 Tayyaba Afsar1,2,†Xiaoyun Huang3,†
Tayyaba Afsar1,2,†Xiaoyun Huang3,† Abid Ali Shah4
Abid Ali Shah4 Safdar Abbas5Shazia Bano6Arif Mahmood3
Safdar Abbas5Shazia Bano6Arif Mahmood3 Junjian Hu7
Junjian Hu7 Suhail Razak1,2*
Suhail Razak1,2* Muhammad Umair2,8*
Muhammad Umair2,8*
- 1Department of Community Health Sciences, College of Applied Medical Sciences, King Saud University, Riyadh, Saudi Arabia
- 2King Salman Center for Disability Research, Riyadh, Saudi Arabia
- 3Department of Neurology, SSL Central Hospital of Dongguan City, Affiliated Dongguan Shilong People’s Hospital of Southern Medical University, Dongguan, China
- 4Center for Medical Genetics and Hunan Key Laboratory of Medical Genetics, School of Life Sciences, Central South University, Changsha, China
- 5Department of Biological Sciences, Dartmouth College, Hanover, NH, United States
- 6Department of Optometry and Vision Sciences, University of Lahore, Lahore, Pakistan
- 7Department of Central Laboratory, SSL Central Hospital of Dongguan City, Affiliated Dongguan Shilong People’s Hospital of Southern Medical University, Dongguan, China
- 8Medical Genomics Research Department, King Abdullah International Medical Research Center (KAIMRC), King Saud Bin Abdulaziz University for Health Sciences, Ministry of National Guard Health Affairs (MNGH), Riyadh, Saudi Arabia
Background: Developmental and epileptic encephalopathies (DEEs) signify a group of heterogeneous neurodevelopmental disorder associated with early-onset seizures accompanied by developmental delay, hypotonia, mild to severe intellectual disability, and developmental regression. Variants in the DNM1 gene have been associated with autosomal dominant DEE type 31A and autosomal recessive DEE type 31B.
Methods: In the current study, a consanguineous Pakistani family consisting of a proband (IV-2) was clinically evaluated and genetically analyzed manifesting in severe neurodevelopmental phenotypes. WES followed by Sanger sequencing was performed to identify the disease-causing variant. Furthermore, 3D protein modeling and dynamic simulation of wild-type and mutant proteins along with reverse transcriptase (RT)–based mRNA expression were checked using standard methods.
Results: Data analysis of WES revealed a novel homozygous non-sense variant (c.1402G>T; p. Glu468*) in exon 11 of the DNM1 gene that was predicted as pathogenic class I. Variants in the DNM1 gene have been associated with DEE types 31A and B. Different bioinformatics prediction tools and American College of Medical Genetics guidelines were used to verify the identified variant. Sanger sequencing was used to validate the disease-causing variant. Our approach validated the pathogenesis of the variant as a cause of heterogeneous neurodevelopmental disorders. In addition, 3D protein modeling showed that the mutant protein would lose most of the amino acids and might not perform the proper function if the surveillance non-sense-mediated decay mechanism was skipped. Molecular dynamics analysis showed varied trajectories of wild-type and mutant DNM1 proteins in terms of root mean square deviation, root mean square fluctuation and radius of gyration. Similarly, RT-qPCR revealed a substantial reduction of the DNM1 gene in the index patient.
Conclusion: Our finding further confirms the association of homozygous, loss-of-function variants in DNM1 associated with DEE type 31B. The study expands the genotypic and phenotypic spectrum of pathogenic DNM1 variants related to DNM1-associated pathogenesis.
Introduction
Developmental and epileptic encephalopathy (DEE) are a group of genetic conditions characterized by severe epilepsy affecting children, resulting in delayed development, neurological and non-neurological comorbidities, and, in rare cases, early mortality (1–3). More than 100 monogenic causes of epilepsies and neurodevelopmental abnormalities have been identified, and genetic factors have since been recognized as significant contributors to DEEs (4, 5).
The majority of genetics-based epilepsies and neurodevelopmental abnormalities are caused by variants in genes involved in synaptic transmission. Several genes involved in synaptic vesicle fission and fusion, such as AP2M1 (MIM: 601024), DNM1 (MIM: 602377), STX1B (MIM: 601485), STXBP1 (MIM: 602926), SNAP25 (MIM: 600322), and VAMP2 (MIM: 185881), have been implicated in neurodevelopmental disorders (6–12). Among these, deleterious variants in the DNM1 gene have been responsible for diverse neurodevelopmental phenotypes and cerebral dysfunctions such as early-onset epileptic encephalopathies. These conditions usually occur at an early period of life accompanying infantile spasms, developmental delay, and movement disorders (11). In general, disease-causing DNM1 variants are responsible for around 2% of patients with infantile spasms or the Lennox–Gastaut syndrome (13).
Dynamin 1 (DNM1; NM_004408), localized to chromosome 9q34.11, encodes an 864 amino acid DNM1 protein, which is a GTPase-binding protein, and performs the function of synaptic vesicle fission for receptor-mediated endocytosis in the presynaptic plasma membrane (14). It consist of five domains: (a) an N-terminal G domain (GTPase domain); (b) a middle domain, involved in oligomerization; (c) a pleckstrin homology (PH) domain; (d) GTPase effector domain (GED); and (e) a proline-rich domain (PRD) (15).
Majority of the de novo deleterious variants occur in the only two GTPase and middle domains manifested in patients with epileptic encephalopathies (11, 16, 17). Only one deleterious variant c.1603A>G (p.Lys535Glu) has been observed in the PH domain in twin siblings positive for intellectual disability (ID) and autistic symptoms but no epileptic encephalopathy (18).
Neurological abnormalities are a highly heterogeneous group of rare genetic disorders that are primarily diagnosed using whole exome sequencing (WES) (19). Here, we describe a patient harboring a novel de novo non-sense pathogenic variant in the middle domain of the DNM1 protein, whose phenotypes included moderate ID, with mild generalized epilepsy with onset in adolescence.
Materials and methods
Ethics statement and study approval
The study presented here describes a proband (IV-2) suffering from a rare neurodevelopmental disorder. The family of the proband showing neurodevelopmental phenotypes was recruited from a remote region of Khyber Pakhtunkhwa (KP), Pakistan. Before the study, the parents were interviewed and patient history was obtained (Figure 1). Informed consent in written form was obtained from the patients and other family members for the publication of the data, results, molecular findings, X-rays, and other related data in this article in compliance with the Helsinki Declaration. Blood samples were collected in Ethylenediaminetetraacetic acid (EDTA) tubes. DNA from blood samples was extracted and quantified using standard methods described previously (20).
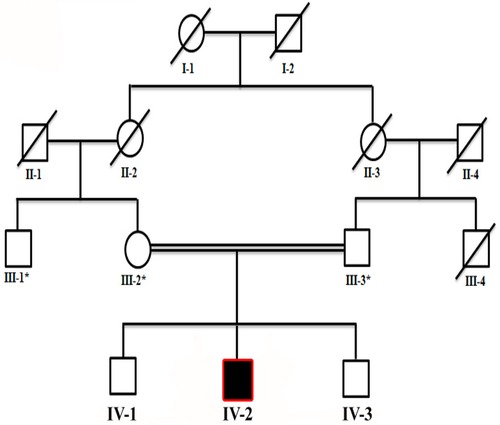
Figure 1. Four-generation pedigree of a family in the present study showing consanguinity (double lines) and autosomal recessive inheritance of the disease. The patient is highlighted with a black square, while an asterisk sign represents individuals subjected for WES. Slashes represent diseased individuals.
Molecular analysis
Chromosomal and cytogenetic analysis
Chromosomal and cytogenetic analysis was performed using standard methods as described previously (21).
Whole exome sequencing
WES was performed using DNA from the affected member (II-I). WES and variants filtering steps were performed as described earlier (22, 23), using the Illumina Hi-seq-2500 platform following the manufacturer instructions. After the standard reaction, The data was obtained and evaluated using the bioinformatics procedure into the variant call format (VCF). The VCF was then uploaded into the professional data analysis software (https://basespace.illumina.com/) to filter out the variants. Standard screening principles were used to search for different functional variants.
Determination of variant pathogenicity
The identified variant was first searched in general population databases such as gnomAD (24) and an in-house database to rule out the occurrence of polymorphism. Second, using different online tools, the disease-causing nature of the variant was verified with tools such as MutationTaster (25), DANN (26), LRT (27), FATHMM (28), EIGEN (29), and BayesDel_addAF (30) along with American College of Medical Genetics (ACMG) guidelines (31).
Variant conservation
The conservation of the variant across different species was checked using the NCBI-HOMOLOGENE database (https://www.ncbinlm.nih.gov/homologene).
Sanger sequencing
The variant obtained was filtered and identified as disease causing using Sanger sequencing following standard methods (21). Genomic sequences of the gene were retrieved from the UCSC genome database browser (http://genome.ucsc.edu/cgi-bin/hgGateway). The primers for Sanger sequencing were designed using the PRIMER3 software (32). The exon 11 containing the identified DNM1 variant was amplified using exon-specific primers through polymerase chain reaction (PCR) following standard protocols. Purification of the PCR-amplified DNA was performed according to the manufacturer's instructions. The data obtained from the Sanger sequencing were analyzed using BIOEDIT tool.
In silico analysis—3D protein structure modeling
The crystal structure of the DNM1 protein was retrieved from RCSB under accession number 4UUK. The preparation module of UCSF Chimera (https://www.cgl.ucsf.edu/chimera/) was used to resolve the missing residues and non-sense mutation was inserted using the mutagenesis tool PyMOL, followed by energy minimization with MMFF94 force field implemented in a molecular operating environment (MOE) (https://www.chemcomp.com/Products.htm). The mutation was induced in the structure using the mutagenesis tool implemented in PyMOL (https://pymol.org/2/) as described by Alotaibi et al. (33).
Modeling and structure minimizations
The crystal structure containing missing loop residues at the N terminal, middle, and C terminal were built by the loop module of Chimera; afterward, the structure was subjected to energy minimization. Then, non-sense mutation was inserted at position 468. Surprisingly upon visual inspections and comparison with the 3D structure of wild-type (WT) DNM1, we found that the novel variant undergoes structural changes in the loop regions of the respective protein.
Molecular dynamics simulation
Molecular dynamics simulation was carried out for the wild-type and mutant proteins to comprehend the dynamic behavior of the mutated and wild states. The protein force field ff14SB implemented in the Amber 20 package was applied (34). To solvate each system, the tip3p water model with an 8.0 box dimension was used. Each system was completely solvated and neutralized by the addition of four Na+ ions, which balanced out the charges in each system. After 6,000 steps of the steepest descent and conjugate gradient minimization technique, the density was then equilibrated for 2 ns. For another 2 ns, the entire system was then equilibrated at constant pressure. The temperature was kept constant at 300 K using the Langevin thermostat. Moreover, a 200 ns molecular dynamics (MD) was run after equilibration (33). For electrostatic interactions, the particle Mesh Ewald (PME) method was used. Hydrogen-containing covalent bonds were handled via the SHAKE algorithm. Finally, at constant pressure and temperature MD simulation was performed using PMEMD (35).
RT-qPCR
In order to functionally validate the variant, total RNA was extracted from all the available family members to investigate the relative mRNA DNM1 expression using GAPDH [DQ403057] as the internal control “house-keeping” gene. cDNA was synthesized using standard methods from total RNA using standard cDNA reverse transcription kit (36). Primer-bank database (https://pga.mgh.harvard.edu/Parabiosys/) was used to design the primer pair and will be provided on request. PCR SYBRGreen Master Mix was used for the qPCR reaction and Quant-Studio 6 Flex Real-Time PCR-System was used. All the reactions were repeated independently, performed in triplicate, and data were analyzed using Expression-Suite software version 1. GAPDH was used as an endogenous control.
Results
Clinical report
The current study reports on a single affected individual (II-1). Parents were first-degree cousins, and the pedigree depicted an autosomal recessive pattern of inheritance (Figure 1). The family had no previous history of such disorder. The parents and two other siblings were normal. The proband revealed features such as neurodevelopmental disorder, mild microcephaly, moderate to severe ID, speech issues, seizures, epileptic encephalopathy, and hypotonia.
His growth parameters at age 9 were as follows: height: 120.6 cm [2 percentile (−2.1 SD)], weight: 22.7 kg [5 percentile (−1.68 SD)], and head circumference (HC): 48.9 cm [<1 percentile (−2.8 SD)]. All the biochemical tests were unremarkable, and no metabolic disorder was observed. Ophthalmological examination showed unremarkable results, including normal funduscopic examination. Electroencephalogram (EEG) revealed multifocal epileptic discharges, thus suggesting epileptic encephalopathy. There were no dysmorphic features observed in the affected individual. He was unable to hold his head without support, roll over, and hold objects properly. Socially, he is shy and avoids making eye contact. Speech and language development were also delayed. The detailed clinical information is mentioned in Table 1.
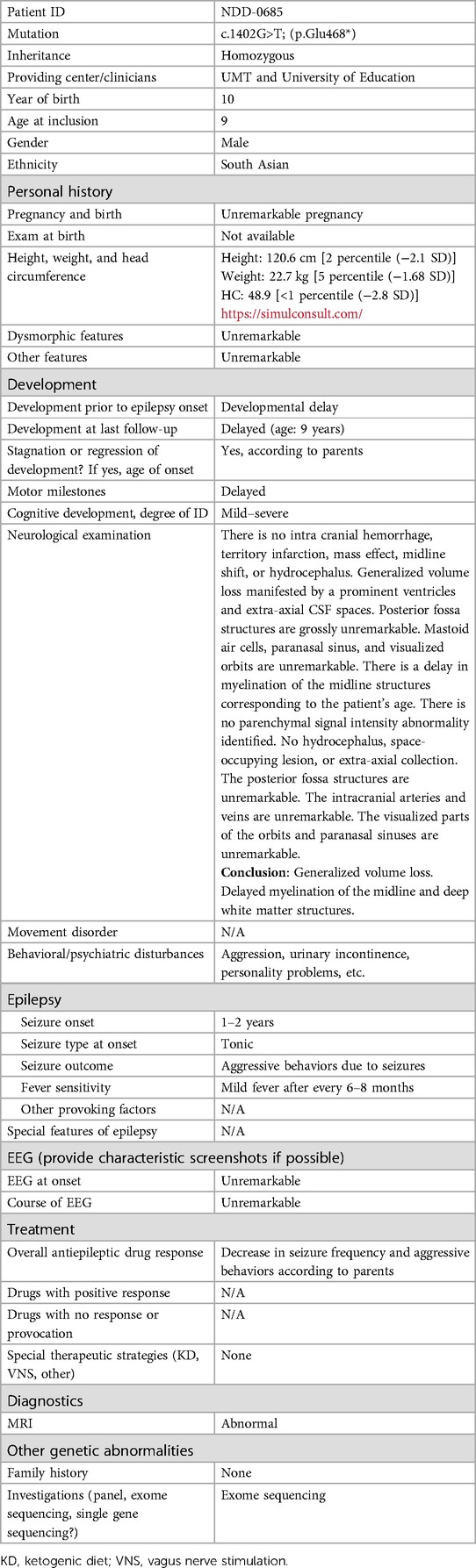
Table 1. Clinical characteristic and general information of a proband with diverse neurodevelopmental phenotypes.
Brain MRI
The brain MRI revealed no territory infarction, intra cranial hemorrhage, mass effect, hydrocephalus, or midline shift. A generalized volume loss was observed manifested by a prominent ventricles and extra-axial cerebrospinal fluid (CSF) spaces (Figure 2). Posterior fossa structures showed unremarkable presentation. Paranasal sinus, mastoid air cells, and visualized orbits were unremarkable. A delay in myelination of the midline structures was observed, corresponding to the patient's age. No abnormality associated with parenchymal signal intensity was identified. No extra-axial collections, hydrocephalus, and/or space-occupying lesions were observed. The posterior fossa structures, intracranial arteries, and veins were unremarkable. The paranasal sinuses and visualized parts of the orbits were unremarkable.
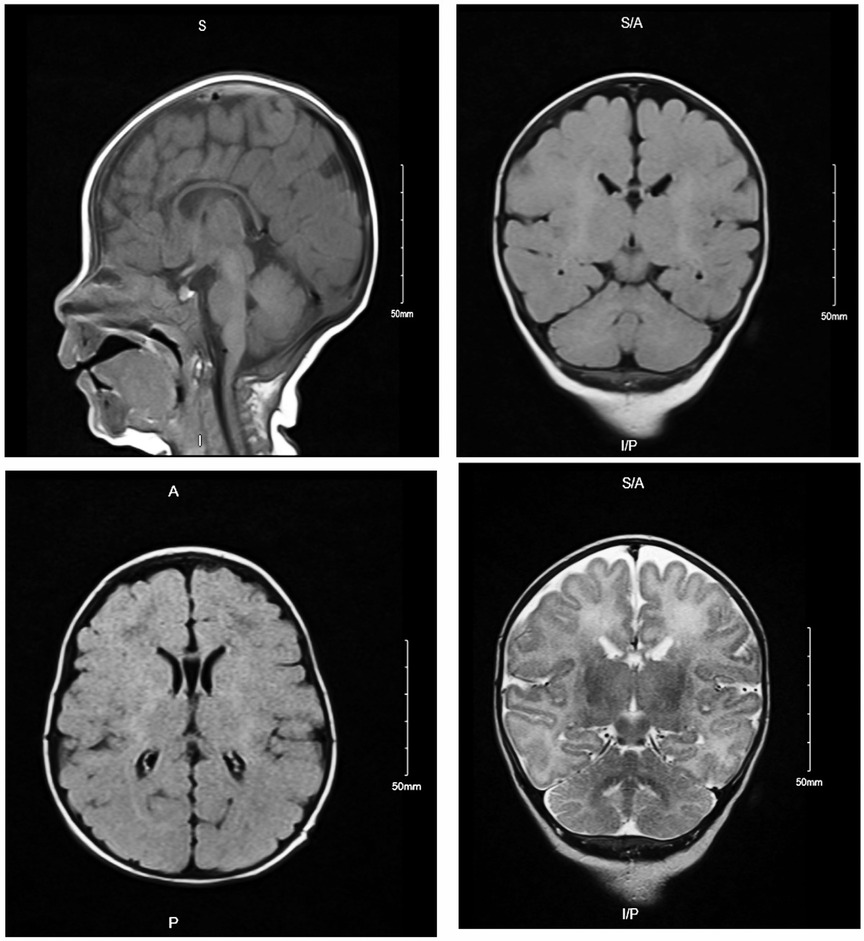
Figure 2. Brain MRI of affected individual (IV-2) revealed generalized volume loss manifested by a prominent ventricles and extra-axial CSF spaces. Posterior fossa structures, paranasal sinus, mastoid air cells, and visualized orbits were unremarkable.
Molecular genetic analysis
Chromosomal and cytogenetic analysis did not reveal any disease-causing genomic alterations that might be the cause of the observed phenotype in the proband. The DNA of the affected individual (II-2) was subjected to WES using standard methods. WES filtration data analysis revealed a novel homozygous non-sense mutation (c.1402G>T; p.Glu468*) in the exon 11 of the DNM1 (NM_004408.4; 9q34.11; ENST000003729) gene. The novelty of the variant was checked in the online literature and HGMD database 2022. The variant results in a premature stop-codon at 468 amino acid position. The mutation might result in a shorter protein that might result in non-sense-mediated decay (NMD).
Sanger sequencing
To consider the segregation of the variant with the disease phenotypes, the identified variant [c.1402G>T] was Sanger sequenced in all family members, which segregated perfectly within the family. The variant was observed in homozygous state in the proband, both the parents (III-2, III-3) and sibling (brother; IV-3) were carriers, and the sibling (brother; IV-1) was wild type (Figures 3A–C).
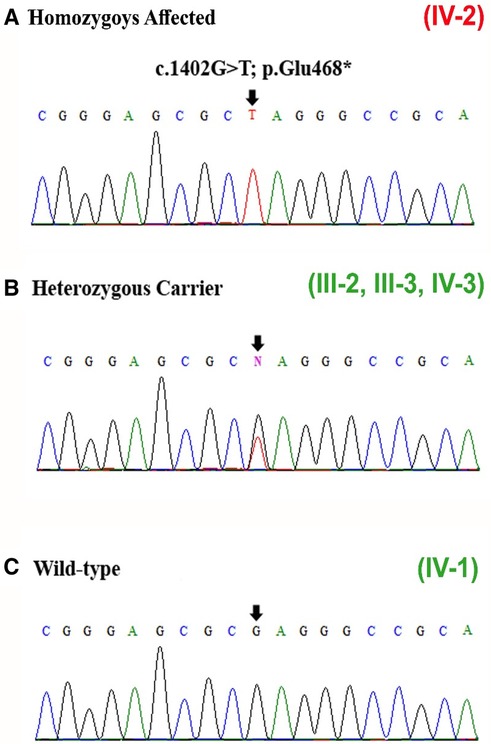
Figure 3. (A–C) Sanger sequencing results of exon 11 of the DNM1 gene showing the homozygous variant [c.1402G>T] identified in the patient (IV-2), carriers (heterozygous) (III-2, III-3, IV-3), and wild type (IV-1). The site of mutation is indicated by black arrows.
Pathogenicity and conservation
The identified non-sense variant (p. Glu468*) was classified as disease causing using different online prediction tools such as MutationTaster (disease-causing [1]), DANN (disease-causing [0.997]), LRT (pathogenic [0]), FATHMM (pathogenic [0.9844]), EIGEN (pathogenic [1.130]), and BayesDel_addAF (pathogenic [0.5774]), and according to the ACMG guidelines, the identified variant was classified as pathogenic (Class I). According to gnomAD, the variant was not observed in homozygous state in the public database thus suggesting a non-polymorphic nature of the variant. Furthermore, the Glu amino acid at position 468 is highly conserved across several species (Figures 4A,B), suggesting key role of this variant in the neurodevelopment.
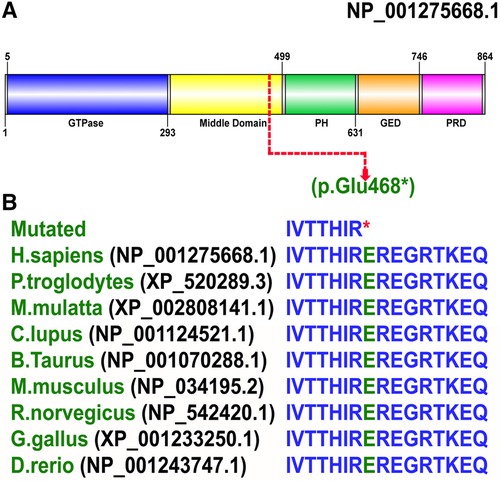
Figure 4. (A) Schematic representation of the DNM1 protein organized into five domains: (a) an N-terminal GTPase domain (5-293 aa); (b) a middle domain (300-499 aa); (c) a PH domain (505-631 aa); (d) a GED (635-746 aa), and (e) a PRD (750-850 aa). The site of mutation p. Glu468* shown in red is in the middle domain (300–499 aa). (B) Conservation of Glu at amino acid 468 in DNM1 in species with a known DNM1 orthologs.
In silico analysis—3D protein modeling
Then non-sense mutation was inserted at position 468 as shown in Figure 4A. Remarkably upon visual inspections and comparison with the 3D structure of wild-type DNM1, we found that the novel variant undergoes structural changes in loop regions of the respective proteins. If the DNM1 protein is not degraded in the NMD, these changes in the final protein structure might lead to a small non-functional protein that might not perform proper function and downstream signaling (Figures 5A,B).
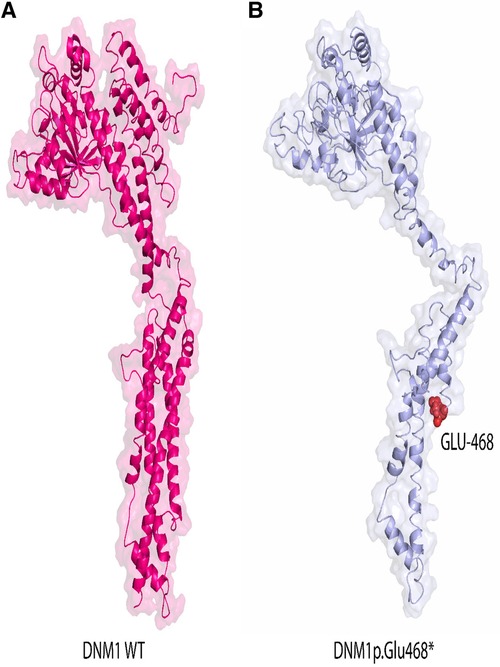
Figure 5. Representation of the 3D structural model of human wild-type DNM1(wt) protein (pink) and mutated protein (light blue). Red color indicates position of novel mutation Glu at position 468 in DNM1p.Glu468* protein identified in this study. Accession number NP_001275668.1 version was used.
RMSD analysis
Root mean square deviation (RMSD) analysis showed that the value of mutant increased rapidly to 4.5 Å from 10 to 15 ns but then decreased to 2 Å for 50 ns. After 50 ns, RMSD slightly increased to 2.5 Å and minor deviations were seen during 65–68 and 85–90 ns, but overall, the structure showed a stable behavior till 100 ns MD run. On the other hand, the wild-type DNM1 revealed a stable behavior during the entire 100 ns MD simulation, only minor deviations were seen during 55–75 ns and the DNM1Wt protein RMSD value of 2.8 Å was attained, and throughout the simulation, it revealed stability. RMSD plots of both the wild-type and the mutated proteins are shown in Figure 6A.
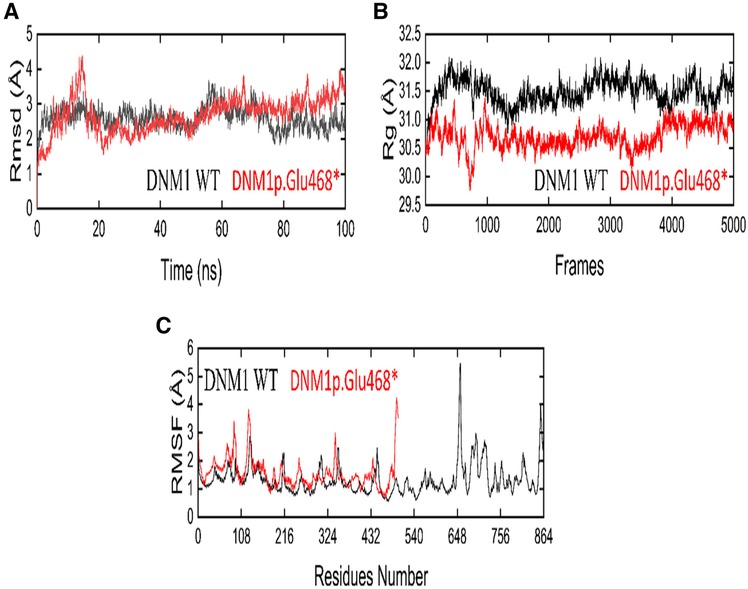
Figure 6. The dynamic behavior of wild-type (WT) and DNM1 (p. Glu468*) mutant variant. (A) RMSD of wild-type DNM1 and DNM1 (p. Glu468*). (B) Radius of gyration for wild-type DNM1 and DNM1(p. Glu468*). (C) RMSF of wild-type DNM1 and DNM1(p. Glu468*), respectively.
Rg analysis
By analyzing the structural compactness along with the stability of the folded or unfolded protein, the radius of gyration (Rg) calculation was carried out. For the mutant and wild states, the average values of Rg were in the range of 29.5–31.0 and 31.0–32.0, respectively. It is clear from the Rg calculation that the wild-type system revealed more compactness as compared to the mutant form during the MD simulation. The Rg plots for both the wild and mutant states are shown in Figure 6B.
RMSF analysis
The root mean square fluctuation (RMSF) was calculated for the C-alpha atoms of both the wild and mutant states in order to explore the fluctuating residues during the 100 ns MD simulation. In the mutant state, the residues 105–107, 110–113, and residue 325 showed more fluctuations during the MD simulation while the residues 114–324 showed stability. On the other hand, the wild state showed major fluctuations at residues 648–652 and the residues 740–750 showed minor fluctuations, while all other residues showed stability during the entire 100 ns MD run. Figure 6C displays the RMSF plots for the wild-type as well as mutant protein.
Non-sense variant in DNM1 reduced mRNA expression
The relative expression data of DNM1 gene in the affected individual, parents, and normal control individuals showed that the proband (IV-2) having the disease-causing homozygous variant (p. Glu468*) had substantial reduction in the DNM1 gene expression as compared to the wild type (control) and carrier (parents) (Figure 7).
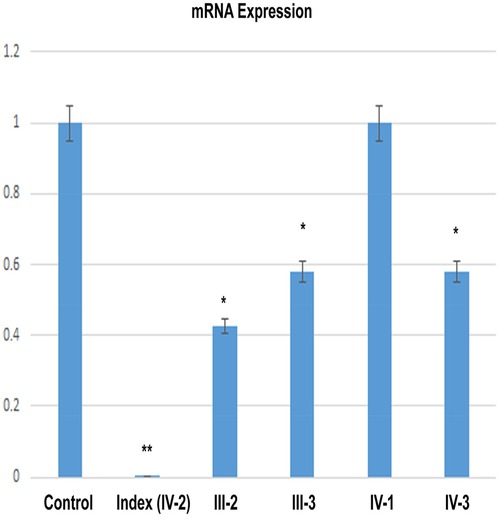
Figure 7. RT-qPCR shows a significant decrease of the DNM1 mRNA expression in an index patient (IV-2) in comparison to control or other members of the family (III-2, III-3, IV-1, and IV-3).
Discussion
Disorders associated with DNM1 pathogenesis have a unique clinical appearance. Patients having disease-causing variants in the DNM1 gene typically reveal profound hypotonia from birth, infantile spasms that might lead to the Lennox–Gastaut syndrome, developmental delay, microcephaly (few patients), cortical visual impairment, speech delay, and intellectual disability (11, 12).
To date, 46 patients with epileptic encephalopathy and DNM1 pathogenic variants have been described in 45 families (37–41).
DNM1 gene encodes dynamin-1, a protein required for clathrin-mediated endocytosis and synaptic vesicle recycling. The fission of clathrin-coated vesicles from the plasma membrane by dynamin-1 oligomers promotes vesicle-mediated neurotransmitter release at the synapse (14). It is only found in neurons in the presynaptic terminal, where it participates in synaptic vesicle endocytosis and membrane recycling after neurotransmitter release (14). In mammals, this gene can produce various isoforms, which are characterized primarily by alternative splicing of the tenth exon—some transcripts have exon 10b, while others contain exon 10c (42).
Herein, we describe a proband having pathogenic Class-I variant in the DNM1 that revealed neurodevelopmental features such as developmental delay, moderate–severe ID, microcephaly, hypotonia, seizures, and speech issues. Features reported previously, such as visual impairment, EEG abnormality, and scoliosis, were not observed in the patient reported here (Table 1). Interestingly, Yigit et al. (41) reported the only two affected individuals having homozygous DNM1 variants [c.97C>T; p.(Gln33*), c.850C>T; p.(Gln284*)] that were having a non-sense mutation and revealed hypotonia, spasticity, dystonia, and feeding abnormalities that overlapped with our patient features (41). However, scoliosis and visual impairment were not observed in our patient.
Previously, only de novo variants in the DNM1 were linked to developmental and epileptic encephalopathy (Table 2; MIM 620352, MIM 616346). Clinically, affected individuals having DNM1 heterozygous missense variants show phenotypic heterogeneity with early-onset epilepsy, hypotonia, and developmental delay (11). These patients have normal MRI analysis with cerebral volume loss over time. Five patients with early-onset DEE (DEE31, OMIM 616246) were the first to have heterozygous missense mutations in the DNM1 gene (13). Since then, many patients having heterozygous, primarily de novo DNM1 mutations, have been reported in the literature (Table 2). Except for one de novo in-frame 06 bp insertion, all observed variants disrupt highly conserved residues and are projected to have a dominant-negative impact on the dynamin-1 function. Detailed clinical comparison of all the patients reported with DNM1 variants have been summarized in Table 3.
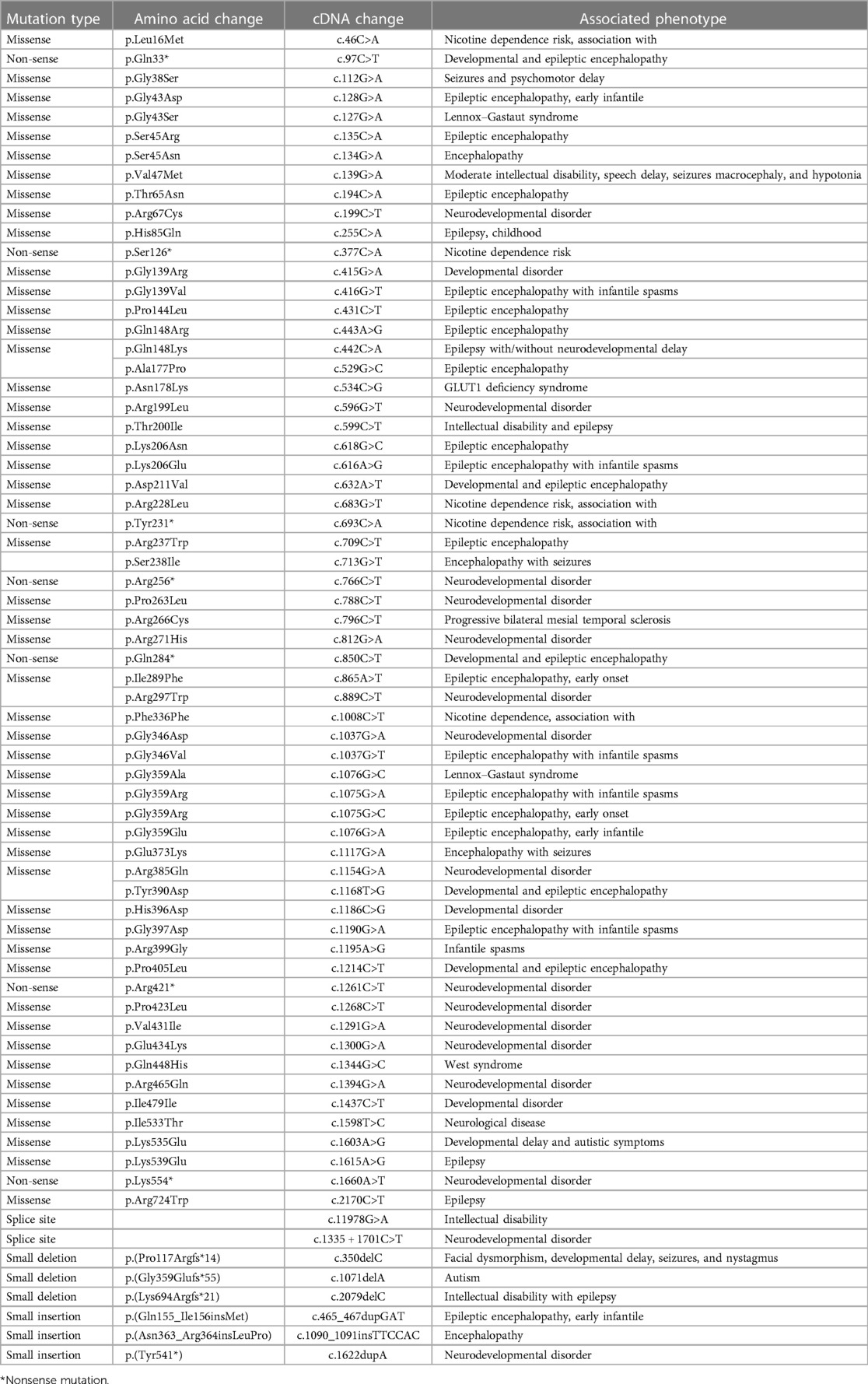
Table 2. Mutations reported in DNM1 associated with neurodevelopmental disorders.
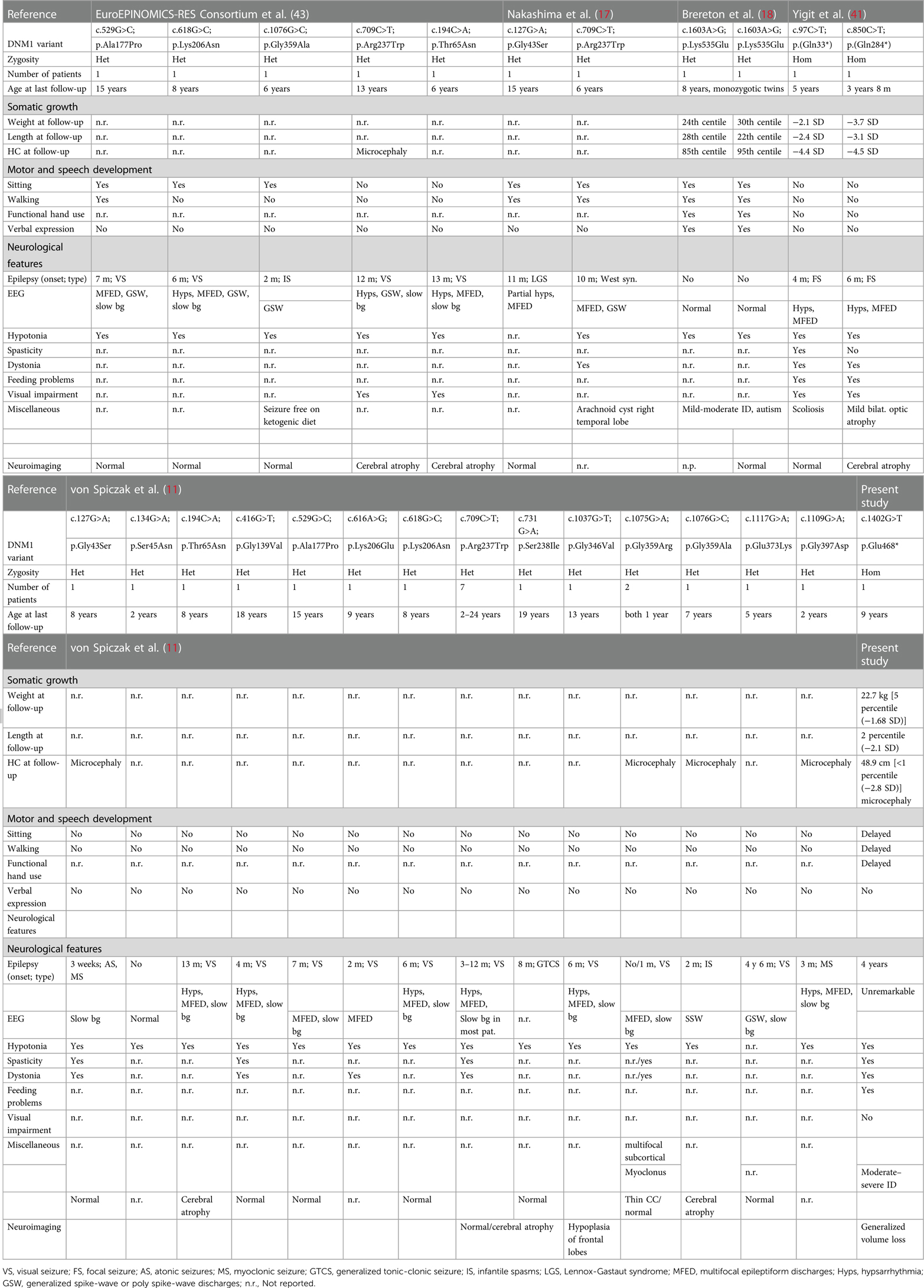
Table 3. Clinical comparison of patients reported with DNM1 mutation.
To date, only 69 variants have been reported in the DNM1 gene including two splice-site variants, three small deletions, three small insertions, seven non-sense, and 54 missense variants (Table 2). All missense variants associated with epileptic encephalopathy cluster in the two major functional domains of the DNM1 protein, the GTPase domain, and the middle domain. In addition, homozygous loss-of-function pathogenic variants in DNM1 have been reported to cause a severe autosomal recessive developmental and epileptic encephalopathy in three unrelated patients (37, 41). Recurrent DNM1 de novo splice-site variant has also been associated with developmental and epileptic encephalopathy with a dominant-negative mechanism (44). The p.Arg237Trp mutation has been observed in approximately one-third of patients. The non-sense variant identified in the present study affects a highly conserved Glu residue within the middle domain that plays a vital role in binding different targets and helps in DNM1 multimerization.
Molecular dynamics simulation is a useful tool to assess the genuine behaviors of biological molecules and their surroundings (19). In the present study, we performed MD simulations on two systems, native and mutant DNM1p.Glu468*. We considered parameters such as RMSD, RMSF, and Rg between the native and mutant p.Glu468* DNM1 protein structures. In terms of RMSD, DNM1 p.Glu468* after initial increase and decrease showed stable behavior till 100 ns MD run while DNM1WT revealed stable behavior during the entire 100 ns MD simulation except for some minor deviations at 55–75 ns of MD run. The RMSF trajectory for DNM1p. Glu468* showed fluctuations at the residues 105–107, 110–113, and 325 while the residues 114–324 showed stability along with native DNM1 that showed major and minor fluctuations at residues 648–652 and 740–750, respectively. Rg denotes the overall spread of the molecule, the average values of Rg were indicated in the range of 29.5–31.0 Å and 31.0–32.0 Å for DNM1 p. Glu468* and wild type, respectively. It is clear from the Rg calculation that the wild-type system revealed more compactness as compared to the mutant form during the MD simulation. Overall, an MD run demonstrated that the non-sense variant in DNM1 protein leads to instability of the p. Glu468* system in comparison to the wild-type protein as manifested by varied trajectories of RMSD, RMSF, and Rg, respectively. In addition, functional validation of this non-sense variant in the DNM1 gene using RT-qPCR approach revealed a decreased expression of the gene in the index patient signifying loss-of-function effect (45). For families impacted by rare inheritable diseases, proper genetic counseling is crucial. Additionally, prenatal genetic screening/diagnosis is the most effective management method for these medical conditions, for which there is no cure at this time (46).
In summary, we reported a novel non-sense variant in the DNM1 leading to severe neurodevelopmental phenotype in the patient that includes early-onset epilepsy, hypotonia, intellectual disability, seizures episodes, and pronounced developmental delay. This study will aid in genetic counseling and bringing awareness regarding the disease in the common population. The study also adds to the variant spectrum of DNM1 and contributes to the evaluation of genotype–phenotype correlation.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article.
Ethics statement
The studies involving humans were approved by Medical Genomics Research Department, King Abdullah International Medical Research Center (KAIMRC), Riyadh, Saudi Arabia. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. The animal study was approved by King Abdullah International Medical Research Center (KAIMRC) Riyadh, Saudi Arabia. The study was conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
TA: Writing – original draft, Writing – review & editing. XH: Writing – original draft, Writing – review & editing. AAS: Writing – original draft, Writing – review & editing, Methodology. SA: Writing – original draft, Writing – review & editing, Data curation. SB: Writing – original draft, Writing – review & editing, Investigation. AM: Writing – original draft, Writing – review & editing, Methodology, Visualization. JH: Writing – original draft, Writing – review & editing. SR: Supervision, Writing – review & editing, Writing – original draft. MU: Conceptualization, Data curation, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
The authors extend their appreciation to the King Salman Center for Disability Research for funding this work through Research Group no KSRG-2023-003.
Acknowledgments
The authors extend their appreciation to the King Salman center for Disability Research for funding this work through Research Group no KSRG-2023-003.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Helbig I, Heinzen EL, Mefford HC. Genetic literacy series: primer part 2—paradigm shifts in epilepsy genetics. Epilepsia. (2018) 59(6):1138–47. doi: 10.1111/epi.14193
2. McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. (2016) 15(3):304–16. doi: 10.1016/s1474-4422(15)00250-1
3. Steward CA, Roovers J, Suner M-M, Gonzalez JM, Uszczynska-Ratajczak B, Pervouchine D, et al. Re-annotation of 191 developmental and epileptic encephalopathy-associated genes unmasks de novo variants in SCN1A. NPJ Genom Med. (2019) 4(1):31. doi: 10.1038/s41525-019-0106-7
4. Heyne HO, Singh T, Stamberger H, Abou Jamra R, Caglayan H, Craiu D, et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat Genet. (2018) 50(7):1048–53. doi: 10.1038/s41588-018-0143-7
5. Helbig I, Lopez-Hernandez T, Shor O, Galer P, Ganesan S, Pendziwiat M, et al. A recurrent missense variant in AP2M1 impairs clathrin-mediated endocytosis and causes developmental and epileptic encephalopathy. Am J Hum Genet. (2019) 104(6):1060–72. doi: 10.1016/j.ajhg.2019.04.001
6. Kaplanis J, Samocha KE, Wiel L, Zhang Z, Arvai KJ, Eberhardt RY, et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. (2020) 586(7831):757–62. doi: 10.1038/s41586-020-2832-5
7. Klöckner C, Sticht H, Zacher P, Popp B, Babcock HE, Bakker DP, et al. De novo variants in SNAP25 cause an early-onset developmental and epileptic encephalopathy. Genet Med. (2021) 23(4):653–60. doi: 10.1038/s41436-020-01020-w
8. Salpietro V, Malintan NT, Llano-Rivas I, Spaeth CG, Efthymiou S, Striano P, et al. Mutations in the neuronal vesicular SNARE VAMP2 affect synaptic membrane fusion and impair human neurodevelopment. Am J Hum Genet. (2019) 104(4):721–30. doi: 10.1016/j.ajhg.2019.02.016
9. Schubert J, Siekierska A, Langlois M, May P, Huneau C, Becker F, et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat Genet. (2014) 46(12):1327–32. doi: 10.1038/ng.3130
10. Verhage M, Sørensen JB. SNAREopathies: diversity in mechanisms and symptoms. Neuron. (2020) 107(1):22–37. doi: 10.1016/j.neuron.2020.05.036
11. von Spiczak S, Helbig KL, Shinde DN, Huether R, Pendziwiat M, Lourenço C, et al. DNM1 encephalopathy: a new disease of vesicle fission. Neurology. (2017) 89(4):385–94. doi: 10.1212/wnl.0000000000004152
12. Xian J, Parthasarathy S, Ruggiero SM, Balagura G, Fitch E, Helbig K, et al. Assessing the landscape of STXBP1-related disorders in 534 individuals. Brain. (2021) 145(5):1668–83. doi: 10.1093/brain/awab327
13. Appenzeller S, Balling R, Barisic N, Baulac S, Caglayan H, Craiu D, et al. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet. (2014) 95(4):360–70. doi: 10.1016/j.ajhg.2014.08.013
14. Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol. (2012) 13(2):75–88. doi: 10.1038/nrm3266
15. Boumil RM, Letts VA, Roberts MC, Lenz C, Mahaffey CL, Zhang ZW, et al. A missense mutation in a highly conserved alternate exon of dynamin-1 causes epilepsy in fitful mice. PLoS Genet. (2010) 6:8. doi: 10.1371/journal.pgen.1001046
16. Allen NM, Conroy J, Shahwan A, Lynch B, Correa RG, Pena SD, et al. Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia. (2016) 57(1):e12–7. doi: 10.1111/epi.13250
17. Nakashima M, Kouga T, Lourenço CM, Shiina M, Goto T, Tsurusaki Y, et al. De novo DNM1 mutations in two cases of epileptic encephalopathy. Epilepsia. (2016) 57(1):e18–23. doi: 10.1111/epi.13257
18. Brereton E, Fassi E, Araujo GC, Dodd J, Telegrafi A, Pathak SJ, et al. Mutations in the PH domain of DNM1 are associated with a nonepileptic phenotype characterized by developmental delay and neurobehavioral abnormalities. Mol Genet Genomic Med. (2018) 6(2):294–300. doi: 10.1002/mgg3.362
19. Khan A, Bruno LP, Alomar F, Umair M, Pinto AM, Khan AA, et al. SPTBN5, Encoding the βV-spectrin protein, leads to a syndrome of intellectual disability, developmental delay, and seizures. Front Mol Neurosci. (2022) 15:877258. doi: 10.3389/fnmol.2022.877258
20. Umair M, Rafique A, Ullah A, Ahmad F, Ali RH, Nasir A, et al. Novel homozygous sequence variants in the GDF5 gene underlie acromesomelic dysplasia type-grebe in consanguineous families. Congenit Anom (Kyoto). (2017) 57(2):45–51. doi: 10.1111/cga.12187
21. Ullah A, Umair M, Majeed AI, Abdullah , Jan A, Ahmad W. A novel homozygous sequence variant in GLI1 underlies first case of autosomal recessive pre-axial polydactyly. Clin Genet. (2019) 95(4):540–1. doi: 10.1111/cge.13495
22. Hayat A, Hussain S, Bilal M, Kausar M, Almuzzaini B, Abbas S, et al. Biallelic variants in four genes underlying recessive osteogenesis imperfecta. Eur J Med Genet. (2020) 63(8):103954. doi: 10.1016/j.ejmg.2020.103954
23. Nøstvik M, Kateta SM, Schönewolf-Greulich B, Afenjar A, Barth M, Boschann F, et al. Clinical and molecular delineation of PUS3-associated neurodevelopmental disorders. Clin Genet. (2021) 100(5):628–33. doi: 10.1111/cge.14051
24. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581(7809):434–43. doi: 10.1038/s41586-020-2308-7
25. Schwarz JM, Cooper DN, Schuelke M. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11(4):361–2. doi: 10.1038/nmeth.2890
26. Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. (2014) 31(5):761–3. doi: 10.1093/bioinformatics/btu703
27. Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. (2009) 19(9):1553–61. doi: 10.1101/gr.092619.109
28. Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. (2013) 34(1):57–65. doi: 10.1002/humu.22225
29. Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. (2016) 48(2):214–20. doi: 10.1038/ng.3477
30. Feng BJ. PERCH: a unified framework for disease gene prioritization. Hum Mutat. (2017) 38(3):243–51. doi: 10.1002/humu.23158
31. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
32. Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. (2000) 132:365–86. doi: 10.1385/1-59259-192-2:365
33. Alotaibi B, Ajmal A, Hakami M, Mahmood A, Wadood A, Hu J. New drug target identification in Vibrio vulnificus by subtractive genome analysis and their inhibitors through molecular docking and molecular dynamics simulations. Heliyon. (2023) 9:e17650. doi: 10.1016/j.heliyon.2023.e17650
34. Wadood A, Ajmal A, Junaid M, Rehman AU, Uddin R, Azam SS, et al. Machine learning-based virtual screening for STAT3 anticancer drug target. Curr Pharm Des. (2022) 28(36):3023–32. doi: 10.2174/1381612828666220728120523
35. Gotz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J Chem Theory Comput. (2012) 8(5):1542–55. doi: 10.1021/ct200909j
36. Shah AA, Zhang G, Li K, Liu C, Kanhar AA, Wang M, et al. Excess of RALGAPB de novo variants in neurodevelopmental disorders. Eur J Med Genet. (2020) 63(11):104041. doi: 10.1016/j.ejmg.2020.104041
37. AlTassan R, AlQudairy H, Alromayan R, Alfalah A, AlHarbi OA, González-Álvarez AC, et al. Clinical, radiological, and genetic characterization of a patient with a novel homoallelic loss-of-function variant in DNM1. Genes (Basel). (2022) 13(12):2252. doi: 10.3390/genes13122252
38. Kolnikova M, Skopkova M, Ilencikova D, Foltan T, Payerova J, Danis D, et al. DNM1 encephalopathy—atypical phenotype with hypomyelination due to a novel de novo variant in the DNM1 gene. Seizure. (2018) 56:31–3. doi: 10.1016/j.seizure.2018.01.020
39. Li H, Fang F, Xu M, Liu Z, Zhou J, Wang X, et al. Clinical assessments and EEG analyses of encephalopathies associated with dynamin-1 mutation. Front Pharmacol. (2019) 10:1454. doi: 10.3389/fphar.2019.01454
40. Mei D, Parrini E, Bianchini C, Ricci ML, Guerrini R. Autism and mild epilepsy associated with a de novo missense pathogenic variant in the GTPase effector domain of DNM1. Am J Med Genet C Semin Med Genet. (2023). doi: 10.1002/ajmg.c.32044
41. Yigit G, Sheffer R, Daana M, Li Y, Kaygusuz E, Mor-Shakad H, et al. Loss-of-function variants in DNM1 cause a specific form of developmental and epileptic encephalopathy only in biallelic state. J Med Genet. (2022) 59(6):549–53. doi: 10.1136/jmedgenet-2021-107769
42. Asinof S, Mahaffey C, Beyer B, Frankel WN, Boumil R. Dynamin 1 isoform roles in a mouse model of severe childhood epileptic encephalopathy. Neurobiol Dis. (2016) 95:1–11. doi: 10.1016/j.nbd.2016.06.014
43. EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet. (2014) 95(4):360–70. doi: 10.1016/j.ajhg.2014.08.013
44. Parthasarathy S, Ruggiero SM, Gelot A, Soardi FC, Ribeiro BFR, Pires DEV, et al. A recurrent de novo splice site variant involving DNM1 exon 10a causes developmental and epileptic encephalopathy through a dominant-negative mechanism. Am J Hum Genet. (2022) 109(12):2253–69. doi: 10.1016/j.ajhg.2022.11.002
45. Umair M. Rare genetic disorders: beyond whole-exome sequencing. J Gene Med. (2023):e3503. doi: 10.1002/jgm.3503
Keywords: DNM1, homozygous variant, non-sense variant, developmental and epileptic encephalopathies (DEEs), novel mutation
Citation: Afsar T, Huang X, Shah AA, Abbas S, Bano S, Mahmood A, Hu J, Razak S and Umair M (2023) Truncated DNM1 variant underlines developmental delay and epileptic encephalopathy. Front. Pediatr. 11:1266376. doi: 10.3389/fped.2023.1266376
Received: 24 July 2023; Accepted: 6 September 2023;
Published: 9 October 2023.
Edited by:
Musharraf Jelani, Islamia College University, PakistanReviewed by:
M. Muaaz Aslam, University of Pittsburgh, United StatesDr. Qamre Alam, King Abdulaziz University, Saudi Arabia
© 2023 Afsar, Huang, Shah, Abbas, Bano, Mahmood, Hu, Razak and Umair. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muhammad Umair a2h1Z29vNHVAeWFob28uY29t Suhail Razak cnVoYWlsMTIzNDVAeWFob28uY29t
†These authors have contributed equally to this work