 Mohammed F. Alosaimi
Mohammed F. Alosaimi Muddathir H. Hamad
Muddathir H. Hamad Muneera J. AlShammari4
Muneera J. AlShammari4- 1Immunology Research Laboratory, Department of Pediatrics, College of Medicine, King Saud University, Riyadh, Saudi Arabia
- 2Allergy and Immunology Unit, Department of Pediatrics, College of Medicine, King Saud University, Riyadh, Saudi Arabia
- 3Division of Neurology, Department of Pediatric Neurology, College of Medicine, King Saud University, Riyadh, Saudi Arabia
- 4Department of Genetics and Metabolic, College of Medicine, King Saud University, Riyadh, Saudi Arabia
- 5Department of Radiology and Medical Imaging, College of Medicine, King Saud University, Riyadh, Saudi Arabia
Background: Bare lymphocyte syndrome type II (BLS II) is a rare form of severe combined immunodeficiency caused by mutations in the CIITA gene, which regulates major histocompatibility complex class II (MHC II) expression.
Objective: We report the case of a Saudi boy with a novel mutation in the CIITA gene who presented with acute and late meningoencephalomyelitis, resulting in severe neurodevelopmental regression.
Methods: We reviewed the patient's clinical and laboratory data obtained from medical records and performed a literature search on BLS II.
Results: The patient presented with acute meningoencephalomyelitis confirmed by MRI findings and was later found to carry a homozygous pathogenic variant in the CIITA gene p.(Leu473Hisfs*15). The patient had no MCH II expression, confirming the genetic diagnosis of autosomal recessive BLS II. Surprisingly, the patient's prior clinical history was unremarkable for significant infections or autoimmunity.
Conclusions: We report a case with a novel CIITA gene mutation presenting atypically with a late and isolated severe infection. Isolated severe meningoencephalomyelitis may be a manifestation of primary immunodeficiency.
Introduction
Bare lymphocyte syndrome type II (BLS II) is a rare inherited form of severe combined immunodeficiency. It was first described in 1978 and is characterized by a lack of major histocompatibility complex class II (MHC II) expression on antigen-presenting cells (1). It manifests with early-onset recurrent infections that mostly affect the respiratory and gastrointestinal systems, resulting in bronchiectasis, malabsorption, and failure to thrive (2). MHC II deficiency is caused by autosomal recessive mutations in genes encoding transcription factors required for MHC II expression. These factors include class II transactivator (CIITA), regulatory factor X, ankyrin repeat-containing (RFXANK), regulatory factor 5 (RFX5), and RFX-associated protein (RFXAP) (3).
CIITA is encoded by a gene with 27 exons that is mapped to chromosome 16p13.13 and consists of 1,130 amino acids (4). CIITA has multiple domains that interact with each other, and it is composed of an amino-terminal acidic domain, proline-, serine-, and threonine-rich (PST) regions, a GTP-binding site, at least one nuclear localization sequence (NLS), and a series of leucine-rich repeats (LRR). A mutation in any of these domains could affect the transcriptional activity of CIITA (5).
Here we report the case of a boy who presented with late and isolated acute meningoencephalomyelitis and was later found to have a novel pathogenic mutation in the CIITA gene that resulted in complete loss of MHC II expression. His atypical presentation presented a challenge in the diagnosis and management of severe combined immunodeficiency.
Case report
We present the case of a 3-year-old Saudi male, who was the offspring of an uncomplicated pregnancy and was born at term by spontaneous vaginal delivery with a birth weight of 2.7 kg. He was in a normal state of health until he presented to our emergency department at the age of 2 years with a 9-day history of fever associated with a runny nose, cough, and vomiting. He also had a 3-day history of progressive lethargy and bilateral lower limb weakness, which was more pronounced on the right side. Furthermore, he had a history of profuse, watery diarrhea 2 days before the presentation. There was no history of abnormal jerky movements, drug ingestion, trauma, or contact with sick patients. He was prescribed oral antibiotics for 1 week without improvement.
The patient's development was appropriate for his age, and he had no history of neurological, visual, or cognitive deficits before this presentation. The patient's past medical history was significant only for COVID-19-induced pneumonia at 6 months of age and a history of acute otitis media at 20 months of age. He received all vaccinations up to one year of age without any complications, which included live vaccines like Bacille Clamette-Guerin(BCG), measles, mumps, rubella (MMR), and varicella.
The patient was the first child of a consanguineous (first-degree) Saudi couple. The father was recently diagnosed with neuromyelitis optica (NMO) and was receiving monthly rituximab therapy. There was no family history of primary immunodeficiency or known genetic disease.
On clinical examination, the patient was febrile, lethargic, and hypoactive. He had no facial asymmetry or dysmorphism; his pupils were reactive bilaterally; and he showed no meningeal signs. Tone and strength were more reduced on the right side of his body (2/5) compared to the left side (3/5). Deep tendon reflexes were normal in the upper limbs and left lower limb and diminished in the right lower limb. Gait could not be assessed since the subject could not bear weight. His systemic examination was otherwise unremarkable, and he had no stigmata of neurocutaneous disease.
An urgent brain CT scan was performed and showed no gross evidence of acute brain injury. A full sepsis work-up and serologic investigation were also performed to look for infectious or autoimmune meningoencephalitis, but the findings were unremarkable (results are shown in Table 1).
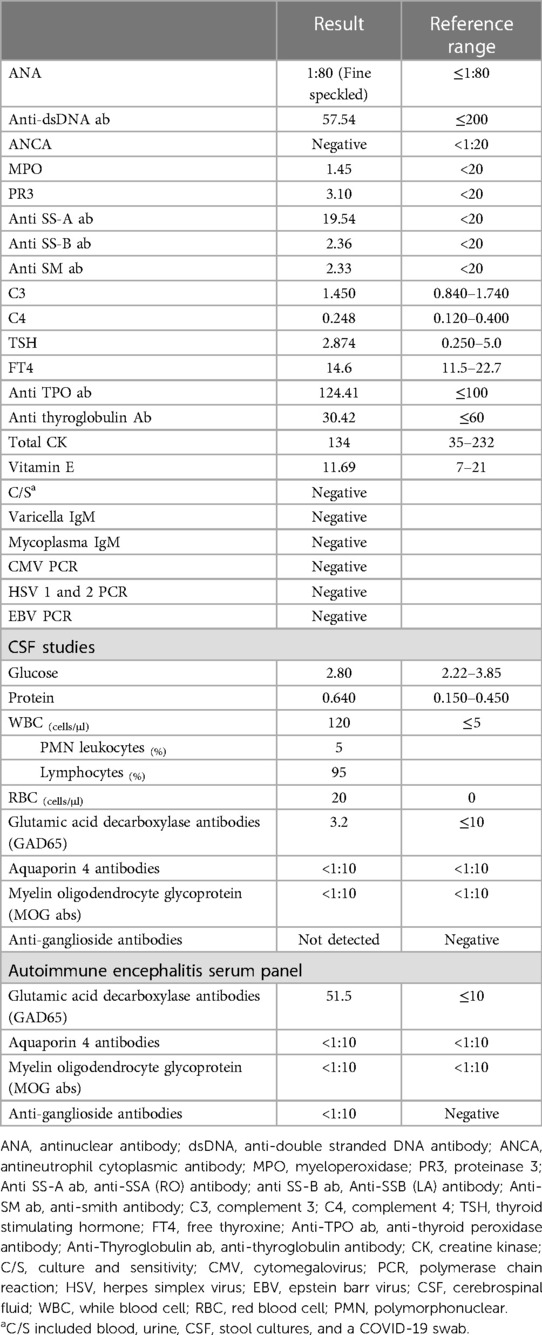
Table 1. Tests performed for the patient.
The patient was admitted to the intensive care unit for neuro-vital monitoring and further management due to progressive weakness. He was started on broad-spectrum antimicrobial and antiviral medications (ceftriaxone, vancomycin, and acyclovir).
Urgent MRI with contrast of the brain and whole spine displayed abnormal T2-weighted-fluid-attenuated inversion recovery (T2-FLAIR) signal intensity and mild diffusion restriction in the midbrain cerebral peduncles bilaterally with leptomeningeal enhancement. Abnormal leptomeningeal enhancement was also seen in the bilateral cerebral hemispheres in the parietal, temporal, and occipital lobes and the pre-chiasmatic segments of the optic nerves. MRI of the spine showed a long segment of abnormal T2 hyperintensity and mild heterogeneous enhancement in the cervical and thoracic spinal cords, along with smooth enhancement along the cauda equina nerve roots (Figure 1). The overall radiologic picture was suggestive of meningoencephalomyelitis. The patient continued to spike a fever for the first 48 hours after admission. However, his fever responded to high-dose intravenous immunoglobulin (IVIG) (1 g/kg of body weight once daily for 2 days). There was no improvement or further deterioration in his neurologic assessment post-IVIG administration. His neuro-ophthalmologic assessment was unremarkable. The patient was discharged home after finishing 3 weeks of antimicrobial therapy. He continued physical therapy with minimal improvement in his lower limb weakness.
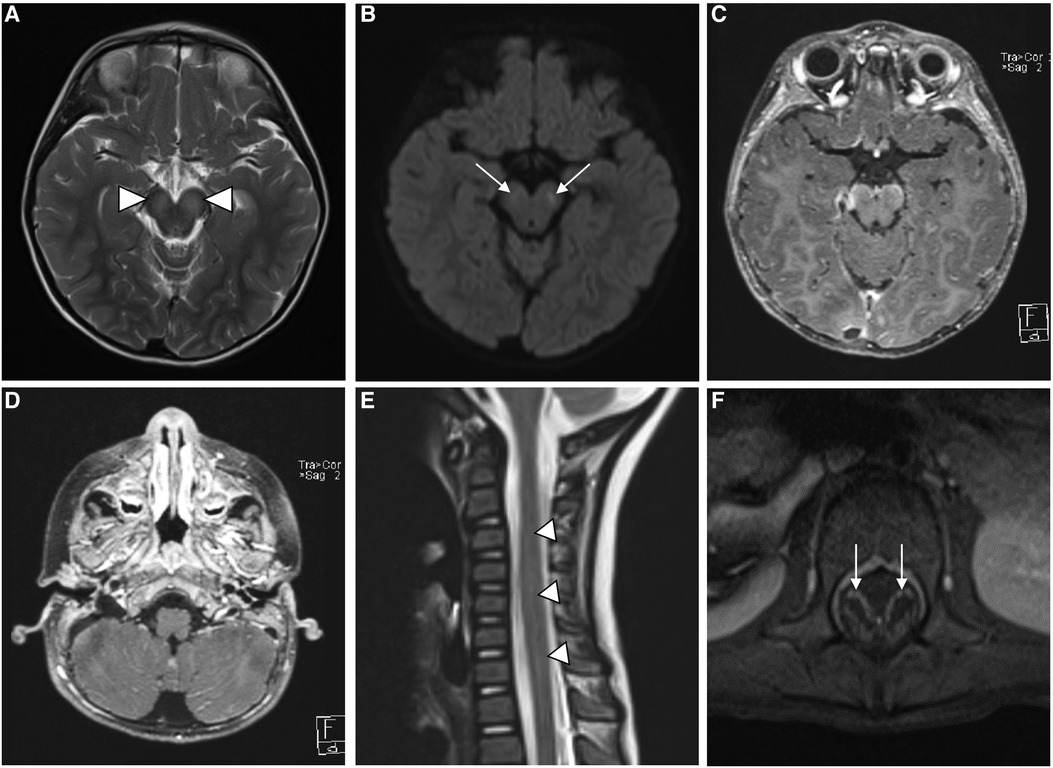
Figure 1. MR imaging of the brain shows bilateral symmetric high signal intensity in the cerebral peduncles (white arrowheads) on T2-weighted images (A), with corresponding faint diffusion restriction (white arrows) on diffusion-weighted images (DWI) (B) axial postcontrast T1-weighted images (C and D) show leptomeningeal enhancement (white lines) along the ventral surface of the midbrain and in the cerebellar folia. Sagittal T2 and axial postcontrast T1-weighted images (E and F) of the spine reveal abnormal hyperintensity of the cervicothoracic spinal cord (white arrow heads in E), along with smooth enhancement of the cuada equina nerve roots (white arrow heads in F).
At a 3-month follow-up clinic visit, the subject was noted to have developmental regression in gross and fine motor skills with difficulty in phonation. His parents reported repeated episodes of choking, and he eventually required nasogastric tube feeding for oropharyngeal dysphagia. On neurologic examination, he was found to have axial hypotonia and diminished deep tendon reflexes. Follow-up MRIs of the brain and whole spine were performed and showed the progression of signal abnormalities in the cerebral peduncles with the development of areas of cavitation and cystic degeneration. New bilateral areas of abnormal T2-FLAIR hyperintensity developed in the medial thalami, globi pallidi, and dorsal pons. The alterations in the cervical and thoracic spinal cord signals developed into focal areas of myelomalacia and cavitation that predominantly involved the anterior horn cells. The cauda equina nerve root enhancement persisted on follow-up MRI (Figure 2). The possibility of vitamin deficiency or riboflavin transporter deficiency was raised. Therefore, the boy was started on a trial of biotin (10 mg daily), thiamine (300 mg daily), and riboflavin (200 mg daily). However, he failed to respond after 4 months of therapy.
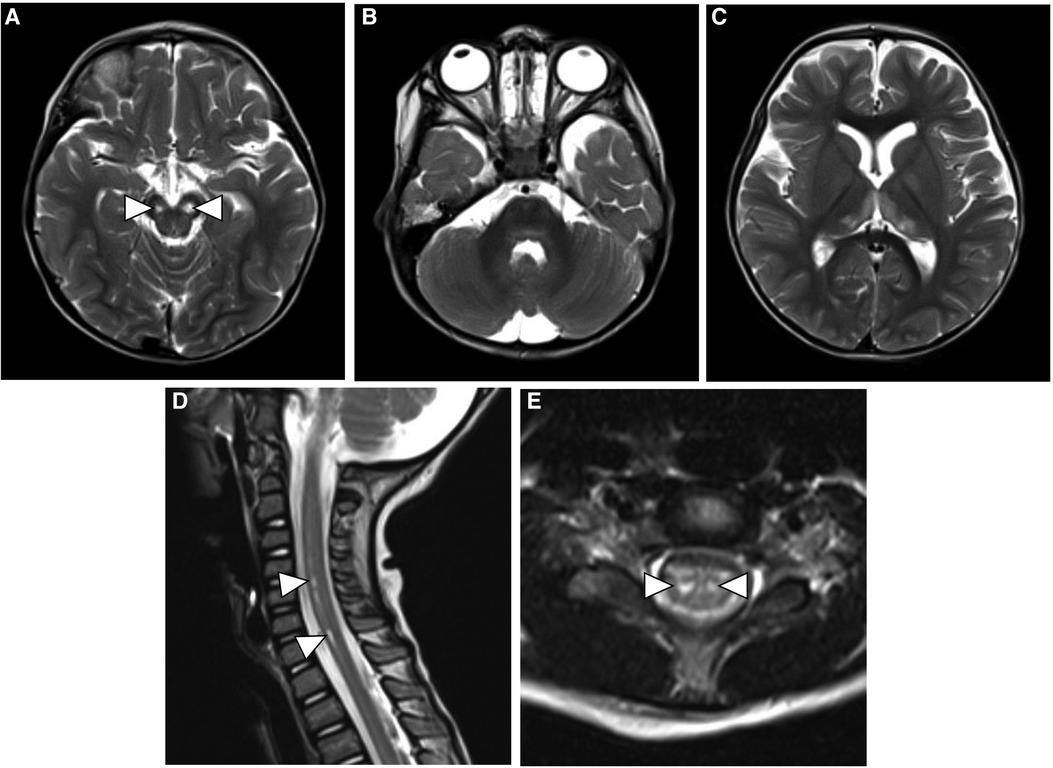
Figure 2. Follow-up axial T2-weighted MR images of the brain (A–C) show cavitary changes in the cerebral peduncles (white arrowheads) with new areas of hyperintensity in the thalami, globi pallidi and dorsal pons. Sagittal and axial T2-weighted images of the spine (D and E) show evolution of the previous spinal cord signal abnormality with development of focal areas of myelomalacia in the anterior horn cells (white arrowheads).
Whole exome sequencing (WES) was ordered clinically to rule out an underlying genetic etiology. It showed that the patient harbored a novel homozygous frameshift variant in the CIITA gene (NM_001286402.1:c.1418_1424del:p.Leu473Hisfs*15). The frameshift mutation resulted in a substitution of the leucine residue at 473 to histidine, followed by 15 novel residues and a premature stop codon. It was predicted to disrupt the GTP-binding site domain. Segregation analysis by Sanger sequencing was performed in a clinical lab and confirmed that the patient was homozygous and both parents were heterozygous for the mutation. The mutation led to a complete absence of MHC II expression in B cells (Table 2). The result is consistent with a genetic diagnosis of autosomal recessive bare lymphocyte syndrome, type II, complementation group A.

Table 2. Immunologic profile of the patient.
Based on this finding, the patient was referred to the immunology clinic, and his immunologic workup showed CD4+ T-cell lymphopenia and panhypogammaglobulinemia (Table 2). The patient was started on trimethoprim/sulfamethoxazole prophylaxis and monthly IVIG and referred to a transplant center for possible hematopoietic stem cell transplantation (HSCT).
Discussion
BLS II is characterized by a lack of MHC II expression, resulting in defective presentation of peptides to CD4+ T helper cells. The immunologic consequences of the loss of MHC II are CD4+ T-cell lymphopenia, hypogammaglobulinemia, and impaired antibody responses to infection or vaccination (7). Consequently, patients with BLS II suffer from severe immunodeficiency, leading to recurrent infections and ultimately death in early childhood (4).
We reported the case of a 3-year-old male patient who was thriving, with a past medical history remarkable only for COVID-19-induced pneumonia and an episode of acute otitis media at 6 and 20 months of age, respectively. He did not suffer from severe recurrent infections or persistent diarrhea, which are characteristic of BLS II. Instead, the young patient presented with severe acute meningoencephalomyelitis. The clinical severity and radiologic findings raised the question of infectious versus autoimmune causes. The boy's autoimmune panel and infectious workup were inconclusive. The possibility of vitamin deficiency or riboflavin transporter deficiency was raised, but he did not respond to replacement therapy. As a last option to reach a diagnosis, a WES was sent clinically and showed a pathogenic variant in CIITA. His immune profile was consistent with BLS II. He had low CD4+ T cells, normal CD8+ T cells, and B cells; reduced levels of IgA, IgM, and IgG; and no MHC II expression on CD19+ cells. Genetic testing showed a novel pathogenic loss-of-function mutation in CIITA [c.1418_1424del p.(Leu473Hisfs*15)] that abrogates MHC II expression.
We looked for possible autoimmune encephalitis or vitamin deficiency causes because the patient had a severe case of meningoencephalomyelitis, but no pathogenic organisms were found. Due to this, we tried high-dose IVIG and vitamin (biotin, thiamine, and riboflavin) replacement therapy empirically, but they had no effect on his condition. His autoimmune panels for both serum and CSF came back negative, except for a small increase in serum anti-glutamic acid decarboxylase (GAD65) antibody levels (51.5). This was inconsistent with autoimmune encephalitis, which often has very high serum titers. A working diagnosis of aseptic meningoencephalomyelitis would therefore be the most likely diagnosis given his presentation. Based on a review of reported cases, there have been only 18 reported BLS II patients with a CIITA mutation, of which 10 had a missense/nonsense mutation, three had a splicing mutation, one was a regulatory mutation, two had a small deletion mutation, and two had gross deletion mutations [Human Gene Mutation Database (HGMD) 2021] (8–21). A literature review of previously reported patients revealed that none had presented with mainly neurologic manifestations, which constituted a challenge in the diagnosis of our patient (8–21).
BLS II patients have a poor prognosis, and they tend to die early because of severe infections. Treatment options include supportive care with prophylactic therapy with IVIG and antibiotics, but the definitive treatment is HSCT, despite a low success rate of only 60% (22, 23). Our patient was deemed ineligible for HSCT due to his poor neurological condition.
In conclusion, this article reports a case of BLS II presenting late with meningoencephalomyelitis, resulting in severe neurodevelopmental regression without a significant prior infectious history. This finding adds to the wide spectrum of clinical and immunologic phenotypes of such a rare disease and promotes awareness of the disease.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
MA: Conceptualization, Data curation, Investigation, Methodology, Project administration, Supervision, Validation, Writing – review & editing. NM: Data curation, Formal Analysis, Methodology, Writing – original draft, Writing – review & editing. MH: Conceptualization, Data curation, Methodology, Writing – review & editing. MA: Data curation, Investigation, Writing – review & editing. DJ: Data curation, Investigation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank the family of the subject for agreeing to participate in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Touraine JL, Betuel H, Souillet G, Jeune M. Combined immunodeficiency disease associated with absence of cell-surface HLA-A and -B antigens. J Pediatr. (1978) 93(1):47–51. doi: 10.1016/S0022-3476(78)80598-8
2. Nekrep N, Fontes JD, Geyer M, Peterlin BM. When the lymphocyte loses its clothes. Immunity. (2003) 18(4):453–7. doi: 10.1016/S1074-7613(03)00086-4
3. Reith W, Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu Rev Immunol. (2001) 19:331–373. doi: 10.1146/annurev.immunol.19.1.331
4. Mach B, Steimle V, Reith W. MHC class II-deficient combined immunodeficiency: a disease of gene regulation. Immunol Rev. (1994) 138:207–21. doi: 10.1111/j.1600-065X.1994.tb00853.x
5. Harton JA, Ting JP. Class II transactivator: mastering the art of major histocompatibility complex expression. Mol Cell Biol. (2000) 20(17):6185–94. doi: 10.1128/MCB.20.17.6185-6194.2000
6. Castano-Jaramillo LM, Bareño-Silva J, Tobon S, Escobar-Gonzalez AF. Meta-analysis of hematopoietic stem cell transplantation in major histocompatibility complex class II deficiency. Pediatr Transplant. (2020) 24(6):e13774. doi: 10.1111/petr.13774
7. Griscelli C, Lisowska-Grospierre B, Mach B. Combined immunodeficiency with defective expression in MHC class II genes. Immunodefic Rev. (1989) 1(2):135–53. PMID: 25172092517209
8. Dimitrova D, Ong PY, O’Gorman MR, Church JA. Major histocompatibility complex class II deficiency complicated by Mycobacterium avium complex in a boy of mixed ethnicity. J Clin Immunol. (2014) 34(6):677–80. doi: 10.1007/s10875-014-0048-x
9. Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell. (1993) 75(1):135–46. doi: 10.1016/S0092-8674(05)80090-X
10. Bontron S, Steimle V, Ucla C, Eibl MM, Mach B. Two novel mutations in the MHC class II transactivator CIITA in a second patient from MHC class II deficiency complementation group A. Hum Genet. (1997) 99(4):541–6. doi: 10.1007/s004390050403
11. Quan V, Towey M, Sacks S, Kelly AP. Absence of MHC class II gene expression in a patient with a single amino acid substitution in the class II transactivator protein CIITA. Immunogenetics. (1999) 49(11-12):957–63. doi: 10.1007/s002510050579
12. Peijnenburg A, Van den Berg R, Van Eggermond MJ, Sanal O, Vossen JM, Lennon AM, et al. Defective MHC class II expression in an MHC class II deficiency patient is caused by a novel deletion of a splice donor site in the MHC class II transactivator gene. Immunogenetics. (2000) 51(1):42–9. doi: 10.1007/s002510050007
13. Wiszniewski W, Fondaneche MC, Le Deist F, Kanariou M, Selz F, Brousse N, et al. Mutation in the class II trans-activator leading to a mild immunodeficiency. J Immunol. (2001) 167(3):1787–94. doi: 10.4049/jimmunol.167.3.1787
14. Dziembowska M, Fondaneche MC, Vedrenne J, Barbieri G, Wiszniewski W, Picard C, et al. Three novel mutations of the CIITA gene in MHC class II-deficient patients with a severe immunodeficiency. Immunogenetics. (2002) 53(10–11):821–9. doi: 10.1007/s00251-001-0395-7
15. Ahmed A, Reith W, Puck JM, Cheng LE. Novel mutation in the class II transactivator associated with immunodeficiency and autoimmunity. J Clin Immunol. (2015) 35(6):521–2. doi: 10.1007/s10875-015-0183-z
16. Yu H, Zhang VW, Stray-Pedersen A, Hanson IC, Forbes LR, de la Morena MT, et al. Rapid molecular diagnostics of severe primary immunodeficiency determined by using targeted next-generation sequencing. J Allergy Clin Immunol. (2016) 138(4):1142–51.e2. doi: 10.1016/j.jaci.2016.05.035
17. Al-Mousa H, Abouelhoda M, Monies DM, Al-Tassan N, Al-Ghonaium A, Al-Saud B, et al. Unbiased targeted next-generation sequencing molecular approach for primary immunodeficiency diseases. J Allergy Clin Immunol. (2016) 137(6):1780–7. doi: 10.1016/j.jaci.2015.12.1310
18. Aluri J, Gupta M, Dalvi A, Mhatre S, Kulkarni M, Hule G, et al. Clinical, immunological, and molecular findings in five patients with major histocompatibility complex class II deficiency from India. Front Immunol. (2018) 9:188. doi: 10.3389/fimmu.2018.00188
19. Hawary RE E, Mauracher AA, Meshaal SS, Eldash A, Abd Elaziz DS, Alkady R, et al. MHC-II deficiency among Egyptians: novel mutations and unique phenotypes. J Allergy Clin Immunol Pract. (2019) 7(3):856–63. doi: 10.1016/j.jaip.2018.07.046
20. Chen QY, Wang WJ, Sun JQ, Hou J, Ying WJ, Wang XC, et al. MHC class II-deficiency caused by CIITA gene mutation: a report of two cases and literature review. Chin J Pract Pediatr. (2018) 33(1):55–9; (In Chinese).
21. Zhang Y, Yokoyama Y, Qing Y, Han C, Zhu J, Yu T, Yin L, Yao R, Wang J. Novel variants in CIITA caused type II bare lymphocyte syndrome: a case report. Asian Pac J Allergy Immunol. (2021). doi: 10.12932/AP-020720-0898. [Epub ahead of print]
22. Damoiseaux M, Damoiseaux J, Pico-Knijnenburg I, van der Burg M, Bredius R, van Well G. Lessons learned from the diagnostic work-up of a patient with the bare lymphocyte syndrome type II. Clin Immunol. (2022) 235:108932. doi: 10.1016/j.clim.2022.108932
Keywords: type II bare lymphocyte syndrome, CIITA gene, novel mutation, MHC II, genetic disorder, meningoencephalomyelitis, neurologic disease
Citation: Alosaimi MF, Hamad MH, AlShammari MJ, Jamjoom DZ and Musibeeh NS (2023) Case report: A late and isolated presentation of meningoencephalomyelitis uncovers a novel pathogenic variant in the CIITA gene. Front. Pediatr. 11:1269396. doi: 10.3389/fped.2023.1269396
Received: 29 July 2023; Accepted: 31 August 2023;
Published: 29 September 2023.
Edited by:
Neslihan Edeer Karaca, Ege University Faculty of Medicine, TürkiyeReviewed by:
Luis Ignacio Gonzalez-Granado, University Hospital October 12, SpainSilvia Clara Giliani, University of Brescia, Italy
© 2023 Alosaimi, Hamad, AlShammari, Jamjoom and Musibeeh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammed F. Alosaimi bWFsb3NhaW1pQGtzdS5lZHUuc2E=
Abbreviations BLS II, bare lymphocyte syndrome type II; MHCII, major histocompatibility complex class II; CIITA, major histocompatibility complex transactivator class II; BCG, bacille clamette-guerin; MMR, measles, mumps, rubella; IVIG, intravenous immunoglobulin; HSC, hematopoietic stem cell transplantation.
†ORCID Mohammed F. Alosaimi orcid.org/0000-0002-8025-349