTKD is based on a positive family history and a detection of the pathogenic variant in one of the genes in an affected individual.
Aim: The aim of our study is to present two case reports of ADTKD with different characteristics (slowly progressive CKD vs. complex clinical presentation with an extrarenal manifestation of the disease) with a literature review.
Methods: A 34-year-old patient with CKD and a positive family history of CKD in whom kidney biopsy showed nonspecific chronic changes, with only genetic analysis confirming the diagnosis of MUC1-related ADTKD. Our second case is of a 17-year-old patient with an unremarkable family history who was initially referred to genetic counseling due to cognitive and motor impairment with long-lasting epilepsy. Extensive workup revealed increased serum creatinine levels with no proteinuria and bland urinary sediment, along with hypomagnesemia. His genetic analysis revealed 17q12 deletion syndrome, causing the loss of one copy of the HNF1B gene, the AATF, and the LHX1 gene.
Conclusion: Autosomal dominant tubulointerstitial kidney diseases are challenging to diagnose due to a lack of typical clinical or histopathological signs as well as an uncharacteristic and versatile clinical presentation. Increased clinical awareness is crucial for the detection of these diseases.
It is unknown how common inherited kidney diseases are, but they are certainly more frequent than diagnosed. There are more than 200 hereditary kidney diseases described worldwide (1, 2). A systematic approach using exome sequencing in more than 3,000 patients with CKD in Germany found diagnostic variants in almost 10% of patients, but this can vary from 7 to 40% depending on population characteristics (3, 4). Among the hereditary tubulointerstitial kidney diseases, the autosomal dominant tubulointerstitial kidney diseases (ADTKD) resulting from mutations in the MUC1, UMOD, HNF1B, REN, SEC61A1, and DNAJB11 genes, are among the commonest. ADTKD-UMOD alone is estimated to be responsible for 3% of the genetic causes of kidney disease worldwide, being the third most common genetic cause of nephropathy after autosomal dominant polycystic kidney disease and Alport syndrome (3). Moreover, ADTKD-MUC1 is the second most prevalent form of ADTKD, although the overall prevalence of ADTKD could be higher considering challenges in identifying pathogenic variants of MUC1 through non-NGS genetic testing (5). While ADTKD is not a common genetic cause of renal impairment, the versatile clinical presentation, unavailability of genetic testing, and relative lack of awareness make it hard to diagnose in everyday clinical practice (6, 7). The aim of our manuscript is therefore to present two case reports of ADTKD with different characteristics (slowly progressive CKD vs. complex clinical presentation with an extrarenal manifestation of the disease) along with a narrative literature review.
There is no typical clinical or histopathological sign of ADTKD. The whole group is characterized by progressive CKD, the findings of tubular atrophy and interstitial fibrosis in kidney biopsy, and an autosomal dominant inheritance pattern (5–10). Usually, there is no significant proteinuria, and the urinary sediment is bland (5, 6). The patients typically do not have severe arterial hypertension early in the course of the disease. There can be a history of early gout, especially with the UMOD gene variants (6). Children have enuresis due to a loss of renal concentration ability.6 ESRD develops by the age of 45, but this can vary among cases (11–13). On ultrasound, the kidneys appear normal or small in size (6, 13). Disease related to variants MUC1, UMOD, SEC61A1, and REN is related to the kidney, whereas disease related to HNF1B has a more complex clinical presentation related to many organs (13). The disease related to DNAJB11 can mimic autosomal dominant polycystic kidney disease (ADPKD). 6 Renal cysts are not pathognomonic for any of the named diseases. Histological changes in the kidney are also not typical—tubular atrophy and fibrosis, lamellation, and thickening of tubular basement membranes, and tubular microcysts can be found. Immunofluorescence for complement and immunoglobulins is negative (13). Multiple names have been used for ADTKD in the past, including ‘Medullary Cystic Kidney Disease type 1’ for the disease caused by MUC1 mutations, and ‘Medullary Cystic Kidney Disease type 2’, ‘Familial Juvenile Hyperuricemic Nephropathy’ for ADTKD-REN, ‘Uromodulin-Associated Kidney Disease’ for UMOD related diseases, which created confusion (13). The gene-based classification, genetic diagnosis, as well as clinical presentation of different types of ADTKD is presented in Table 1. Each one of the associated genes encodes a protein with a specific function related to the normal performance of the kidney and other organs. In short, the MUC1 gene produces mucin-1, the UMOD gene encodes uromodulin, also known as the Tamm-Horsfall protein, the HNF1B gene produces hepatocyte nuclear factor 1 beta, the SEC61A1 gene produces translocon subunit SEC61A, and the DNAJB11 gene produces the cofactor of GRP78/BiP (6–10).
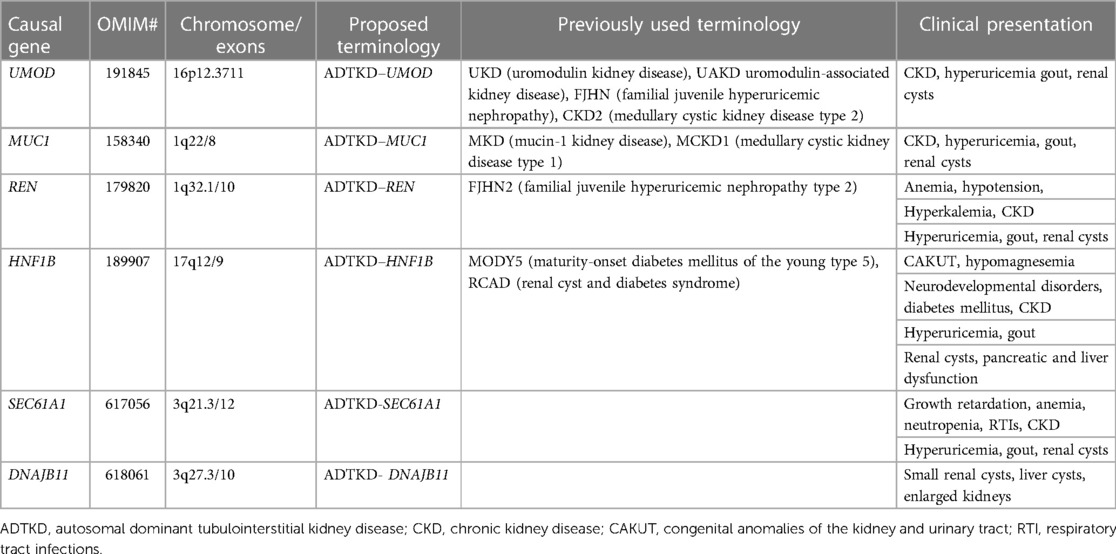
Table 1. The gene-based classification, genetic diagnosis, and clinical presentation of different types of ADTKD.
In ADTKD related to variants in the UMOD gene, the accumulation of the disarranged uromodulin protein inside the endoplasmic reticulum of epithelial cells in the renal tubules leads to tubular cell death. This process leads to chronic kidney failure (14). ADTKD-UMOD patients have slowly progressive kidney damage, usually in the second or third decade of life, with bland urinary sediment and slightly elevated proteinuria (1, 6, 13). The ability to maintain urinary concentration is impaired, but not clinically relevant. Changes in kidney histology are unspecific, but can mimic focal segmental glomerulosclerosis (FSGS) (14). However, electron microscopy can visualize uromodulin accumulation as intracellular deposits of amorphous or fibrillar material in the endoplasmatic reticulum of Henle's loop (15, 16). Kidney ultrasound may reveal kidney cysts, and kidneys can be small or normal in size. 1 Hyperuricemia is often present, usually precedes CKD, and is more prevalent in men (17). The diagnosis is established by genetic testing, revealing a heterozygous pathogenic variant in the UMOD gene. More than 130 pathogenic variants have been described, predominantly missense changes clustering in exons 3 and 4, leading to the misfolding of the uromodulin protein (5, 18, 19). In the ADTKD cohort, the median age of ESRD onset was 54 years, and women seem to have better renal survival than men. 17 No specific treatment is available for ADTKD-UMOD. Usual therapy is similar to treatment for other forms of CKD. Hyperuricemia is treated with allopurinol or febuxostat, which can prevent gout attacks. However, there is no data suggesting that therapy with this agent could slow down the progression of CKD (1, 13). Interestingly, disease progression was slowed in an animal model of ADTKD-UMOD after treatment with a tumor necrosis factor-α inhibitor, emphasizing a potential role of inflammation in ADTKD-UMOD development (20).
The MUC1 gene is located on chromosome 1q21, and it encodes mucin 1, a transmembrane protein, expressed on the apical surface of many cells. Mucin 1 provides a protective cellular lair but is also involved in cellular signaling (18). In the kidney, mucin 1 is expressed in the distal tubule and collecting duct (18). Most MUC1 mutations are caused by cytosine duplication within a seven-cytosine stretch in the variable-number tandem repeat (VNTR) region, that usually produces a frameshift mutation (21). Other mutations include the addition of a guanosine residue or the loss of two cytosine residues. The mutated gene produces an abnormal protein, and retention of the changed protein in the renal tubular epithelial cells leads to kidney damage. A positive staining of abnormal proteins in the urinary exfoliated cells could help diagnose the condition (21). The true prevalence of ADTKD-MUC1 is unknown, as pathogenic variants in MUC-1 are not detected by NGS analysis. The diagnostic procedure requires specialized diagnostic testing, which is not routinely performed. The main clinical features of ADTKD-MUC1 are the same as in ADTKD-UMOD, i.e., chronic kidney disease, hyperuricemia, gout, bland urinary sediment, and dominant inheritance. The renal disease appears to be more aggressive in patients with ADTKD-MUC1 in comparison to ADTKD-UMOD with an earlier onset of ESDR (mean age 45 years) (17). There is no specific therapy for ADTKD-MUC1. However, the newly identified molecule BRD4780 induces the removal of changed proteins and could be a promising solution for toxic proteinopathies, such as ADTKD-MUC1 or ADTKD-UMOD (22).
The autosomal dominant tubulointerstitial kidney disease related to REN variants was previously known as Familial Juvenile Hyperuricemic Nephropathy Type 2. The REN gene is located on chromosome 1, and the disease is mostly caused by mutations in the first exon of the REN gene. The type of mutation is either a missense change or an in-frame deletion. These mutations lead to improper renal development (23). Renin has a vital role in the renin-angiotensin-aldosterone system, which has a crucial role in a lot of biological functions, e.g., sodium and potassium balance, vascular tone modification, etc (24). ADTKD-REN has an early onset during childhood, and rarely presents as a milder form of the disease in adulthood.6 Similar to other forms of ATDKD, ADTKD-REN doesn't have any specific clinical or pathohistological features, except for decreased immunohistochemical staining for renin in the juxtaglomerular apparatus (18). However, due to decreased secretion of renin and aldosterone, hypotension and hyperkalemia can be observed (21, 25). Patients suffering from ADTKD—REN are susceptible to acute renal injury in setting of hypovolemia and NSAID use (18, 24). Despite early onset and reduced GFR in childhood, kidney function remains stable during adolescence (26). Another sign of ADTKD-REN is hypoproliferative anemia, which is probably secondary to low erythropoietin levels. This type of anemia disappears in the adolescence, which could be explained with increased production of sex steroids resulting in increased erythropoietin production (18, 25, 26).
Regarding the treatment of patients with ADTKD-REN, alongside the treatment of hyperuricemia and CKD, hypoproliferative anemia is treated with erythropoietin, hypotension and hyperkalemia can be treated with fludrocortisone (18, 25). Moreover, the potential role of BRD4780 can be taken into account, due to the fact that prorenin is accumulated in the kidney, similar to ADTKD-MUC1 (22).
Autosomal dominant tubulointerstitial kidney disease is also related to HNF1B, a gene that is located on chromosome 17q12 and is expressed in multiple tissues with an important role in early embryonic development, which could explain the complex presentations of the HNF1B-related disorders. They can affect many organs: the kidney and genitourinary tract, pancreas, and the liver. The loss of HNF1B gene function is one of the features of the 17q12 deletion syndrome. Studies suggest that a loss of one copy of the HNF1B gene in each cell causes the kidney and urinary tract abnormalities, as well as abnormalities of the pancreas that underlie diabetes (27–29). It was found that loss of HNF1B induces epithelial-mesenchymal transition and leads to kidney fibrosis (30). The loss of one copy of LHX1, also located on chromosome 17q12, is the likely cause of intellectual disability, behavioral and psychiatric conditions (27–31). Thus, ADTK-HNF1B, unlike other ADTKD forms presenting with CKD and gout, has a complex, syndromic presentation involving multiple organic systems. The MAGIC LUCID is helpful mnemonic which summarizes the clinical features of ADTKD-HNF1B, both renal and extrarenal: M = Hypomagnesemia; A = Autosomal dominant; G = Genital tract abnormalities, including bicornuate uterus, absent uterus, vaginal hypoplasia; I = Incomplete penetrance; C = Cysts of the kidney and other structural abnormalities, including multicystic kidneys, fetal bilateral hyperechogenic kidneys, kidney agenesis, and hypoplastic kidneys; L = Liver test abnormalities; U = Uric acid elevation; C = Chronic kidney disease; I = Inherited; D = Diabetes and pancreatic anomalies (32).
The autosomal dominant tubulointerstitial kidney disease-SEC61A1 is a disease associated with pathogenic variants, i.e., missense mutations in SEC61A1, a gene responsible for the encoding of the alpha subunit of the endoplasmatic reticular membrane translocon SEC61. The function of the SEC61 complex is crucial for the kidney development (10). The pathogenic variants cause SEC61-channelopathy due to the change in selectivity and permeability of the translocon channel. Moreover, the changed SEC61α accumulates in the endoplasmatic reticulum, leading to alterations in post-translational modifications and folding of various secretory and transmembrane proteins (uromodulin, mucin-1, renin) (10, 33). These alterations are causing endoplasmatic reticulum dysfunction and apoptosis, which leads to interstitial fibrosis (10). No specific treatment is available for ADTKD-SEC61A1, but recent data show that sodium phenylbutyrate (a small molecule approved for treatment of urea cycle disorders) may reverse the impairment of renin transport in ADTKD-SEC61A1 (34).
ADTKD-DNAJB11 is a very rare form of ADTKD with an atypical clinical presentation. Recently, five different heterozygous pathogenic variants in the DNAJB11 gene were identified in seven families, consistent with autosomal dominant inheritance. The remarkable feature of ADTKD—DNAJB11 is that the renal phenotype and clinical presentation markedly overlap with autosomal dominant polycystic kidney disease with small bilateral renal cysts, slightly enlarged kidneys, liver cysts, and slowly progressive renal failure. ESRD develops later than in other ADTKD forms, most commonly in the sixth or seventh decade (35). Thus, the diagnosis can only be made by performing genetic analysis and confirming the presence of a pathogenic variant in the DNAJB11 gene (35). The DNAJB11 gene produces a cofactor of GRP78/BiP, a major endoplasmatic reticulum protein, that has multiple functions in maintaining endoplasmatic reticulum homeostasis. Interestingly, histologic analysis of renal biopsy samples from affected patients also showed intracellular depositions of uromodulin and mucin-1 (35). There is no specific therapy available for ADTKD-DNAJB11 (6).
A 34-year-old patient was admitted to our department due to kidney injury. His serum creatinine levels were elevated (140–150 μmol/L, reference range 60–104 μmol/L, GFR 50 ml/min/1.73 m2). He had bland urinary sediment and no significant proteinuria (0.1 g in daily urine). Despite his slightly reduced GFR, he had hypoproliferative anemia with a hemoglobin level of 116 g/L. His blood pressure was slightly elevated, up to 145/95 mmHg. The kidneys appeared normal on the ultrasound exam. He had no history of other diseases, and he was taking no medication. His father and aunt also had chronic kidney disease. They both developed end-stage renal disease in adulthood (at 36 and 50 years of age, respectively), but the exact cause of ESRD was not established. We performed a kidney biopsy, which showed nonspecific chronic changes in the glomeruli and tubulointerstitium. Small foci of fibrosis in the tubulointerstium were observed, with stratified tubular basal membranes (Figures 1, 2). One small segment of glomerular sclerosis was found. The immunofluorescence finding was nonspecific, and electron microscopy showed no significant changes other than focal loss of podocyte foot processes, thus mimicking focal segmental glomerulosclerosis (FSGS). Considering positive family history, clinical data, and histopathological findings in the kidney, genetic analysis was performed in order to confirm ADTKD. DNA was extracted from an EDTA peripheral blood sample. A molecular genetic analysis of the genes UMOD, HNF1 B, REN, and PAX2, as well as parts of the MUC1 gene, was without pathological findings. Targeted polymerase chain reaction (PCR) amplification of the VNTR-region between exons 2 and 3 of the MUC1 gene was performed. Subsequently, amplicons were digested with Mwol and a proper mini-sequencing approach (SnaPshot Multiplex Kit, Life Technologies) was used to specifically prove the presence of the ADTKD-causing cytosine duplication c.428dupC in the MUC1 gene (OMIM 158340; RefSeq NM_002456.5) (36). The generated fragments were separated with an automatic sequencing system (Applied Biosystems) and analyzed using the GeneMapper 4.1 software (37, 38). To determine the pathogenicity of the variant, the results were compared across public databases: OMIM (Online Mendelian Inheritance in Man), Database of Single Nucleotide Polymorphisms (dbSNP), US National Library of Medicine (Pubmed), Exome Variant Server (EVS), Exome Aggregation Consortium Browser (ExAC), Genome Aggregation Database Browser (gnomAD), Human Gene Mutation Database (HGMD), and Leiden Open Variation Database (LOVD) (36, 39–45). The detected sequence variant c.428dupC within the MUC1 VNTR (variable number of tandem repeats) leads to a shift in the reading frame, causing an early stop codon shortly beyond the VNTR domain, resulting in a protein lacking the transmembrane region and the cytoplasmatic domain, and is the prototypic pathogenic mutation in ADTKD-MUC1 (class 5-variant; HGMD-ID CP132082; Pubmed PMIDs 23396133, 24670410). The classification of sequence variants was done according to Wallis et al. (46). The finding of a pathogenic variant in MUC-1 confirmed the ADTKD-MUC1 diagnosis in our patient. The patient was treated with allopurinol, calcium channel blocker and an ACE inhibitor. In a six-year follow-up period, his kidney function was stable in stage G3. The clinical data are summarized in Table 2.
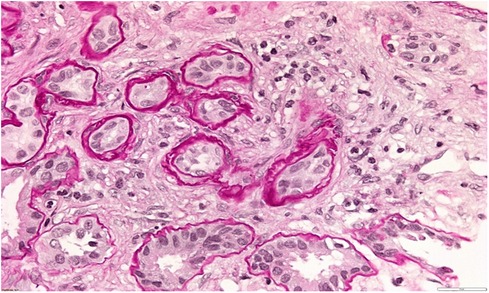
Figure 1. Kidney biopsy showing small foci of fibrosis in tubulointerstitium and stratified tubular basal membranes.
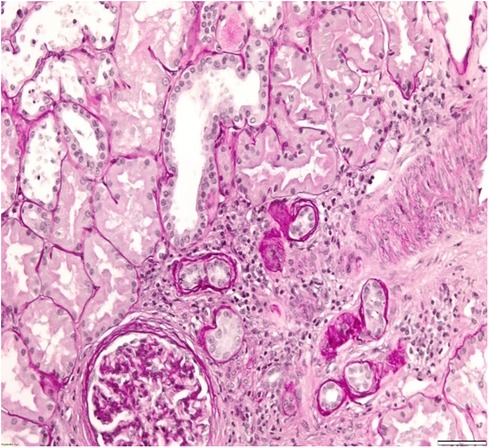
Figure 2. Kidney biopsy showing small foci of fibrosis in tubulointerstitium.

Table 2. Clinical course of ADTKD-MUC1 patient.
A 17-year-old male patient was referred to genetic counseling due to dysmorphic facial features, intellectual disability, and epilepsy. He had significant delays in motor and speech development and had been treated with valproic acid due to epilepsy since childhood. On physical examination, he had a height of 177 cm (50th centile), a weight of 69 kg (46th centile), a head circumference of 56 cm (25th centile), a long and narrow face appearance, a protruding chin, and prominent ears. His intellectual efficiency was between moderate and severe. Family history was negative for mental or kidney diseases. Additional workup was done, revealing hypomagnesemia (Mg 0.45 mmol/L, reference range 0.74–0.97 mmol/L). He had slightly elevated serum creatinine levels (92 μmol/L, reference range 55–90 μmol/L) with bland urinary sediment and no proteinuria—0.04 g in daily urine (Table 3). On ultrasound, the kidneys were of normal size but with impaired corticomedullar differentiation, hyperechoic parenchyma, and dilatated pyelons. The genomic DNA of the proband and, subsequently, that of his parents and sister was extracted from an EDTA peripheral blood sample. Array comparative genomic hybridization was performed using the SurePrint G3 Human 8 × 60 k Agilent oligo-microarray chip (Agilent Technologies, Santa Clara, CA, USA). Normal human male DNA (Agilent Technologies) was used as a reference. DNA labeling, slide hybridization, and washing were performed following the standard protocol provided by Agilent (47). A microarray slide was scanned in the Agilent Microarray Scanner System, and image data were extracted using the Agilent Feature Extraction software (v12.0.31). Agilent Cytogenomics software (v5.1.1.15) was used for the analysis of the results to determine copy number variations (CNVs). Genomic positions refer to the UCSC Genome Browser, February 2009, and the NCBI Build 37 reference sequence (GRCh37/hg19) (48).
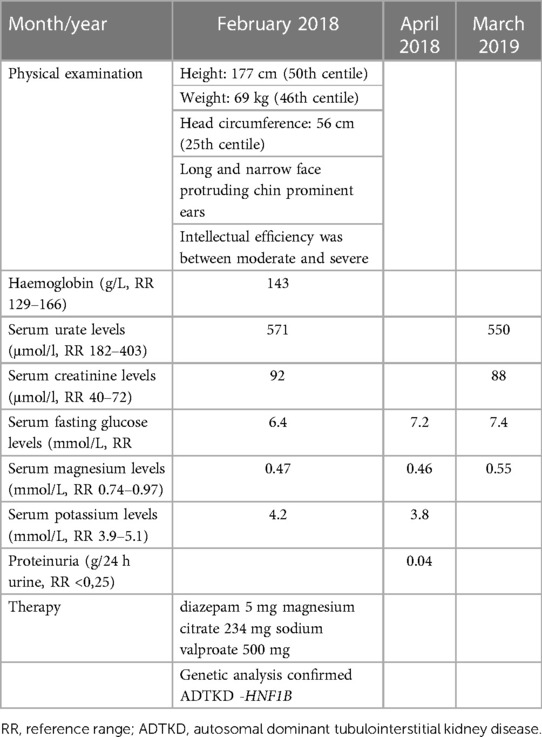
Table 3. Clinical course of ADTKD-HNF1B patient.
Array-CGH data were confirmed by using MLPA (Multiplex Ligation Probe Amplification) with the SALSA MLPA kit P297-B2 Microdeletion Syndromes-2 (49). The MLPA reaction was performed according to the manufacturer's standard protocol and reagents (MRC Holland, Amsterdam, the Netherlands) (50). Amplified products were recognized and quantified by capillary electrophoresis on an ABI 3,130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). For MLPA analysis, raw data were exported into the software Coffalyser (MRC- Holland). To determine the pathogenicity of CNVs, the microarray results were compared across public databases: DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans Using Ensemble Resources), OMIM (Online Mendelian Inheritance in Man), ClinGen (Clinical Genome Resource and ISCA), PubMed, and population database DGV (Database of Genomic Variants) (36, 40, 51–53). Array-CGH analysis revealed a male molecular karyotype with a 1,392 kb heterozygous interstitial deletion on the long arm of chromosome 17 involving region q12 [hg 19 coordinates chr17: 34,856,055-36,248,918] according to the UCSC Genome Browser (GRCh37/hg19 assembly) (Figure 3). Copy number variations (CNVs) reported in the Database of Genomic Variants were excluded from further analysis. The deleted region 17q12 contains 19 protein-coding genes, three of which are reported in the OMIM Morbid Map: Hepatocyte nuclear factor-1-beta HNF1B (OMIM ID 189907), Acetyl-CoA carboxylase alpha ACACA (OMIM ID 200350), and phosphatidylinositol glycan anchor biosynthesis class W protein PIGW (OMIM ID 610275). The deletion was confirmed by MLPA (Multiplex Ligation Probe Amplification) using SALSA MLPA probemix P297-B2 Microdeletion Syndromes-2 (MRC-Holland). MLPA analysis revealed the loss of LHX1, AATF, and HNF1B genes in the patient, his mother, and his sister (Figure 4), and a normal copy number for all genes of this probemix in the father (54). The patient was treated with magnesium suspension therapy, diazepam, and valproate. During the follow-up period, he didn't develop any new health issues, and his kidney function remained stable. In the meantime, his mother developed MODY5 diabetes. She had discreetly reduced renal function with mild hydronephrosis of the right kidney and multiple kidney cysts. Medical records of the patient's sister were not available for this report.
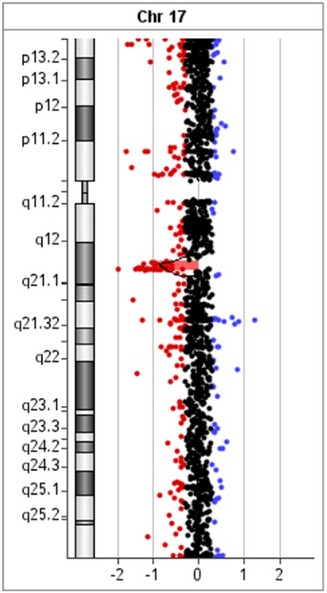
Figure 3. Array comparative genomic hybridization of the proband showed a 1.39 Mb heterozygous interstitial deletion at 17q12 arr[GRCh37] 17q12(34856055_36248918)x1.
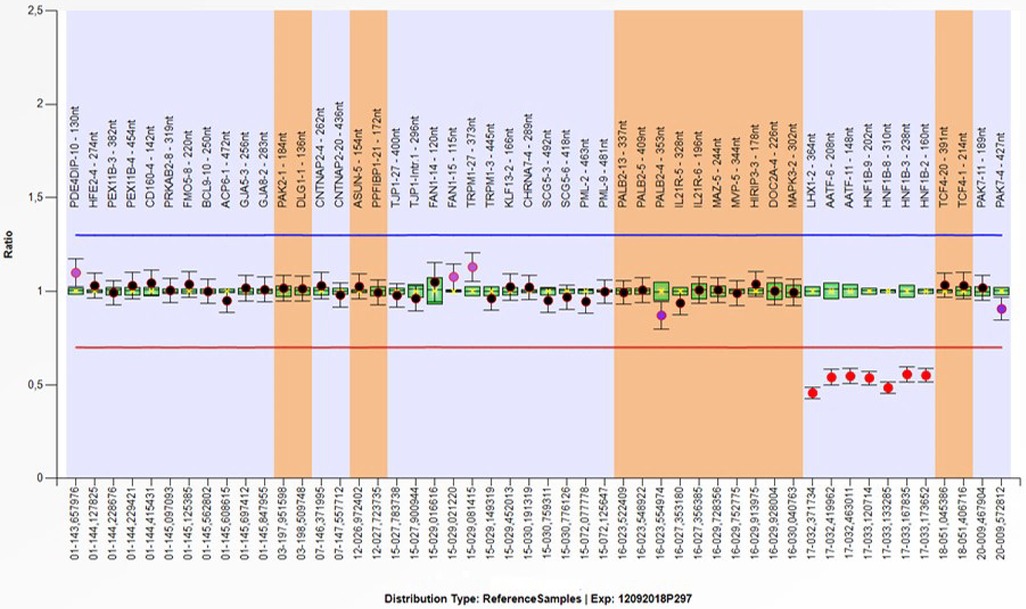
Figure 4. MLPA (Multiplex ligation probe amplification) SALSA MLPA probemix P297-B2 microdeletion syndromes -2 (MRC-holland): rsa 17q12(LHX1,AATF,HNF1B)x1 mat.
ADTKD includes different genetic forms (ADTKD-UMOD, ADTKD-MUC1, ADTKD-REN, ADTKD-HNF1B, ADTKD-SEC61A1, and ADTKD-DNAJB11) of slowly progressive kidney disease. The positive family history compatible with autosomal dominant inheritance and CKD with the clinical characteristics of tubulointerstitial kidney disease should raise suspicion of ADTKD (13). If there is no family history of CKD, clinical suspicion should be raised based on clinical characteristics, including early onset of hyperuricemia or gout, and compatible histology on a kidney biopsy (if available). In the case of HNF1B-related disease, there should be extrarenal manifestations of the disease. The diagnosis is based on a positive family history and compatible kidney histology in at least one affected family member, with a demonstration of a pathogenic variant in one of the aforementioned genes in an affected individual or at least one family member (13). The role of kidney biopsy in diagnosing ADTKD is controversial since there is no pathognomonic finding and histological changes can mimic focal segmental glomerulosclerosis (FSGS), much like in our ADTKD-MUC1 patient. The clinical findings (reduced GFR of 50 ml/min/1.73 m2, bland urinary sediment with no significant proteinuria) were not consistent with the diagnosis of FSGS, which made us consider genetic analysis in order to establish the correct diagnosis. Although hypo proliferative anemia is more common in ADTKD-REN, our patient had low values of hemoglobin at diagnosis (116 g/L). It is reported that ADTKD-MUC1 has a more aggressive clinical course in comparison to ADTKD-UMOD, with an earlier onset of ESDR (mean age 45 years) (17).
Our ATDKD-MUC1 patient, however, has a stable kidney function in stage G3 at age 40.
Regarding our patient with ADTKD-HNF1B, he had classic clinical features of the disease (CKD without significant proteinuria, bland urinary sediment, hypomagnesemia, neurologic disturbances, and kidney structural abnormalities). Family history seemed to be negative; however, the genetic analysis revealed the diagnosis and AD inheritance consistent with ADTKD-HNF1B disease.
Deletions within the 17q12 region, which contains the HNF1B, ACACA, and LHX1 genes, are consistent with a diagnosis of recurrent 17q12 deletion syndrome. We have been searching in the Decipher and other databases for relatively similar-sized deletions at 17q12 and have identified over 70 cases with certain pathogenicity, pointing to the mostly characteristic phenotype of this syndrome. In this way, the recurrent nature of deletions and duplications is thought to be mediated by flanking segmental duplications or low-copy repeats (LCRs) that increase the likelihood of copy number variation through nonallelic homologous recombination (NAHR). The presence of region-specific low-copy repeats (LCRs) is commonly caused by NAHR between paralogous genomic segments (55)..
HNF1B is a transcription factor-2 (TCF2), whose expression was detected in the kidney, liver, bile ducts, thymus, genital tract, pancreas, lung, and gut (56). Haploinsufficiency of the HNF1B gene is responsible for most of the features associated with the 17q12 microdeletion syndrome. LHX1 plays a role in the differentiation of neural cells and the transcriptional control of axonal guidance and has been associated with epilepsy, autism, and intellectual disability (57). The ACACA gene encodes acetyl-CoA carboxylase, an important enzyme in fatty acid synthesis. The recurrent 17q12 deletion is a pathogenic variant with incomplete penetrance and variable expressivity. 30 As reported, the estimation risk for DD/ID of penetrance was about 34.4% for the 17q12 deletion, including the HNF1B gene (58). There is no specific treatment for any form of ADTKD. The data about the possible beneficial effect of treatment with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers on CKD progression in ADTKD patients is lacking. Patients with UMOD-related disease who develop gout will probably have relapses and therefore should be treated with allopurinol or febuxostat (13, 59). It is not known if a strict diet with low purine content is beneficial in patients with hyperuricemia with UMOD mutations. Diuretics should be prescribed cautiously in all patients with ADTKD, as they may worsen hyperuricemia and volume depletion (60). Increased fluid intake is recommended to compensate for possible urine concentration dysfunction. A low-sodium diet is not recommended for ADTKD-UMOD and ADTKD-REN patients, as decreased salt intake may worsen hyperuricemia and volume depletion. 13 Non-steroidal anti-inflammatory drugs should be avoided in all patients with ADTKD, especially in patients with REN mutations who are very susceptible to acute deterioration of renal function. In patients with ADTKD-REN, erythropoietin and fludrocortisone may be used for the treatment of anemia and symptomatic hypotension, respectively (13, 61). Fludrocortisone should not be prescribed in patients with deteriorating renal function, arterial hypertension, hypokalemia, and edema (61). All ADTKD patients are good candidates for kidney transplantation since there is no disease recurrence in the graph (6).
In summary, our two cases emphasize the importance of considering ADTKD within the wide range of differential diagnoses when examining a patient presenting with CKD, even in the absence of family history and other suggestive symptoms and signs. We believe that further reports of ADTKD cases might aid in improving the recognition and care of this intriguing disease group.
The ADTKD—MUC1 patient has been worried about his health since he was young, due to an unknown kidney disease present in the family. Although the diagnosis was established, he is still troubled by the fact that his future remains uncertain.
The mother of the ADTKD-HNF1B patient consulted a pediatrician due to the suspicion of a hereditary disease in her son, although the family history was not clearly positive. The diagnostic work-up confirmed her suspicions. Although her son's condition is now stable, he needs constant care and attention.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethical approval was not required for the studies involving humans. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants' legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
MF: Writing – original draft, Writing – review & editing. SH: Writing – original draft, Writing – review & editing. BB: Formal Analysis, Writing – original draft, Writing – review & editing. IT: Formal Analysis, Writing – original draft, Writing – review & editing. SB: Formal Analysis, Writing – original draft, Writing – review & editing. MC: Formal Analysis, Writing – original draft, Writing – review & editing. LL: Writing – original draft, Writing – review & editing. EI: Writing – review & editing. IV: Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Devuyst O, Knoers N, Remuzzi G, Schaefer F. Board of the working group for inherited kidney diseases of the European Renal Association and European dialysis and transplant association. Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet Lond Engl. (2014) 383(9931):1844–59. doi: 10.1016/S0140-6736(14)60659-0
2. Rasouly H, Groopman E, Heyman-Kantor R, Fasel D, Mitrotti A, Westland R, et al. The burden of candidate pathogenic variants for kidney and genitourinary disorders emerging from exome sequencing. Ann Intern Med. (2019) 170(1):11. doi: 10.7326/M18-1241
3. Groopman E, Marasa M, Cameron-Christie S, Petrovski S, Aggarwal V, Milo-Rasoulyet H, et al. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med. (2019) 380(2):142–51. doi: 10.1056/NEJMoa1806891
4. Cocchi E, Nestor JG, Gharavi AG. Clinical genetic screening in adult patients with kidney disease. Clin J Am Soc Nephrol CJASN. (2020) 15(10):1497–510. doi: 10.2215/CJN.15141219
5. Mabillard H, Sayer JA, Olinger E. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease. Nephrol Dial Transplant. (2021) 10:1093. doi: 10.1093/ndt/gfab268
6. Econimo L, Schaeffer C, Zeni L, Cortinovis R, Alberici F, Rampoldi L, et al. Autosomal dominant tubulointerstitial kidney disease (ADTKD): an emerging cause of genetic chronic kidney disease. Kidney Int Rep. (2022) 7(11):2332–44. doi: 10.1016/j.ekir.2022.08.012
7. Bleyer AJ, Kmoch S. Autosomal dominant tubulointerstitial kidney disease: of names and genes. Kidney Int. (2014) 86:459–61. doi: 10.1038/ki.2014.125
8. Faguer S, Decramer S, Chassaing N, Bellanné-Chantelot C, Calvas P, Beaufils S, et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int. (2011) 80:768–76. doi: 10.1038/ki.2011.225
9. Medicine M-N. Online Mendelian inheritance in man. An online catalog of human genes and genetic disorders [Website]. John Hopkins University: OMIM; 2018
10. Bolar NA, Golzio C, Živna M, Hayot G, Van Hemelrijk C, Schepers D, et al. Heterozygous loss-of-function SEC61A1 mutations cause autosomal-dominant tubulo-interstitial and glomerulocystic kidney disease with anemia. Am J Hum Genet. (2016) 99:174–87. doi: 10.1016/j.ajhg.2016.05.028
11. Bleyer AJ, Kmoch S, Antignac C, Robins V, Kidd K, Kelsoeet J, et al. Variable clinical presentation of an MUC1 mutation causing medullary cystic kidney disease type 1. Clin J Am Soc Nephrol. (2014) 9:527–35. doi: 10.2215/CJN.06380613
12. Bollée G, Dahan K, Flamant M, Morinière V, Pawtowski A, Heidet L, et al. Phenotype and outcome in hereditary tubulointerstitial nephritis secondary to UMOD mutations. Clin J Am Soc Nephrol. (2011) 6:2429–38. doi: 10.2215/CJN.01220211
13. Eckardt KU, Alper S, Antignac C, Bleyer A, Chauveau D, Dahan K, et al. Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management—a KDIGO consensus report. Kidney Int. (2015) 88(4):676–83. doi: 10.1038/ki.2015.28
14. Chun J, Wang M, Wilkins M, Knob A, Benjamin A, Bu L, et al. Autosomal dominant tubulointerstitial kidney disease-uromodulin misclassified as focal segmental glomerulosclerosis or hereditary glomerular disease. Kidney Int Rep. (2020) 5:519–29. doi: 10.1016/j.ekir.2019.12.016
15. Scolari F, Caridi G, Rampoldi R, Tardanico R, Izzi C, Pirulli D, et al. Uromodulin storage diseases: clinical aspects and mechanisms. Am J Kidney Dis. (2004) 44:987–99. doi: 10.1053/j.ajkd.2004.08.021
16. Nasr S, Lucia J, Galgano S, Markowitz G, D’Agati V. Uromodulin storage disease. Kidney Int. (2008) 73:971–6. doi: 10.1038/sj.ki.5002679
17. Olinger E, Hofmann P, Kidd K, Dufour I, Belge H, Schaefferet C, et al. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease due to mutations in UMOD and MUC1. Kidney Int. (2020) 88:717–31. doi: 10.1016/j.kint.2020.04.038
18. Devuyst O, Olinger E, Weber S, Eckardt KU, Kmoch S, Rampoldi R, et al. Autosomal dominant tubulointerstitial kidney disease. Nat Rev Dis Primers. (2019) 25:1–20. doi: 10.1038/s41572-019-0109-9
19. Kidd K, Vyletal P, Schaeffer C, Olinger E, Živná M, Hodaňováe K, et al. Genetic and clinical predictors of age of ESKD in individuals with autosomal dominant tubulointerstitial kidney disease due to UMOD mutations. Kidney Int Rep. (2020) 5(9):1472–85. doi: 10.1016/j.ekir.2020.06.029
20. Johnson B, Dang L, Marsh G, Roach M, Levine Z, Monti A, et al. Uromodulin p.Cys147Trp mutation drives kidney disease by activating ER stress and apoptosis. J Clin Invest. (2017) 127:3954–396. doi: 10.1172/JCI93817
21. Wang G, Rui H, Dong H, Sun L, Yang M, Wang Y, et al. SMRT Sequencing revealed to be an effective method for ADTKD-MUC1 diagnosis through follow-up analysis of a Chinese family. Sci Rep. (2020) 10(1):8616. doi: 10.1038/s41598-020-65491-2
22. Dvela-Levitt M, Kost-Alimova M, Emani M, Kohnert E, Thompson R, Sidhom E, et al. Small molecule targets TMED9 and promotes lysosomal degradation to reverse proteinopathy. Cell. (2019) 178:521–35. doi: 10.1016/j.cell.2019.07.002
23. Schaeffer C, Izzi C, Vettori A, Pasqualetto E, Cittaro D, Lazarevic D, et al. Autosomal dominant tubulointerstitial kidney disease with adult onset due to a novel renin mutation mapping in the mature protein. Sci Rep. (2019) 9(1):11601. doi: 10.1038/s41598-019-48014-6
24. Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. (2006) 86:747–803. doi: 10.1152/physrev.00036.2005
25. Zivna M, Hulkova H, Matignon M, Hodaňová K, Vylet'al P, Kalbáčová M, et al. Dominant renin gene mutations associated with early-onset hyperuricemia, anemia, and chronic kidney failure. Am J Hum Genet. (2009) 85:204–13. doi: 10.1016/j.ajhg.2009.07.010
26. Živná M, Kidd K, Zaidan M, Vyleťal P, Barešová V, Hodaňová K, et al. An international cohort study of autosomal dominant tubulointerstitial kidney disease due to REN mutations identifies distinct clinical subtypes. Kidney Int. (2020) 98:1589–604. doi: 10.1016/j.kint.2020.06.041
27. Laffargue F, Bourthoumieu S, Llanas B, Baudouin V, Lahoche A, Morin D, et al. Towards a new point of view on the phenotype of patients with a 17q12 microdeletion syndrome. Arch Dis Child. (2015) 100(3):259–64. doi: 10.1136/archdischild-2014-306810
28. Mefford H, Clauin S, Sharp A, Moller R, Ullmann R, Kapur R, et al. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am J Hum Genet. (2007) 81(5):1057–69. doi: 10.1086/522591
29. Moreno-De-Luca D, Mulle JG, Kaminsky EB, Sanders SJ, Myers SM, Adam MP, et al. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am J Hum Genet. (2010) 87(5):618–30. doi: 10.1016/j.ajhg.2010.10.004
30. Nagamani S, Erez A, Shen J, Li C, Roeder E, Cox S, et al. Clinical spectrum associated with recurrent genomic rearrangements in chromosome 17q12. Eur J Hum Genet. (2010) 18(3):278–84. doi: 10.1038/ejhg.2009.174
31. Izzi C, Dordoni C, Econimo L, Delbarba E, Grati F, Martin E, et al. Variable expressivity of HNF1B nephropathy, from renal cysts and diabetes to medullary sponge kidney through tubulo-interstitial kidney disease. Kidney Int Rep. (2020) 5(12):2341–50. doi: 10.1016/j.ekir.2020.09.042
32. Bleyer A, Wolf W, Kidd K, Zivna M, Kmochet S. Autosomal dominant tubulointerstitial kidney disease: more than just HNF1β. Pediatr Nephrol. (2021) 37(5):933–46. doi: 10.1007/s00467-021-05118-4
33. Lang S, Pfeffer S, Lee P, Cavalié A, Helms V, Förster F, et al. An update on Sec61 channel functions, mechanisms, and related diseases. Front Physiol. 2017;8:887. doi: 10.3389/fphys.2017.00887
34. Sicking M, Živná M, Bhadra P, Barešová V, Tirincsi A, Hadzibeganovic D, et al. Phenylbutyrate rescues the transport defect of the Sec61α mutations V67G and T185A for renin. Life Sci Alliance. (2022) 5(4):e202101150. doi: 10.26508/lsa.202101150
35. Cornec-Le Gall E, Olson R, Besse W, Heyer C, Gainullin W, Smithet J, et al. Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet. (2018) 102(5):832–44. doi: 10.1016/j.ajhg.2018.03.013
36. Available at: http://www.omim.org (Accessed May 15, 2023).
37. Kirby A, Gnirke A, Jaffe DB, Barešová V, Pochet N, Blumenstiel B, et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet. (2013) 45(3):299–303. doi: 10.1038/ng.2543
38. Ekici A, Hackenbeck T, Moriniere V, Pannes A, Buettner M, Uebe S, et al. Renal fibrosis is the common feature of autosomal dominant tubulointerstitial kidney diseases caused by mutations in mucin 1 or uromodulin. Kidney INT. (2014) 86(3):589–99. doi: 10.1038/ki.2014.72
39. Available at: http://www.ncbi.nlm.nih.gov/SNP
40. Available at: http://www.ncbi.nlm.nih.gov/pubmed
41. Available at: http://evs.gs.washington.edu/EVS
42. Available at: http://exac.broadinstitute.org
43. Available at: http://gnomad.broadinstitute.org
44. Available at: http://www.hgmd.org
45. Available at: http://www.lovd.nl/3.0/home
46. Wallis Y, Payne S, Mcanulty C, Bodmer D, Sister-mans E, Robertson K, et al. Practice Guidelines for the Evaluation of Pathogenicity and the Reporting of Sequence Variants in Clinical Molecular Genetics, (2013).
47. Available at: https://www.agilent.com/cs/library/usermanuals/public
48. Available at: http://genome.ucsc.edu/index.html
49. Available at: https://www.mrcholland.com/product/P297/2912
51. Available at: https://decipher.sanger.ac.uk
52. Available at: http://www.clinicalgenome.org
53. Available at: http://dgv.tcag.ca/dgv/app/home
54. McGowan-Jordan J, Hastings RJ, Moore S, editors. ISCN 2020: An international system for human cytogenomic Nomenclature. Basel: Karger (2020). p. 225–6.
55. Stankiewicz P, Lupski J. Genome architecture, rearrangements and genomic disorders. Trends Genet. (2002) 18:74–82. doi: 10.1016/S0168-9525(02)02592-1
56. Edghill E, Bingham C, Ellard S, Hattersley A. Mutations in hepatocyte nuclear factor-1-beta and their related phenotypes. J. Med. Genet. (2006) 43:84–90. doi: 10.1136/jmg.2005.032854
57. Rasmussen M, Vestergaard EM, Graakjaer J, Petkov Y, Bache I, Fagerberg C, et al. 17q12 Deletion and duplication syndrome in Denmark: a clinical cohort of 38 patients and review of the literature. Am J Med Genet A. (2016) 170(11):2934–42. doi: 10.1002/ajmg.a.37848
58. Rosenfeld J, Coe B, Eichler E, Cuckle H, Shaffer L. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet Med. (2013) 15(6):478–81. doi: 10.1038/gim.2012.164
59. Faruque LI, Ehteshami-Afshar A, Wiebe N, Tjosvold L, Homik J, Tonelli M, et al. A systematic review and meta-analysis on the safety and efficacy of febuxostat versus allopurinol in chronic gout. Semin Arthritis Rheum. (2013) 43:367–75. doi: 10.1016/j.semarthrit.2013.05.004
60. Labriola L, Olinger E, Belge H, Pirson Y, Dahan K, Devuyst O. Paradoxical response to furosemide in uromodulin-associated kidney disease. Nephrol Dial Transplant. (2015) 30:30–335. doi: 10.1093/ndt/gfu389
Keywords: hereditary kidney disease, autosomal dominant inheritance, tubulointerstitial kidney disease, kidney cyst, renal failure, case report
Citation: Fistrek Prlic M, Huljev Frkovic S, Beck B, Tonkovic Durisevic I, Bulimbasic S, Coric M, Lamot L, Ivandic E and Vukovic Brinar I (2023) Two sides of the same coin: a complex presentation of autosomal dominant tubulointerstitial kidney diseases: a literature review and case reports. Front. Pediatr. 11:1283325. doi: 10.3389/fped.2023.1283325
Received: 25 August 2023; Accepted: 25 October 2023;
Published: 10 November 2023.
Edited by:
Constantinos J. Stefanidis, "Mitera" Children’s Hospital, GreeceReviewed by:
Marijan Saraga, University of Split, Croatia© 2023 Fistrek Prlic, Huljev Frkovic, Beck, Tonkovic Durisevic, Bulimbasic, Coric, Lamot, Ivandic and Vukovic Brinar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivana Vukovic Brinar aXZlbWVkZXhAeWFob28uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.