
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 21 November 2023
Sec. Pediatric Neurology
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1251914
 T. Foiadelli1
T. Foiadelli1 A. Santangelo2*
A. Santangelo2* G. Costagliola3
G. Costagliola3 E. Costa4M. Scacciati4
E. Costa4M. Scacciati4 A. Riva5,6G. Volpedo5,6M. Smaldone4A. Bonuccelli2
A. Riva5,6G. Volpedo5,6M. Smaldone4A. Bonuccelli2 A. M. Clemente1
A. M. Clemente1 A. Ferretti7
A. Ferretti7 S. Savasta8
S. Savasta8 P. Striano5,6
P. Striano5,6 A. Orsini2
A. Orsini2
Status epilepticus (SE) is a medical emergency resulting from the failure of the mechanisms involved in seizure termination or from the initiation of pathways involved in abnormally prolonged seizures, potentially leading to long-term consequences, including neuronal death and impaired neuronal networks. It can eventually evolve to refractory status epilepticus (RSE), in which the administration of a benzodiazepine and another anti-seizure medications (ASMs) had been ineffective, and super-refractory status epilepticus (SRSE), which persists for more than 24 h after the administration of general anesthesia. Objective of the present review is to highlight the link between inflammation and SE. Several preclinical and clinical studies have shown that neuroinflammation can contribute to seizure onset and recurrence by increasing neuronal excitability. Notably, microglia and astrocytes can promote neuroinflammation and seizure susceptibility. In fact, inflammatory mediators released by glial cells might enhance neuronal excitation and cause drug resistance and seizure recurrence. Understanding the molecular mechanisms of neuroinflammation could be crucial for improving SE treatment, wich is currently mainly addressed with benzodiazepines and eventually phenytoin, valproic acid, or levetiracetam. IL-1β signal blockade with Anakinra has shown promising results in avoiding seizure recurrence and generalization in inflammatory refractory epilepsy. Inhibiting the IL-1β converting enzyme (ICE)/caspase-1 is also being investigated as a possible target for managing drug-resistant epilepsies. Targeting the ATP-P2X7R signal, which activates the NLRP3 inflammasome and triggers inflammatory molecule release, is another avenue of research. Interestingly, astaxanthin has shown promise in attenuating neuroinflammation in SE by inhibiting the ATP-P2X7R signal. Furthermore, IL-6 blockade using tocilizumab has been effective in RSE and in reducing seizures in patients with febrile infection-related epilepsy syndrome (FIRES). Other potential approaches include the ketogenic diet, which may modulate pro-inflammatory cytokine production, and the use of cannabidiol (CBD), which has demonstrated antiepileptic, neuroprotective, and anti-inflammatory properties, and targeting HMGB1-TLR4 axis. Clinical experience with anti-cytokine agents such as Anakinra and Tocilizumab in SE is currently limited, although promising. Nonetheless, Etanercept and Rituximab have shown efficacy only in specific etiologies of SE, such as autoimmune encephalitis. Overall, targeting inflammatory pathways and cytokines shows potential as an innovative therapeutic option for drug-resistant epilepsies and SE, providing the chance of directly addressing its underlying mechanisms, rather than solely focusing on symptom control.
According to the International League Against Epilepsy (ILAE), Status epilepticus (SE) is a condition resulting either from the failure of the mechanisms responsible for seizure termination or from the initiation of mechanisms which lead to abnormally prolonged seizures. It is a condition that can have long-term consequences, including neuronal death, neuronal injury, and alteration of neuronal networks, depending on the type and duration of seizures (1). Based on the clinical presentation, SE can be classified into convulsive SE (featured by motor symptoms and impairment of consciousness), and non-convulsive SE (2). Given the severity of SE and the potential development of irreversible brain damage, there is an urgent need to dissect its pathogenesis to find new potential therapeutic targets. Specifically, the most relevant therapeutic challenges are represented by refractory status epilepticus (RSE), in which the administration of a benzodiazepine bolus and another anti-seizure medication (ASM) does not resolve the clinical picture (3), and super-refractory status epilepticus (SRSE), which persists for more than 24 h after the administration of general anesthesia (2).
During the last years, an increasing interest has been posed on the involvement of neuroinflammation in epileptogenesis and in the pathogenesis of developmental and epileptic encephalopathies (4). Neuroinflammation is also implicated in enhancing and maintaining the pathogenic mechanism of SE. Therefore, the use of drugs acting on the inflammatory response (especially, anti-cytokine agents) has been empirically introduced in patients with RSE to achieve seizure control, while preclinical studies have focused on the identification of potential targets to regulate neuroinflammation in epilepsy and SE.
In this paper, we will present the main molecular mechanisms responsible for neuroinflammation in SE and the effect of currently available therapeutic strategies for convulsive SE on the inflammatory response. Furthermore, the potential new therapeutic agents targeting neuroinflammation will be presented, focusing on data deriving from preclinical and clinical studies.
In this narrative review, we performed comprehensive analysis of existing literature through three reputable databases: PubMed, Embase, and Cochrane. Our search was carefully guided by a thoughtfully curated set of keywords, designed to ensure a thorough investigation of pertinent research. These keywords included terms such as “epilepsy,” “inflammation,” “neuro-inflammation,” “status epilepticus,” “FIRES,” “NORSE,” “RSRE,” and “SRSE.” To maintain consistency, we specifically focused on articles published in the English language.
Our inclusion criteria were designed to encompass a wide spectrum of studies that delved into various facets of the relationship between status epilepticus (SE) and neuroinflammation. This included both clinical and preclinical investigations, studies exploring the role of cytokines and biomarkers, and an examination of how current and future SE therapies intersect with inflammatory pathways. Studies that exclusively concentrated on clinical aspects of SE or those that did not directly contribute to our primary research question were excluded.
To ensure a rigorous and methodical approach to our review, two independent reviewers conducted an initial screening by evaluating study titles and abstracts. This initial screening aimed to assess the relevance of each study to our research question. Subsequently, full-text articles of potentially relevant studies underwent a more detailed review to determine their suitability for inclusion in our narrative review. In instances where differences or uncertainties arose during the review process, a third reviewer was engaged to facilitate discussion and achieve consensus. This collaborative effort was instrumental in maintaining the quality and consistency of study selection throughout our review process.
The main results of our research are listed in Table 1. SE can lead to significant morbidity and mortality, and the mechanisms underlying its development and progression are complex and not fully understood. However, several studies suggest that inflammation plays a significant role in the pathogenesis of SE.
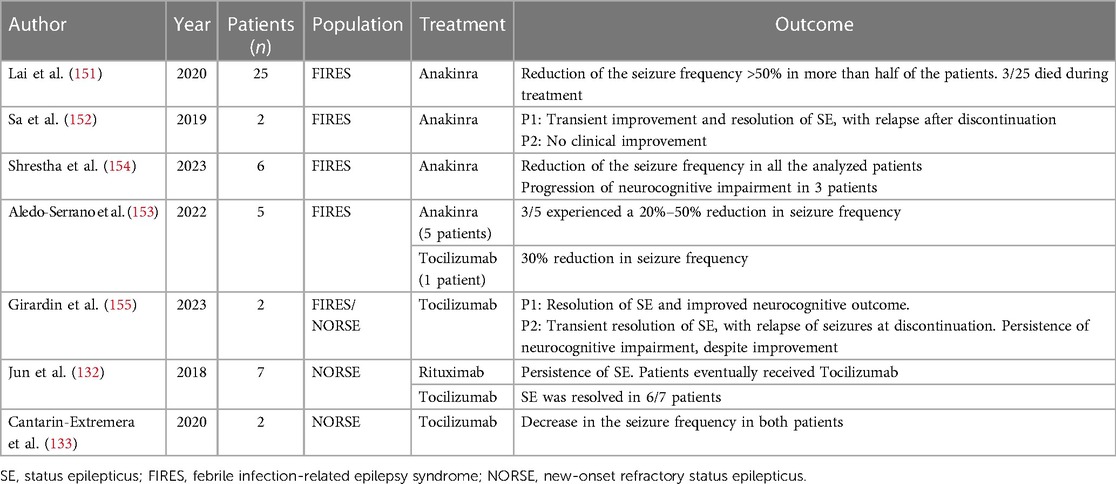
Table 1. Overview of the main clinical studies involving monoclonal antibodies targeting neuroinflammation in status epilepticus.
Higher levels of pro-inflammatory cytokines, such as interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α), have been reported in SE (4). These molecules can be secreted by activated glial cells, such as microglia and astrocytes, and can be associated with the release of pro-inflammatory molecules by damaged neurons, including high-mobility group box 1 protein (HMGB1) and damage-associated molecular patterns [DAMPs (5)]. This molecular cascade has been suggested to trigger glia-mediated neuroinflammation. On the other hand, different authors have suggested that changes in neuronal activity and energy metabolism caused by SE could also activate microglia and astrocytes. For example, prolonged seizures can induce an increase in extracellular potassium, which eventually lead astrocytes to release neurotransmitters that, in turn, can modulate synaptic activity (6). Seizures may also directly activate microglia and astrocytes by triggering receptors on their cell surfaces. For instance, seizures activate the P2X7 receptor of microglia, leading to the release of pro-inflammatory cytokines (7).
Activated microglia and astrocytes have been shown to express IL-1β (8), which might promote seizures through the upregulation of NMDA receptors on postsynaptic cells (9). Interestingly, the IL-1β antagonist, Anakinra, has been shown to ameliorate long-term potentiation impairment (10). Furthermore, preclinical studies have showed that TNF-α is also released by activated glial cells. It has been reported to regulate N-cadherin (11), which in turn plays a pivotal role in modulating the organization of excitatory and inhibitory synapses. Moreover, TNF-α could potentially promote seizures by increasing microglial glutamate release through the upregulation of glutaminase enzyme (12), as well as by enhancing the expression of AMPA receptors (13). Finally, it has also been shown that TNF-α affects inhibitory neurotransmission by promoting GABA receptor endocytosis (14).
Astrocytes and microglia could also increase the levels of IL-6 within the central nervous system (CNS), which eventually reduce long-term potentiation and hippocampal neurogenesis, while promoting gliosis. These effects could contribute to creating a subset for epilepsy (15).
The blood-brain barrier (BBB) is a critical component of the immune control within the brain, moduling the access of immune cells and cytokines. In SE, the BBB might become dysfunctional, allowing immune cells and cytokines to access the brain more easily. Such disruption eventually participates in the development of neuroinflammation and neuronal damage (16). Inflammatory mediators, namely IL-1, IL-6, and TNF alpha, can contribute to the BBB breakdown, leading to increased permeability and leukocyte infiltration (17). Increased levels of white blood cells into the hippocampus, such as neutrophils, have been associated with neurodegeneration and temporal lobe epilepsy (18). Moreover, it has been hypothesized that the upregulation of adhesion molecules promoted by seizures, such as VCAM-1 and CD44, might contribute to BBB permeability, neuro-inflammation, and subsequent seizure generation (19).
Inflammatory response in SE can also lead to the activation of the complement system, a key component of the immune response. Notably, the activation of C1q-C3 signaling pathway has been observed in animal models and in humans with SE (20). The activation of the complement system can concur to the recruitment of immune cells and the development of inflammation in the brain.
Finally, the activation of the inflammasome, which plays a pivotal role in innate immune response, has also been implicated in the development of SE. In fact, the inflammasome is activated in response to danger signals, such as those produced during seizures, leading to the release of IL-1β (5). In animal models of SE, blocking inflammasome activation has been proved to reduce seizure activity and improve epilepsy outcomes (21).
In addition to the excitatory processes mentioned earlier, inhibitory neurotransmission appears to play a significant role in the complex inflammatory mechanisms associated with the development of epilepsy. Notably, elevated levels of IL-1β have been correlated with a reduced GABA-A currents in cases of temporal lobe epilepsy. Furthermore, TNF-α has been observed to promote the endocytosis of GABA receptors.
Nonetheless, the role of anti-inflammatory molecules has been increasingly studied during the last years, including COX-2-selective and nonselective inhibitors (22, 23).
These findings underscore the intricate interplay between inflammatory cytokines and the regulation of inhibitory neurotransmission in the pathogenesis of epilepsy. Understanding these multifaceted processes holds promise for advancing our knowledge of epilepsy development and potentially uncovering new avenues for therapeutic intervention.
Preclinical studies with transgenic murine models clearly show that neuroinflammation can promote the generation and recurrence of seizures by increasing neuronal excitability and therefore lowering seizure threshold (13, 22, 24). For instance, overexpression of caspase-1 in subicular pyramidal neurons was sufficient to induce pharmacoresistant temporal lobe epilepsy in rats (25). On the other hand, preventing activation mediated by the IL-1 receptor 1 (IL-1R1)/TLR4 signaling pathway both pharmacologically or via genetic intervention, leads to a decrease in both acute and chronic seizure frequency (26). IL-1R1 co-localizes with the N-methyl-d-aspartate (NMDA) receptor, which is involved in excitotoxicity and seizures (27–29). Induction of the IL-1β/IL-1R1 cascade causes post-translational modifications in NMDAR, resulting in NMDA-induced Ca2 + influx, neuronal activation, and hippocampal kainate (KA) seizures in mice (9, 30, 31). HMGB1 activation of TLR4 exerts similar effects (32) and TLR4 KO mice show reduced KA-induced seizure activity (33). TNF-α also plays a pro-inflammatory role and mice with excessively high levels of this cytokine are at high risk of epilepsy (34). However, this signaling cascade plays a dual role in seizure susceptibility in mice, depending on whether TNF-α acts through TNFR1 or 2 (35). While TNFR1 KO mice show a decreased incidence of seizure, TNFR2 KO mice display increased seizure behavior (36).
Glial cells surrounding neurons in the brain are the main source of these inflammatory cytokines and their functions affect neuroinflammation and susceptibility to seizures. Microglia are a CNS-resident macrophage population which can have inflammatory properties during infection or sterile insult to the brain (37). The communication between microglia and neurons is important to maintain homeostasis and has been demonstrated to be neuroprotective in epilepsy. In fact, mice lacking the microglial P2Y2 receptor display worsened seizure behavior (38). Furthermore, inhibiting mTOR leads to delayed microglial activation in the hippocampus of mice with KA-induced epilepsy (39). Another type of glia, the astrocytes, perform metabolic, structural, homeostatic, and neuroprotective tasks in the CNS. One of their functions is to regulate extracellular glutamate levels through the glutamate uptake system. Inhibition of the astrocyte glutamate transporter GLT-1 in mice with cortical dysplasia lowers seizure threshold and enhances neuronal excitability (40). Furthermore, activation of the astrocytic TLR4-MyD88-ERK1/2 pathway in mice leads to over-excitation at the neuronal level and increased density of excitatory synapses following LPS injection, which could increase seizure susceptibility (41).
Interestingly, it is not always neuroinflammation that causes seizures, but also the other way around. Seizures can induce brain injury, which in turn can activate microglia and astrocytes to release a wide spectrum of inflammatory mediators with neurotoxic properties. High levels of inflammatory mediators can lead to enhanced neuronal excitation and BBB dysfunction in the mouse brain, ultimately causing drug resistance in epilepsy and recurrence of seizures, giving rise to a vicious cycle (42). Thus, understanding the unique molecular mechanisms of neuroinflammation in seizure disorders is crucial to identify inflammatory mediators and pathways which could act as biomarkers for development and severity of epilepsy, as well as becoming novel therapeutic targets. Pharmacological immunomodulation of these pathways might provide a novel avenue for treating epilepsy (42). For instance, it has been shown that inhibiting leukotriene D4 signaling attenuates seizure development in a murine model of chemically induced kindled seizure (43). Similar results were obtained by inhibiting IL-1β biosynthesis (44), as well as by administering an agonist of the prostaglandin E2 EP1 receptor and inhibiting COX-2 (45). Administration of different COX-2 inhibitors lead to affect the status epilepticus (SE) in rats. Furthermore, IL-1β, TLR4, HMGB1, P2X7 receptor, and EP2/PGE2 receptor antagonists have also been shown to modify SE in mice and rats (46). Lastly, a recent study in a mouse model of pentylenetetrazole-induced epileptic seizures also showed that administration of selenium nanoparticles had an anticonvulsant effect, which was mediated by decreased oxidative stress and neurotoxicity (47).
Due to the profound association between neuroinflammation and epileptogenesis, it is crucial to find diagnostic and monitoring biomarkers to provide biological insights on the role of inflammatory metabolism in neurological conditions and to develop novel diagnostic and therapeutic strategies to manage these disorders. In particular, cerebrospinal fluid (CSF) levels of neopterin, quinolinic acid, kynurenine, and tryptophan have been identified as potential biomarkers for neuroinflammation. Neopterin is a byproduct of the tetrahydrobiopterin de novo pathway synthesized by myeloid cells upon IFN-γ stimulation and it is found at increased levels in the CSF of individuals with neuroinflammatory disorders (48, 49). Furthermore, a preclinical study found increased levels of neopterin in the supernatant of primary rat astrocytes and mouse hippocampal slices following oxidative stress. These observations suggest that neopterin can also be produced by nerve cells under stress conditions (50). Another CSF biomarker of neuroinflammation is quinolinic acid (QA), a metabolite of the kynurenine pathway of tryptophan metabolism (49). QA has been implicated in the pathogenesis of neurological diseases in humans due to its potency as an excitotoxin. Elevated levels of QA can lead to oxidative stress, cytoskeletal disruption, behavioral alteration, and even cell death. At least some of the effects of QA can be attributed to its activation of the NMDA receptors (51). Kynurenine levels can also act as biomarkers for neuroinflammation and the activation of the kynurenine pathway of tryptophan metabolism can lead to immune suppression and neurotoxicity. Interestingly, it has been shown that modulation of this pathway can limit neurodegeneration in a murine multiple sclerosis model (52). Furthermore, the kynurenine/tryptophan ratio is used as a measurement for activity of indoleamine 2,3-dioxygenase, which is usually low under basal condition but can greatly increase during immune activation (49, 53). The downregulation of indoleamine 2,3-dioxygenase and the upregulation of hippocampal kynurenic acid lead to antiepileptic effects in a brain injury neonatal rat model of infantile spasms treated with antibiotics in combination with a ketogenic diet (54). Another recent study using a temporal lobe epilepsy rat model also showed reduced levels of tryptophan in the hippocampus and the anterior temporal lobe, which led to an enhanced frequency and amplitude of spontaneous excitatory postsynaptic currents in these brain regions (55). Because of their involvement in neuroinflammation, these metabolites can be used as reliable biomarkers in preclinical and clinical models. In particular, CSF neuropteran has an 82% sensitivity for defining neuro inflammation, followed by quinolinic acid with a 57% sensitivity, the kynurenine/tryptophan ratio with a 47% sensitivity, and finally kynurenine alone with 37% sensitivity in a pediatric cohort (49).
In a study published by Hanin et al. involving 51 patients with new-onset refractory status epilepticus (NORSE), the levels of 12 cytokines/chemokines were measured in serum or cerebrospinal fluid (CSF). A comparison was made between patients with and without SE, as well as between the 51 patients with cryptogenic NORSE (cNORSE) and the 47 patients with known-etiology refractory status epilepticus (RSE).
Interestingly, The results showed a significant increase in the levels of IL-6, TNF-α, CXCL8/IL-8, CCL2, MIP-1α, and IL-12p70 pro-inflammatory cytokines/chemokines in patients with SE compared to those without SE, both in serum and CSF. Among patients with cNORSE, the serum levels of innate immunity pro-inflammatory cytokines/chemokines (CXCL8, CCL2, and MIP-1α) were significantly higher compared to patients with non-cryptogenic RSE (56).
In a separate study involving 85 children with idiopathic epilepsy, the concentrations of CSF neuron specific enolase (NSE), IL-1β, and EPO were measured. The epileptic groups showed a significant increase in the mean concentrations of CSF NSE, IL-1β, and EPO compared to the control groups (P < 0.01). Additionally, positive correlations were observed between the levels of IL-1β, NSE, and EPO (57).
Febrile infection-related epilepsy syndrome (FIRES) is a condition where individuals experience a NORSE following a febrile illness that occurred within two weeks to 24 h before the onset of refractory SE. In this case, febrile illness may or may not be accompanied by fever at SE onset. Seizures in FIRES are often resistant to treatment and can lead to long-term cognitive and neurological impairments. Understanding the underlying mechanisms and immune responses associated with FIRES is crucial for developing targeted therapeutic approaches. Kothur et al. have analyzed 32 cytokines and chemokines in CSF of pediatric patients with different epilepsy syndromes, including FIRES/FIRES-related disorders (FRD), febrile/afebrile status epilepticus (FSE and ASE), and chronic epilepsy with frequent daily seizures. Surprisingly, the elevation of such molecules was higher in FIRES, and in FSE, when compared to chronic epilepsy and controls without neurological or immunological disorders. Moreover, in FIRES Th1-associated cytokines and chemokines, as well as IL-6, CCL2, CCL19, and CXCL1, resulted elevated when compared to the levels observed in encephalitis, which involved a broader network of cytokines/chemokines. In FSE, CXCL9, CXCL10, CXCL11, and CCL19 were elevated compared to ASE, despite similar median seizure duration and timing of CSF testing in relation to seizures (58).
According to the American Epilepsy Society (AES) guidelines (59) published in 2016, the management of convulsive status epilepticus should start in the first 5 min with an early stabilization phase consisting in primary first aid for seizures based on the “ABC” approach, followed by administration of benzodiazepines (BDZs) (60–62).
About the role of BDZ in contrasting neuroinflammation, one of the first evidences in literature dates to 1996, when Park and colleagues demonstrated that BZDs could produce anti-inflammatory effects binding to microglial cells (63). According to this, Midazolam and Diazepam would be able to reduce the synthesis and release of proinflammatory and neurotoxic molecules generated by activated microglia (64) and to inhibit microglial activation and proliferation itself.
Moreover, diazepam seems to be able of inducing a state of cellular inactivation defining a reduced transcription factors activity and chemotactic aptitude, inhibition of Ca2C-mediated signaling and a diminished production of cytokines (65). If the seizures continue beyond 20 min, second-line therapy will be started.
Giving the lack of evidence about a better approach option, choices are often dictated by local availability, cost, and patient-specific factors. The options are IV phenytoin or Fosphenytoin, Valproic acid, or Levetiracetam (66). If none of the above suggested therapies are obtainable, IV phenobarbital could be considered (67).
Phenytoin can reduce the activation of m-TOR Pathway, decreasing the levels of proinflammatory cytokines such as IL-1β, IL-6 and TNF-α (68). VPA has antioxidant properties, inducing the suppression of lipid peroxidation and oxidative DNA damage, as well as anti-inflammatory effects, resulting in diminution of MPO permeation and microglial activation (69). It also promotes a decrease in brain inflammation and degeneration by regulation the NF-κB pathway (70) and discourages lipopolysaccharide-induced production of TNF-α and IL-6 (71, 72).
In the end, recent evidence suggests that Levetiracetam exerts neuroprotective effects via anti-inflammatory actions (73). It seems to be able to suppress the expression of proinflammatory molecules, such as TNF- α, IL-6 and IL-1β (74) and to reduce mononuclear phagocyte-mediated phagocytosis (75).
The third therapy stage should be contemplated when the seizure duration reaches 40 min and status epilepticus became refractory. Possibilities include repeating a second-line medication or resorting to an anesthetic drug (59).
Usually, the most used anesthetic agents include Midazolam, short-acting barbiturates (Pentobarbital/Thiopentone), and propofol. Currently, Midazolam is perhaps the most used due to faster onset of action and short duration of effect (76). Midazolam is recently reported to exert a neuroprotective effect by inhibiting inflammation. Through the regulation on the RhoA/ROCK2 pathway, it ameliorates the impairment of the blood–brain barrier against LPS (77). Then, based on recent findings, it is conceivable that midazolam might inhibit IL-1b-induced STAT3 phosphorylation and IL-6 release suppressing ROS production (78). However, further investigation will be required to clarify this concept.
According to some preclinical studies, use of anesthetics such as Propofol moderates the stimulation and minimizes the secretion of proinflammatory cytokines (79–81). Newly, Lu and colleagues validated that the favorable effects of Propofol are mediated by the JAK1/STAT3 way and that it could reveal anti-neuroinflammatory action by repression of proinflammatory mediators from microglial cells (82). However, its use in children should be limited because of the increased risk of Propofol infusion syndrome (PRIS), a life-threatening state characterized by rhabdomyolysis, arrhythmias, metabolic acidosis, myocardial and renal failure that can occur using doses greater than 65 mcg/kg/min for 48 or more hours (83).
As about one-third of the patients continue seizing despite these treatment lines, thus evolving to refractory SE, and half of these subsequently develop super-refractory SE, it seems important to consider further treatment alternatives (84).
Ketamine recently emerged as a promising treatment, due to advantageous hemodynamics and a singular mechanism of action than conventional anesthetics (85). Its great lipid solubility establishes a rapid CNS uptake and onset of action (86), moreover in late stages of SE, there is a decrease in the number of effective GABA-A receptors and up-regulated glutamate NMDA receptors that potentiate its action (87). It has also been shown that Ketamine may reduce neuroinflammation by diminishing the quantity of microglia and active macrophages in cerebral cortical tissues as well as TNF- α production (88, 89).
A variety of immunomodulatory treatments have been proposed over the past years. The most used include corticosteroids, IV immunoglobulin (IVIG) and plasmapheresis. Their application is sustained by modern findings on immunologic (antibodies against neural receptors such as voltage-gated potassium channels and NMDA receptors) and inflammatory (stimulation of inflammatory signaling pathways such as Interleukin-1 receptor/toll-like receptor pathway) actions that may provide to their basic pathophysiology (90–92).
The outcomes of these cures are inconstant, and researches are yet to have not confirmed a clear a certain efficacy response. They probably may be beneficial in recognized autoimmune epilepsies or entities with supposed immunological basis, such as febrile-infection related epilepsy syndrome (FIRES) (60), but further studies are needed necessaries.
Finally, therapeutic hypothermia could be pondered as an adjunctive therapy for RSE/SRSE (93). In fact, multiple case reports have shown its efficacy in resolution of RSE in children (94, 95) and this outcome seems to be due to its ability to diminish cerebral metabolic rate, cerebral edema, inflammation, oxidative stress, and glutaminergic inducement (96, 97).
Assuming its possible feasible role in the pathophysiology of epilepsy, targeting affecting the epileptogenic proconvulsant-convulsant effect of inflammatory cytokines appears as an innovative therapeutic option in drug resistant epilepsies and SEs. In this way we have the possibility of acting directly on the pathogenetic mechanisms rather than only on symptom control (98).
Many pharmacological studies involving IL-1β/IL-1R1, HMGB1/TLR4, COX-2/ prostaglandins or the complement system have revealed that these inflammatory pathways suggestively provide to the beginning and/or recurrence of SE and that their targeting may be potentially disease- modifying (99–103).
The 2022 International consensus recommendations for management of new onset refractory status epilepticustreatment (104) supports the use of the human recombinant interleukin 1 (IL-1) receptor antagonist (Anakinra) in refractory SE.
Interleukin 1β (IL-1β) is a proinflammatory cytokine released by glial cells promoting neuroinflammation, enhancing neuronal excitability and contributing to refractories of seizures (105). Recent studies have shown that Anakinra could have a therapeutic role in controlling seizure recurrence and generalization in inflammatory refractory epilepsy (106–108), emerging as a suitable option for the treatment of SE of unknown cause in the early stages.
Moreover, the interleukin converting enzyme (ICE)/caspase-1, able to inhibit the conversion of pro-IL-1b to the pro-convulsant IL- 1b, has been recently contemplated as a possible target for the management of drug-resistant epilepsies (109). Pralnacasan (inhibitors of the IL-1b converting enzyme) and Belnacasan (elective inhibitor of caspases from the ICE/caspase-1 family) are currently undergoing phase III clinical trial, and the preliminary results are promising: In mice, intracerebroventricular administration of Pralnacasan and intraperitoneal administration of Belnacasan, appears to reduce 50% seizure duration (110, 111). Furthermore, a recent phase 2b double-blind randomized controlled trial involving the selective inhibitor of interleukin converting enzyme VX765, showed that in the 60 patients undergoing the therapy the percentage of responder-rate, of patients who were seizure-free for 2 weeks, and percentage of reduction in seizure rates ranged from 13% to 19% (112).
HMGB and TLR4 antagonists are also a potential novel anti-convulsive strategy (113, 114).
Resveratrol, for example, a type of natural phenol, has shown anti-inflammatory and neuroprotective properties (115). It has been recognized that it can suppress NFkB induced by TLRs 3 and 4 and the expression of INF-beta (116). Moreover, according to some research, this phenol reduces microglial activation and cyclooxygenases stimulation, often involved in epileptogenesis (117, 118). A study has also defined an antioxidant effect of Resveratrol against epileptogenic oxidative stress in the brain (119).
The ATP-gated purinergic P2X7 receptor (P2X7R) is an ion channel receptor situated on the superficial of microglia that can be activated by the ATP effluence following exposure to an exogenous stimulus, such as seizures (120–122). This stimulates the P2X7R-mediated NLRP3 inflammasome and the successive release of inflammatory molecules (123). According to recent data, demonstrating the defection in P2X7R expression after SE in both experimental animals and patients, the development of P2X7R antagonists could be useful in the treatment of refractory SE. Most reports (124–129) have suggested that Astaxanthin (AST), a molecule belonging to the carotenoid family, could be helpful in attenuating neuroinflammation in SE by inhibiting the ATP-P2X7R signal.
It has been demonstrated that it can reduce the extracellular ATP concentration, thereby constraining P2X7R activation and upregulation, causing the inhibition of the inflammatory signaling pathway. It can also considerably suppress the expression of inflammatory cytokine genes such as TNF-α, Cox-2, and IL-1β and it has strong antioxidant properties.
Increased serum and CFS levels of IL-6, an inflammatory cytokine having a pivotal role in enhancing and maintaining the inflammatory response and activating adaptive immunity, have been demonstrated in patients with refractory epilepsy (130, 131).
Supporting its involvement in these conditions, clinical and experimental data have reported some cases responding to treatment with Tocilizumab.
In 2018, Jun et al. (132) investigated the therapeutic potential Tocilizumab in 7 patients with new onset refractory status epilepticus (NORSE), reporting a resolution of SE after 1 or 2 doses of therapy in 6 patients with a median interval of 3 days from the initiation and no recurrence of SE during the observation period. Furthermore, in two children with Refractory Febrile Infection-Related Epilepsy Syndrome (FIRES), a decrease in seizures after Tocilizumab administration was documented with no side effects (133). The same favorable outcome was reported in a 6-year-old boy with Anakinra-Refractory FIRES (134).
Ketogenic diet (KD) represents a promising approach: it is a therapeutic dietary characterized by low-calorie, low-carbohydrate, high-fat, and standard protein intake which collectively sustain a state of ketosis, closely resembling the metabolic state induced by fasting.
Although literature data about its anti-inflammatory properties are poor, recent studies (135) have shown that it may influence neurotransmitter levels related to seizure onset. Specifically, the KD appears to enhance the activity of the inhibitory neurotransmitter gamma-aminobutyric acid (GABA). This effect is achieved through different pathways, including the activation of glutamic acid decarboxylase and the inhibition of transaminase activity. Furthermore, the KD could elevate the epileptic threshold by increasing ATP-sensitive potassium channels (136, 137). Beyond these seizure-related benefits, the KD may also exert a neuroprotective action. This includes the upregulation of calbindin, the inhibition of apoptotic factors like caspase 3, and an increase in the concentration of kynurenic acid. Additionally, the diet reduces the presence of oxygen free radicals (ROS) through the elevation of polyunsaturated fatty acids and neuronal uncoupled proteins (138–140).
Moreover, several studies have consistently observed that the ketogenic diet (KD) influences the diversity of the microbiome, leading to changes in the production of gut metabolites. In the context of epilepsy, it is noteworthy that children with this condition exhibit alterations in their gut microbiota, potentially contributing to the development or severity of seizures. Some research has postulated that changes in the expression of short-chain fatty acids (SCFA), which are gut metabolites capable of crossing the blood-brain barrier, may provide valuable insights into the modulatory effects of the KD on certain diseases. However, the precise mechanisms through which SCFA may influence disease expression remain to be fully elucidated (138, 141).
The management of epilepsy with cannabidiol (CBD), a cannabis derivative, has engendered impressive enthusiasm in recent years.
Numerous experimental reports indicate that CBD can diminish seizure occurrence and length with standard and unconventional antiepileptic properties, along with a neuroprotective and anti-inflammatory function (142). Several preclinical studies suggested that CBD revealed strong inhibitory effects of neurotoxic molecules and inflammatory cytokines, emphasizing its therapeutic potential for the treatment of refractory SE (143). Although CBD's effects on neuroinflammation appear to be still poorly understood, its molecular mechanism of action seems to be related to the downregulation of NADPH oxidase-mediated ROS, TLR4-NFκB and IFN-β-JAK-STAT pathways (144, 145). Gofshteyn et al. (142) reported the potential therapeutic effect of cannabidiol for FIRES in a series of 7 pediatric patients. Moreover, Rajsekar R. Rajaraman et al. (146) described the situation of a child with long-standing super-refractory status epilepticus (SRSE) who exhibited quick and complete resolution of SRSE upon exposure to pure cannabidiol. These evidences suggest that CBD can be considered as a potential treatment in SRSE.
The involvement of the inflammatory response in SE led to the search for therapeutic strategies targeting inflammatory mediators in this condition (2, 147). Currently, clinical experience is limited to the use in isolated case reports or series of the anti-cytokine agents Anakinra and Tocilizumab (148). However, this field is in continuous expansion, since studies on animal models of epilepsy and SE have led to the identification of potential new therapeutic targets.
Both Anakinra and Tocilizumab are currently used in the treatment of several autoinflammatory disorders (such as familial Mediterranean fever), other rheumatic diseases (arthritis, vasculitis), and cytokine-release syndromes (CRS) (3, 149, 150).
In SE, the most well-recognized application of Anakinra is the administration in patients with the febrile infection-related epilepsy syndrome (FIRES), and other reports describe its use in new-onset refractory status epilepticus (NORSE) or refractory/super refractory status epilepticus (SRSE) (148) To date, in the published cases Anakinra has been administered with different regimens (timing for drug initiation, posology, treatment duration) and resulted in a clinically relevant reduction of seizures in more than half of the reported patients (148, 151–154), while data on the long-term neuropsychological outcome have to be better defined7,10. Concerning the safety profile of Anakinra in epileptic individuals, the occurrence of infections has been reported in about 30% of the patients, but drug withdrawal due to severe adverse events is only rarely reported (148, 151–154).
The experience with the use of Tocilizumab in SE is limited to less than 50 patients, mostly suffering from NORSE or SRSE. Similarly to Anakinra, Tocilizumab has shown a clinical effect of reduction/arrest of seizures in most of the patients, while the safety profile showed the development of clinically relevant infections in about 20% of the described patients (132, 133, 148, 153, 155).
Interestingly, Tocilizumab has been effectively used to treat a patient with COVID-19-associated SE, as well as patients with CRS following chimeric-antigen T cell receptor (CAR-T) cells administration (156, 157).
Although the role of Anakinra and Tocilizumab in SE is promising, the interpretation of their efficacy is complicated by the heterogeneity of the available studies, together with the frequent concomitant administration of other treatments (immunosuppression, ASMs). Also, the use of biomarkers, such as the analysis of serum and CSF cytokines, has been performed only in a reduced percentage of patients, and the correlation between their trend and the clinical outcome is not defined (148).
The role of other biologic agents, including the anti- TNF- α Etanercept and the anti-CD20 antibody Rituximab, is limited to patients with specific etiologies of epilepsy and SE. Targeting TNF- α represents a promising strategy for patients suffering from Rasmussen encephalitis, in which the administration of Etanercept has shown a decrease in seizure frequency, although there are no specific data regarding its use in SE (158, 159). On the other hand, the application of Rituximab is limited to patients with drug-resistant epilepsy in the context of autoimmune encephalitis (160), while the use in cryptogenic RSE did not provide a clinically relevant effect in a recent small case series published by Jun et al. (132). Regarding therapies targeting integrins, a recent trial on the administration of the anti-α4-integrin antibody Natalizumab evidenced a reduction in the seizure frequency in patients with drug-resistant epilepsy (161), although the drug has not yet been investigated in SE.
SE represents a hard therapeutic challenge, and there is currently no standard approach to refractory or super-refractory cases. The use of animal models of SE, as well as the in vivo determination of serum and inflammatory biomarkers and the analysis of bioptic/surgical specimens have pointed out the central role of neuroinflammation in initiating and perpetrating the pathogenic process leading to SE. Therefore, there is an urgent need to develop therapeutic strategies targeting neuroinflammation in this category of patients. Although the clinical experience with drugs targeting neuroinflammation in SE is currently limited to the use of anti-IL-1 and anti-IL-6 agents, preclinical research is rapidly progressing and will hopefully lead to the identification of new targeted therapies (Figure 1), including chemokines and their receptors, and intracellular signaling molecules.
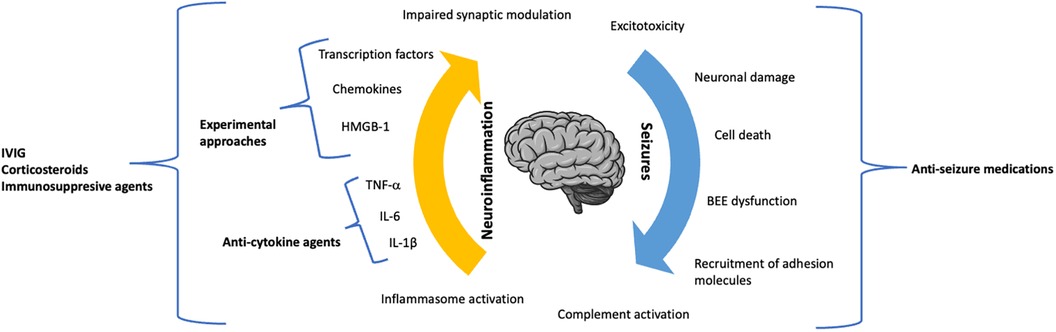
Figure 1. The dynamic role of neuroinflammation and seizure activity in the pathogenic process of status epilepticus.
Conceptualization, TF, AO, AS, and PS; Methodology, resources, data curation, writing – original draft preparation, writing – review and editing, TF, AS, GC, EC, MS, AR, GV, MSm AB, AF, SS, PS, AO, and AMC; supervision and project administration: TF, AO, and PS. All authors contributed to the article and approved the submitted version.
Research supported by PNRR-MUR-M4C2 PE0000006 Research Program “MNESYS”—A multiscale integrated approach to the study of the nervous system in health and disease. IRCCS ‘G. Gaslini’ is a member of ERN-Epicare.
The reviewer EP declared a shared affiliation with the author AF to the handling Editor at the time of review.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Trinka E, Cock H, Hesdorffer D, Rossetti AO, Scheffer IE, Shinnar S, et al. A definition and classification of status epilepticus–report of the ILAE task force on classification of Status epilepticus. Epilepsia. (2015) 56(10):1515–1523. doi: 10.1111/epi.13121
2. Rana A, Musto AE. The role of inflammation in the development of epilepsy. J Neuroinflammation. (2018) 15(1):144. doi: 10.1186/s12974-018-1192-7
3. Maniscalco V, Abu-Rumeileh S, Mastrolia MV, Marrani E, Maccora I, Pagnini I, et al. The off-label use of anakinra in pediatric systemic autoinflammatory diseases. Ther Adv Musculoskelet Dis. (2020) 12:1759720X20959575. doi: 10.1177/1759720X20959575
4. Orsini A, Foiadelli T, Costagliola G, Michev A, Consolini R, Vinci F, et al. The role of inflammatory mediators in epilepsy: focus on developmental and epileptic encephalopathies and therapeutic implications. Epilepsy Res. (2021) 172:106588. doi: 10.1016/j.eplepsyres.2021.106588
5. Vezzani A, Fujinami RS, White HS, Preux PM, Blümcke I, Sander JW, et al. Infections, inflammation and epilepsy. Acta Neuropathol. (2016) 131(2):211–234. doi: 10.1007/s00401-015-1481-5
6. Wallraff A, Köhling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. (2006) 26(20):5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006
7. Engel T, Jimenez-Pacheco A, Miras-Portugal MT, Diaz-Hernandez M, Henshall DC. P2x7 receptor in epilepsy; role in pathophysiology and potential targeting for seizure control. Int J Physiol Pathophysiol Pharmacol. (2012) 4(4):174–187. 3544219.23320131
8. Alyu F, Dikmen M. Inflammatory aspects of epileptogenesis: contribution of molecular inflammatory mechanisms. Acta Neuropsychiatr. (2016) 29(01):1–16. doi: 10.1017/neu.2016.47
9. Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. (2003) 23:8692–700. doi: 10.1523/JNEUROSCI.23-25-08692.2003
10. Hoshino K, Uchinami Y, Uchida Y, Saito H, Morimoto Y. Interleukin-1β modulates synaptic transmission and synaptic plasticity during the acute phase of sepsis in the senescence-accelerated mouse hippocampus. Front Aging Neurosci. (2021) 13:637703. (Published 2021 Feb 10). doi: 10.3389/fnagi.2021.637703
11. Kubota K, Inoue K, Hashimoto R, Kumamoto N, Kosuga A, Tatsumi M, et al. Tumor necrosis factor receptor-associated protein 1 regulates cell adhesion and synaptic morphology via modulation of N-cadherin expression. J Neurochem. (2009) 110(2):496–508. doi: 10.1111/j.1471-4159.2009.06099.x
12. Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, et al. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. (2006) 281(30):21362–8. doi: 10.1074/jbc.M600504200
13. Galic MA, Riazi K, Pittman QJ. Cytokines and brain excitability. Front Neuroendocrinol. (2012) 33(1):116–25. doi: 10.1016/j.yfrne.2011.12.002
14. Stellwagen D. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. (2005) 25(12):3219–28. doi: 10.1523/JNEUROSCI.4486-04.2005
15. Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci. (2012) 8(9):1254–66. doi: 10.7150/ijbs.4679
16. Marchi N, Granata T, Ghosh C, Janigro D. Blood-brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia. (2012) 53(11):1877–1886. doi: 10.1111/j.1528-1167.2012.03637.x
17. Wilson C, Finch C, Cohen H. Cytokines and cognition-the case for a head-to-toe inflammatory paradigm. J Am Geriatr Soc. (2002) 50(12):2041–56. doi: 10.1046/j.1532-5415.2002.50619.x
18. Zattoni M, Mura ML, Deprez F, Schwendener RA, Engelhardt B, Frei K, et al. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. (2011) 31(11):4037–50. doi: 10.1523/JNEUROSCI.6210-10.2011
19. Fabene PF, Navarro Mora G, Martinello M, Rossi B, Merigo F, Ottoboni L, et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med. (2008) 14(12):1377–83. doi: 10.1038/nm.1878
20. Schartz ND, Wyatt-Johnson SK, Price LR, Colin SA, Brewster AL. Status epilepticus triggers long-lasting activation of complement C1q-C3 signaling in the hippocampus that correlates with seizure frequency in experimental epilepsy. Neurobiol Dis. (2018) 109(Pt A):163–173. doi: 10.1016/j.nbd.2017.10.012
21. Liu J, Si Z, Li S, Huang Z, He Y, Zhang T, et al. The calcineurin inhibitor FK506 prevents cognitive impairment by inhibiting reactive astrogliosis in pilocarpine-induced status epilepticus rats. Front Cell Neurosci. (2018) 11:428. doi: 10.3389/fncel.2017.00428
22. Rojas A, Jiang J, Ganesh T, Yang MS, Lelutiu N, Gueorguieva P, et al. Cyclooxygenase-2 in epilepsy. Epilepsia. (2014) 55(1):17–25. doi: 10.1111/epi.12461
23. Dey A, Kang X, Qiu J, Du Y, Jiang J. Anti-inflammatory small molecules to treat seizures and epilepsy: from bench to bedside. Trends Pharmacol Sci. (2016) 37(6):463–484. doi: 10.1016/j.tips.2016.03.001
24. Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. (2015) 96(Pt A):70–82. doi: 10.1016/j.neuropharm.2014.10.027
25. Vezzani A, Balosso S, Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol. (2019) 15(8):459–472. doi: 10.1038/s41582-019-0217-x
26. Xu C, Zhang S, Gong Y, Nao J, Shen Y, Tan B, et al. Subicular caspase-1 contributes to pharmacoresistance in temporal lobe epilepsy. Ann Neurol. (2021) 90(3):377–390. doi: 10.1002/ana.26173
27. Villasana-Salazar B, Vezzani A. Neuroinflammation microenvironment sharpens seizure circuit. Neurobiol Dis. (2023) 178:106027. doi: 10.1016/j.nbd.2023.106027
28. Gardoni F, Boraso M, Zianni E, Corsini E, Galli CL, Cattabeni F, et al. Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1β and NMDA stimulation. J Neuroinflammation. (2011) 8(1):14. doi: 10.1186/1742-2094-8-14
29. Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. (2013) 14(6):383–400. doi: 10.1038/nrn3504
30. Ghasemi M, Schachter SC. The NMDA receptor complex as a therapeutic target in epilepsy: a review. Epilepsy Behav. (2011) 22(4):617–40. doi: 10.1016/j.yebeh.2011.07.024
31. Kovac S, Domijan AM, Walker MC, Abramov AY. Seizure activity results in calcium- and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis. (2014) 5(10):e1442. doi: 10.1038/cddis.2014.390
32. Balosso S, Maroso M, Sanchez-Alavez M, Ravizza T, Frasca A, Bartfai T, et al. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain. (2008) 131(Pt 12):3256–65. doi: 10.1093/brain/awn271
33. Balosso S, Liu J, Bianchi ME, Vezzani A. Disulfide-containing high mobility group box-1 promotes N-methyl-D-aspartate receptor function and excitotoxicity by activating toll-like receptor 4-dependent signaling in hippocampal neurons. Antioxid Redox Signal. (2014) 21(12):1726–40. doi: 10.1089/ars.2013.5349
34. Iori V, Maroso M, Rizzi M, Iyer AM, Vertemara R, Carli M, et al. Receptor for advanced glycation endproducts is upregulated in temporal lobe epilepsy and contributes to experimental seizures. Neurobiol Dis. (2013) 58:102–14. doi: 10.1016/j.nbd.2013.03.006
35. van Vliet EA, Aronica E, Vezzani A, Ravizza T. Review: neuroinflammatory pathways as treatment targets and biomarker candidates in epilepsy: emerging evidence from preclinical and clinical studies. Neuropathol Appl Neurobiol. (2018) 44(1):91–111. doi: 10.1111/nan.12444
36. Balosso S, Ravizza T, Aronica E, Vezzani A. The dual role of TNF-α and its receptors in seizures. Exp Neurol. (2013) 247:267–71. doi: 10.1016/j.expneurol.2013.05.010
37. Yin J, Valin KL, Dixon ML, Leavenworth JW. The role of microglia and macrophages in CNS homeostasis, autoimmunity, and cancer. J Immunol Res. (2017) 2017:5150678. doi: 10.1155/2017/5150678
38. Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu LJ. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J Neurosci. (2014) 34(32):10528–40. doi: 10.1523/JNEUROSCI.0416-14.2014
39. Yang MT, Lin YC, Ho WH, Liu CL, Lee WT. Everolimus is better than rapamycin in attenuating neuroinflammation in kainic acid-induced seizures. J Neuroinflammation. (2017) 14(1):15. doi: 10.1186/s12974-017-0797-6
40. Campbell SL, Hablitz JJ. Decreased glutamate transport enhances excitability in a rat model of cortical dysplasia. Neurobiol Dis. (2008) 32(2):254–61. doi: 10.1016/j.nbd.2008.07.003
41. Henneberger C, Steinhäuser C. Astrocytic TLR4 at the crossroads of inflammation and seizure susceptibility. J Cell Biol. (2016) 215(5):607–609. doi: 10.1083/jcb.201611078
42. Vishwakarma S, Singh S, Singh TG. Pharmacological modulation of cytokines correlating neuroinflammatory cascades in epileptogenesis. Mol Biol Rep. (2022) 49(2):1437–1452. doi: 10.1007/s11033-021-06896-8
43. Rehni AK, Singh TG. Modulation of leukotriene D4 attenuates the development of seizures in mice. Prostaglandins Leukot Essent Fatty Acids. (2011) 85(2):97–106. doi: 10.1016/j.plefa.2011.04.003
44. Maroso M, Balosso S, Ravizza T, Iori V, Wright CI, French J, et al. Interleukin-1β biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics. (2011r) 8(2):304–15. doi: 10.1007/s13311-011-0039-z
45. Fischborn SV, Soerensen J, Potschka H. Targeting the prostaglandin E2 EP1 receptor and cyclooxygenase-2 in the amygdala kindling model in mice. Epilepsy Res. (2010) 91(1):57–65. doi: 10.1016/j.eplepsyres.2010.06.012
46. Aronica E, Bauer S, Bozzi Y, Caleo M, Dingledine R, Gorter JA, et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia. (2017) 58(Suppl 3):27–38. doi: 10.1111/epi.13783
47. Molero-Luis M, Casas-Alba D, Orellana G, Ormazabal A, Sierra C, Oliva C, et al. Cerebrospinal fluid neopterin as a biomarker of neuroinflammatory diseases. Sci Rep. (2020) 10(1):18291. doi: 10.1038/s41598-020-75500-z
48. Yan J, Kothur K, Mohammad S, Chung J, Patel S, Jones HF, et al. CSF Neopterin, quinolinic acid and kynurenine/tryptophan ratio are biomarkers of active neuroinflammation. EBioMedicine. (2023) 91:104589. doi: 10.1016/j.ebiom.2023.10458
49. Ghisoni K, Martins R, Barbeito L, Latini A. Neopterin as a potential cytoprotective brain molecule. J Psychiatr Res. (2015) 71:134–139. doi: 10.1016/j.jpsychires.2015.10.003
50. Lugo-Huitrón R, Ugalde Muñiz P, Pineda B, Pedraza-Chaverrí J, Ríos C, Pérez-de la Cruz V. Quinolinic acid: an endogenous neurotoxin with multiple targets. Oxid Med Cell Longevity. (2013) 2013:104024. doi: 10.1155/2013/104024
51. Sundaram G, Lim CK, Brew BJ, Guillemin GJ. Kynurenine pathway modulation reverses the experimental autoimmune encephalomyelitis mouse disease progression. J Neuroinflammation. (2020) 17(1):176. doi: 10.1186/s12974-020-01844-y
52. Badawy AA, Guillemin G. The plasma [kynurenine]/[tryptophan] ratio and indoleamine 2,3-dioxygenase: time for appraisal. Int J Tryptophan Res. (2019) 12:1178646919868978. doi: 10.1177/1178646919868978
53. Mu C, Choudhary A, Mayengbam S, Barrett KT, Rho JM, Shearer J, et al. Seizure modulation by the gut microbiota and tryptophan-kynurenine metabolism in an animal model of infantile spasms. EBioMedicine. (2022) 76:103833. doi: 10.1016/j.ebiom.2022.103833
54. Dey S, Dubey V, Dixit AB, Tripathi M, Chandra PS, Banerjee J. Differential levels of tryptophan-kynurenine pathway metabolites in the hippocampus, anterior temporal lobe, and neocortex in an animal model of temporal lobe epilepsy. Cells. (2022) 11(22):3560. doi: 10.3390/cells11223560
55. Hanin A, Cespedes J, Dorgham K, Pulluru Y, Gopaul M, Gorochov G, et al. Cytokines in new-onset refractory Status epilepticus predict outcomes. Ann Neurol. (2023) 94(1):75–90. doi: 10.1002/ana.26627
56. Shi LM, Chen RJ, Zhang H, Jiang CM, Gong J. Cerebrospinal fluid neuron specific enolase, interleukin-1β and erythropoietin concentrations in children after seizures. Childs Nerv Syst. (2017) 33(5):805–811. doi: 10.1007/s00381-017-3359-4
57. Kothur K, Bandodkar S, Wienholt L, Chu S, Pope A, Gill D, et al. Etiology is the key determinant of neuroinflammation in epilepsy: elevation of cerebrospinal fluid cytokines and chemokines in febrile infection-related epilepsy syndrome and febrile status epilepticus. Epilepsia. (2019) 60(8):1678–88. doi: 10.1111/epi.16275
58. Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American epilepsy society. Epilepsy Curr. (2016) 16(1):48–61. doi: 10.5698/1535-7597-16.1.48
59. Samanta D, Garrity L, Arya R. Refractory and super-refractory Status epilepticus. Indian Pediatr. (2020) 57(3):239–253. doi: 10.1007/s13312-020-1759-0
60. Silbergleit R, Durkalski V, Lowenstein D, Conwit R, Pancioli A, Palesch Y, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med. (2012) 366:591–600. doi: 10.1056/NEJMoa1107494
61. Brigo F, Nardone R, Tezzon F, Trinka E. Nonintravenous midazolam versus in- travenous or rectal diazepam for the treatment of early status epilepticus: a sys- tematic review with meta-analysis. Epilepsy Behav. (2015) 49:325–36. doi: 10.1016/j.yebeh.2015.02.030
62. Park CH, Carboni E, Wood PL, Gee KW. Characterization of peripheral benzodiazepine type sites in a cultured murine BV-2 microglial cell line. Glia. (1996) 16:65–70. doi: 10.1002/(SICI)1098-1136(199601)16:1%3C65::AID-GLIA7%3E3.0.CO;2-A
63. Wilms H, Claasen J, Rohl C, Sievers J, Deuschl G, Lucius R. Involvement of benzodiazepine receptors in neuroinflammatory and neurodegenerative diseases: evidence from activated microglial cells in vitro. Neurobiol. Dis. (2003) 14:417–424. doi: 10.1016/j.nbd.2003.07.002
64. Lokensgard JR, Hu S, Hegg CC, Thayer SA, Gekker G, Peterson PK. Diazepam inhibits HIV-1 tat-induced migration of human microglia. J. Neurovirol. (2001) 7:481–486. doi: 10.1080/135502801753170345
65. Vossler DG, Bainbridge JL, Boggs JG, Novotny EJ, Loddenkemper T, Faught E, et al. Treatment of refractory convulsive status epilepticus: a comprehensive review by the American epilepsy society treatments committee. Epilepsy Curr. (2020) 20(5):245–264. doi: 10.1177/1535759720928269
66. Amengual-Gual M, Fischer SN, Gloss DS, Olson DM, Towne AR, Naritoku D, et al. Treatment of refractory convulsive Status epilepticus: a comprehensive review by the American epilepsy society treatments committee. Epilepsy Curr. (2020) 20(5):245–264. doi: 10.1177/1535759720928269
67. Crawshaw AA, Cock HR. Medical management of status epilepticus: emergency room to intensive care unit. Seizure. (2020) 75:145–152. (Erratum in: Seizure. 2020 Aug; 80:282). doi: 10.1016/j.seizure.2019.10.006
68. Pottoo FH, Salahuddin M, Khan FA, Alomar F, Al Dhamen MA, Alhashim AF, et al. Thymoquinone potentiates the effect of phenytoin against electroshock-induced convulsions in rats by reducing the hyperactivation of m-TOR pathway and neuroinflammation: evidence from in vivo. In Vitro and Computational Studies. Pharmaceuticals (Basel). (2021) 14(11):1132. doi: 10.3390/ph14111132
69. Suda S, Katsura K, Kanamaru T, Saito M, Katayama Y. Valproic acid attenuates ischemia-reperfusion injury in the rat brain through inhibition of oxidative stress and inflammation. Eur J Pharmacol. (2013) 707(1–3):26–31. doi: 10.1016/j.ejphar.2013.03.020
70. Chang P, Williams AM, Bhatti UF, Biesterveld BE, Liu B, Nikolian VC, et al. Valproic acid and neural apoptosis, inflammation, and degeneration 30 days after traumatic brain injury, hemorrhagic shock, and polytrauma in a swine model. J Am Coll Surg. (2019) 228(3):265–275. doi: 10.1016/j.jamcollsurg.2018.12.026
71. Ichiyama T, Okada K, Lipton JM, Matsubara T, Hayashi T, Furukawa S. Sodium valproate inhibits production of TNF-alpha and IL-6 and activation of NF-kappaB. Brain Res. (2000) 857:246–251. doi: 10.1016/s0006-8993(99)02439-7
72. Peng GS, Li G, Tzeng NS, Chen PS, Chuang DM, Hsu YD, et al. Valproate pretreatment protects dopaminergic neurons from LPS- induced neurotoxicity in rat primary midbrain cultures: role of microglia. Brain Res Mol Brain Res. (2005) 134:162–169. doi: 10.1016/j.molbrainres.2004.10.021
73. Contreras-García IJ, Cárdenas-Rodríguez N, Romo-Mancillas A, Bandala C, Zamudio SR, Gómez-Manzo S, et al. Levetiracetam mechanisms of action: from molecules to systems. Pharmaceuticals (Basel). (2022) 15(4):475. doi: 10.3390/ph15040475
74. Itoh K, Ishihara Y, Komori R, Nochi H, Taniguchi R, Chiba Y, et al. Levetiracetam treatment influences blood-brain barrier failure associated with angiogenesis and inflammatory responses in the acute phase of epileptogenesis in post-status epilepticus mice. Brain Res. (2016) 1652:1–13. doi: 10.1016/j.brainres.2016.09.038
75. Itoh K, Taniguchi R, Matsuo T, Oguro A, Vogel CFA, Yamazaki T, et al. Suppressive effects of levetiracetam on neuroinflammation and phagocytic microglia: a comparative study of levetiracetam, valproate and carbamazepine. Neurosci Lett. (2019) 708:134363. doi: 10.1016/j.neulet.2019.134363
76. Abend NS, Dlugos DJ. Treatment of refractory status epilepticus: literature review and a proposed protocol. Pediatr Neurol. (2008) 38:377–90. doi: 10.1016/j.pediatrneurol.2008.01.001
77. Zheng J, Zhang W, Kang P, Zheng X, He K, Bai H, et al. Midazolam ameliorates impairment of the blood-brain barrier (BBB) against LPS. Neurotox Res. (2022) 40(3):751–762. doi: 10.1007/s12640-022-00508-4
78. Tanabe K, Kozawa O, Iida H. Midazolam suppresses interleukin-1β-induced interleukin-6 release from rat glial cells. J Neuroinflamm. (2011) 8:68. doi: 10.1186/1742-2094-8-68
79. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. (2005) 8:752–758. doi: 10.1038/nn1472
80. Kannan G, Kambhampati SP, Kudchadkar SR. Effect of anesthetics on microglial activation and nanoparticle uptake: implications for drug delivery in traumatic brain injury. J Control Release. (2017) 263:192–199. doi: 10.1016/j.jconrel.2017.03.032
81. Ye X, Lian Q, Eckenhoff MF, Eckenhoff RG, Pan JZ. Differential general anesthetic effects on microglial cytokine expression. PLoS One. (2013) 8:e52887. doi: 10.1371/journal.pone.0052887
82. Lu Y, Gu Y, Ding X, Wang J, Chen J, Miao C. Intracellular Ca2 + homeostasis and JAK1/STAT3 pathway are involved in the protective effect of propofol on BV2 microglia against hypoxia-induced inflammation and apoptosis. PLoS One. (2017) 12:e0178098. doi: 10.1371/journal.pone.0178098
83. Fodale V, La Monaca E. Propofol infusion syndrome: an overview of a perplexing disease. Drug Saf. (2008) 31:293–303. doi: 10.2165/00002018-200831040-00003
84. Vezzani A, Dingledine R, Rossetti AO. Immunity and inflammation in status epilepticus and its sequelae: possibilities for therapeutic application. Expert Rev Neurother. (2015) 15(9):1081–92. doi: 10.1586/14737175.2015.1079130
85. Vasquez A, Farias-Moeller R, Tatum W. Pediatric refractory and super-refractory status epilepticus. Seizure. (2019) 68:62–71. doi: 10.1016/j.seizure.2018.05.012
86. Bertrand HG, Ellen YC, O’Keefe S, Flecknell PA. Comparison of the effects of ketamine and fentanyl-midazolam-medetomidine for sedation of rhesus macaques (Macaca mulatta). BMC Vet. Res. (2016) 12:93. doi: 10.1186/s12917-016-0721-9
87. Keros S, Buraniqi E, Alex B, Antonetty A, Fialho H, Hafeez B, et al. Increasing ketamine use for refractory status epilepticus in US pediatric hospitals. J Child Neurol. (2017) 32:638–46. doi: 10.1177/0883073817698629
88. Chang EI, Zarate MA, Rabaglino MB, Richards EM, Arndt TJ, Keller-Wood M, et al. Ketamine decreases inflammatory and immune pathways after transient hypoxia in late gestation fetal cerebral cortex. Physiol. Rep. (2016) 4(6):e12741. doi: 10.14814/phy2.12741
89. Groetzinger LM, Rivosecchi RM, Bain W, Bahr M, Chin K, McVerry BJ, et al. Ketamine infusion for adjunct sedation in mechanically ventilated adults. Pharmacotherapy. (2018) 38:181–188. doi: 10.1002/phar.2065
90. Shorvon S, Ferlisi M. The treatment of super-refractory status epilepticus: a critical review of available therapies and a clinical treatment protocol. Brain. (2011) 134(Pt 10):2802–18. doi: 10.1093/brain/awr215
91. Vezzani A, Ruegg S. The pivotal role of immunity and inflammatory processes in epilepsy is increasingly recognized: introduction. Epilepsia. (2011) 52(Suppl.3):1–4. doi: 10.1111/j.1528-1167.2011.03028.x
92. Vezzani A, Balosso S, Aronica E, Ravizza T. Basic mechanisms of status epilepticus due to infection and inflammation. Epilepsia. (2009) 50(Suppl.12):56–7. doi: 10.1111/j.1528-1167.2009.02370.x
93. Guilliams K, Rosen M, Buttram S, Zempel J, Pineda J, Miller B, et al. Hypothermia for pediatric refractory status epilepticus. Epilepsia. (2013) 54:1586–94. doi: 10.1111/epi.12331
94. Lin JJ, Lin KL, Hsia SH, Wang HS. Therapeutic hypothermia for febrile infection-related epilepsy syndrome in two patients. Pediatr Neurol. (2012) 47:448–50. doi: 10.1016/j.pediatrneurol.2012.08.013
95. Shein SL, Reynolds TQ, Gedela S, Kochanek PM, Bell MJ. Therapeutic hypothermia for refractory status epilepticus in a child with malignant migrating partial seizures of infancy and SCN1A mutation: a case report. Ther Hypothermia Temp Manage. (2012) 2:144–9. doi: 10.1089/ther.2012.0013
96. Corry JJ, Dhar R, Murphy T, Diringer MN. Hypothermia for refractory status epilepticus. Neurocrit Care. (2008) 9:189–97. doi: 10.1007/s12028-008-9092-9
97. Motamedi GK, Lesser RP, Vicini S. Therapeutic brain hypothermia, its mechanisms of action, and its prospects as a treatment for epilepsy. Epilepsia. (2013) 54:959–70. doi: 10.1111/epi.12144
98. Matin N, Tabatabaie O, Falsaperla R, Lubrano R, Pavone P, Mahmood F, et al. Epilepsy and innate immune system: a possible immunogenic predisposition and related therapeutic implications. Hum Vaccin Immunother. (2015) 11(8):2021–9. doi: 10.1080/21645515.2015.1034921
99. Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun. (2011) 25(7):1281–9. doi: 10.1016/j.bbi.2011.03.018
100. Rojas A, Jiang J, Ganesh T, Yang MS, Lelutiu N, Gueorguieva P, et al. Cyclooxygenase-2 in epilepsy. Epilepsia. (2014) 55(1):17–25. doi: 10.1111/epi.12461
101. Xiong ZQ, Qian W, Suzuki K, McNamara JO. Formation of complement membrane attack complex in mammalian cerebral cortex evokes seizures and neurodegeneration. J Neurosci. (2003) 23(3):955–60. doi: 10.1523/JNEUROSCI.23-03-00955.2003
102. Ravizza T, Balosso S, Aronica E, Vezzani A. Brain inflammation and epilepsy. In: Rho JM, Sankar R, Strafstrom CE, editors. Epilepsy: Mechanisms, models and translational perspectives. Boca Raton, FL, USA: CRC Press (2010). p. 45–59.
103. Kulkarni SK, Dhir A. Cyclooxygenase in epilepsy: from perception to application. Drugs Today (Barc). (2009) 45(2):135–54. doi: 10.1358/dot.2009.45.2.1322481
104. Wickstrom R, Taraschenko O, Dilena R, Payne ET, Specchio N, Nabbout R, et al. International consensus recommendations for management of new onset refractory Status epilepticus (NORSE) incl. Febrile infection-related epilepsy syndrome (FIRES): statements and supporting evidence. Epilepsia. (2022) 63(11):2840–64. doi: 10.1111/epi.17397
105. Vezzani A, Baram TZ. New roles for interleukin-1 Beta in the mechanisms of epilepsy. Epilepsy Curr. (2007) 7(2):45–50. doi: 10.1111/j.1535-7511.2007.00165.x
106. Dilena R, Mauri E, Aronica E, Bernasconi P, Bana C, Cappelletti C, et al. Therapeutic effect of anakinra in the relapsing chronic phase of febrile infection-related epilepsy syndrome. Epilepsia Open. (2019) 4(2):344–350. doi: 10.1002/epi4.12317
107. Kenney-Jung DL, Vezzani A, Kahoud RJ, LaFrance-Corey RG, Ho ML, Muskardin TW, et al. Febrile infection-related epilepsy syndrome treated with anakinra. Ann Neurol. (2016) 80:939e45. doi: 10.1002/ana.24806
108. Westbrook C, Subramaniam T, Seagren RM, Tarula E, Co D, Furstenberg-K nauff M, et al. Febrile infection-related epilepsy syndrome treated successfully with anakinra in a 21- year- old woman. WMJ. (2019) 118:135–9. 7082129.31682750
109. Randle JC, Harding MW, Ku G, Schonharting M, Kurrle R. ICE/Caspase-1 inhibitors as novel anti- inflammatory drugs. Expert Opin Invest Drugs. (2001) 10:1207–9. doi: 10.1517/13543784.10.7.1207
110. Wannamaker W, Davies R, Namchuk M, Pollard J, Ford P, Ku G, et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro- phenyl)-methanoyl]-amino}-3, 3-dimethyl-butanoyl)- pyrrolidine-2-carboxylic acid ((2R, 3S)-2-ethoxy-5- oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/ caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1b and IL-18. J Pharmacol Exp Ther. (2007) 321:509–16. doi: 10.1124/jpet.106.111344
111. Ravizza T, Lucas SM, Balosso S, Bernardino L, Ku G, Noe F, et al. Inactivation of caspase-1 in rodent brain: a novel anticonvulsive strategy. Epilepsia. (2006) 47:1160–8. doi: 10.1111/j.1528-1167.2006.00590.x
112. Incorporated VP. Study of VX-765 in subjects with treatment-resistant partial epilepsy. (Study code-clini-caltrials.gov identifier: NCT01048255)).
113. Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, et al. Toll-like receptor 4 and high- mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. (2010) 16:413–9. doi: 10.1038/nm.2127
114. Zurolo E, Iyer A, Maroso M, Carbonell C, Anink JJ, Ravizza T, et al. Activation of toll-like receptor, RAGE and HMGB1 signalling in malformations of cortical devel- opment. Brain. (2011) 134:1015–32. doi: 10.1093/brain/awr032
115. Albani D, Polito L, Signorini A, Forloni G. Neuroprotective properties of resveratrol in different neurodegenerative disorders. Biofactors. (2010) 36:370–6. doi: 10.1002/biof.118
116. Youn HS, Lee JY, Fitzgerald KA, Young HA, Akira S, Hwang DH. Specific inhibition of MyD88-independent signaling pathways of TLR3 and TLR4 by resveratrol: molecular targets are TBK1 and RIP1 in TRIF complex. J Immunol. (2005) 175:3339–46. doi: 10.4049/jimmunol.175.5.3339
117. Zhang F, Liu J, Shi J-S. Anti-inflammatory activities of resveratrol in the brain: role of resveratrol in micro- glial activation. Eur J Pharmacol. (2010) 636:1–7. doi: 10.1016/j.ejphar.2010.03.043
118. Subbaramaiah K, Chung WJ, Michaluart P, Telang N, Tanabe T, Inoue H, et al. Resveratrol inhibits cyclooxygenase-2 transcription and activity in phorbol ester-treated human mammary epithelial cells. J Biol Chem. (1998) 273:21875–82. doi: 10.1074/jbc.273.34.21875
119. Bhat KP, Kosmeder JW, Pezzuto JM. Biological effects of resveratrol. Antioxid Redox Signal. (2001) 3:1041–64. doi: 10.1089/152308601317203567
120. Christian CA. Persistent protection against pathology and paroxysms by P2X7R antagonism. Epilepsy Currents. (2018) 18(1):42–44. doi: 10.5698/1535-7597.18.1.42
121. Janks L, Sharma CVR, Egan TM. A central role for P2X7 receptors in human microglia. J Neuroinflamm. (2018) 15(1):325. doi: 10.1186/s12974-018-1353-8
122. Beamer E, Fischer W, Engel T. The ATP-gated P2X7 receptor as a target for the treatment of drug-resistant epilepsy. Front Neurosci. (2017) 11:21. doi: 10.3389/fnins.2017.00021
123. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity. (2017) 47(1):15–31. doi: 10.1016/j.immuni.2017.06.020
124. Yu Y, Feng S, Wei S, Zhong Y, Yi G, Chen H, et al. Extracellular ATP activates P2X7R-NF -κB (p65) pathway to promote the maturation of bone marrow- derived dendritic cells of mice. Cytokine. (2019) 119:175–181. doi: 10.1016/j.cyto.2019.03.019
125. Terrone G, Frigerio F, Balosso S, Ravizza T, Vezzani A. Inflammation and reactive oxygen species in status epilepticus: bio- markers and implications for therapy. Epilepsy Behav. (2019) 101:106275. doi: 10.1016/j.yebeh.2019.04.028
126. Hormozi M, Ghoreishi S, Baharvand P. Astaxanthin induces apopto- sis and increases activity of antioxidant enzymes in LS-180 cells. Artif Cells Nanomed Biotechnol. (2019) 47(1):891–895. doi: 10.1080/21691401.2019.1580286
127. Lu Y, Wang X, Feng J, Xie T, Si P, Wang W. Neuroprotective effect of astaxanthin on newborn rats exposed to prenatal maternal seizures. Brain Res Bull. (2019) 148:63–69. doi: 10.1016/j.brainresbull.2019.03.009
128. Deng X, Wang M, Hu S, Feng Y, Shao Y, Xie Y, et al. The neuroprotective effect of astaxanthin on pilocarpine-induced status epilepticus in rats. Front Cell Neurosci. (2019) 13:123. doi: 10.3389/fncel.2019.00123
129. Wang M, Deng X, Xie Y, Chen Y. Astaxanthin attenuates neuroinflammation in Status epilepticus rats by regulating the ATP-P2X7R signal. Drug Des Devel Ther. (2020) 14:1651–1662. doi: 10.2147/DDDT.S249162
130. Li G, Bauer S, Nowak M, Norwood B, Tackemberg B, Rosenow F, et al. Cytokines and epilepsy. Seizure. (2011) 20:249–56. doi: 10.1016/j.seizure.2010.12.005
131. Lorigados Pedre L, Morales Chacón LM. Follow-up of peripheral IL-1β and IL-6 and relation with apoptotic death in drug-resistant temporal lobe epilepsy patients submitted to surgery. Behav Sci. (2018) 8:21. doi: 10.3390/bs8020021
132. Jun JS, Lee ST, Kim R, Chu K, Lee SK. Tocilizumab treatment for new onset refractory status epilepticus. Ann Neurol. (2018) 84(6):940–945. doi: 10.1002/ana.25374
133. Cantarín-Extremera V, Jiménez-Legido M, Duat-Rodríguez A, García-Fernández M, Ortiz-Cabrera NV, Ruiz-Falcó-Rojas ML, et al. Tocilizumab in pediatric refractory status epilepticus and acute epilepsy: experience in two patients. J Neuroimmunol. (2020) 340:577142. doi: 10.1016/j.jneuroim.2019.577142
134. Stredny CM, Case S, Sansevere AJ, Son M, Henderson L, Gorman MP. Interleukin-6 blockade with tocilizumab in anakinra-refractory febrile infection-related epilepsy syndrome (FIRES). Child Neurol Open. (2020) 7:2329048X20979253. doi: 10.1177/2329048X20979253
135. Koh S, Dupuis N, Auvin S. Ketogenic diet and neuroinflammation. Epilepsy Res. (2020) 167:106454. doi: 10.1016/j.eplepsyres.2020.106454
136. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. (2015) 21:263–269. doi: 10.1038/nm.3804
137. Rahman M, Muhammad S, Khan MA, Chen H, Ridder DA, Muller-Fielitz H, et al. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat Commun. (2014) 5:3944. doi: 10.1038/ncomms4944
138. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut Microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. (2016) 167(1469–1480):e1412. doi: 10.1016/j.cell.2016.11.018
139. Kim DY, Simeone KA, Simeone TA, Pandya JD, Wilke JC, Ahn Y, et al. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann Neurol. (2015) 78:77–87. doi: 10.1002/ana.24424
140. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. (2015) 21:263–269. doi: 10.1038/nm.3804
141. Santangelo A, Corsello A, Spolidoro GCI, Trovato CM, Agostoni C, Orsini A, et al. The influence of ketogenic diet on gut Microbiota: potential benefits, risks and indications. Nutrients. (2023) 15(17):3680. doi: 10.3390/nu15173680
142. Gofshteyn JS, Wilfong A, Devinsky O, Bluvstein J, Charuta J, Ciliberto MA, et al. Cannabidiol as a potential treatment for febrile infection-related epilepsy syndrome (FIRES) in the acute and chronic phases. J. Child Neurol. (2017) 32(1):35–40. doi: 10.1177/0883073816669450
143. Yousaf M, Chang D, Liu Y, Liu T, Zhou X. Neuroprotection of cannabidiol, its synthetic derivatives and combination preparations against microglia-mediated neuroinflammation in neurological disorders. Molecules. (2022) 27(15):4961. doi: 10.3390/molecules27154961
144. Ben-Shabat S, Hanus LO, Katzavian G, Gallily R. New cannabidiol derivatives: synthesis, binding to cannabinoid receptor, and evaluation of their antiinflammatory activity. J Med Chem. (2006) 49(3):1113–7. doi: 10.1021/jm050709m
145. Yousaf M, Chang D, Liu Y, Liu T, Zhou X. Neuroprotection of cannabidiol, its synthetic derivatives and combination preparations against microglia-mediated neuroinflammation in neurological disorders. Molecules. (2022) 27(15):4961. doi: 10.3390/molecules27154961
146. Rajaraman RR, Sankar R, Hussain SA. Successful use of pure cannabidiol for the treatment of super-refractory status epilepticus. Epilepsy Behav Case Rep. (2018) 10:141–144. doi: 10.1016/j.ebcr.2018.07.004
147. Lee SK. Diagnosis and treatment of Status epilepticus. J Epilepsy Res. (2020) 10(2):45–54. doi: 10.14581/jer.20008
148. Costagliola G, Depietri G, Michev A, Riva A, Foiadelli T, Savasta S, et al. Targeting inflammatory mediators in epilepsy: a systematic review of its molecular basis and clinical applications. Front Neurol. (2022) 13:741244. doi: 10.3389/fneur.2022.741244
149. Yamanaka G, Ishida Y, Kanou K, Suzuki S, Watanabe Y, Takamatsu T, et al. Towards a treatment for neuroinflammation in epilepsy: interleukin-1 receptor antagonist, anakinra, as a potential treatment in intractable epilepsy. Int J Mol Sci. (2021) 22(12):6282. doi: 10.3390/ijms22126282
150. Sheppard M, Laskou F, Stapleton PP, Hadavi S, Dasgupta B. Tocilizumab (Actemra). Hum Vaccin Immunother. (2017) 13(9):1972–1988. doi: 10.1080/21645515.2017.1316909
151. Lai YC, Muscal E, Wells E, Shukla N, Eschbach K, Hyeong Lee K, et al. Anakinra usage in febrile infection related epilepsy syndrome: an international cohort. Ann Clin Transl Neurol. (2020) 7(12):2467–2474. doi: 10.1002/acn3.51229
152. Sa M, Singh R, Pujar S, D'Arco F, Desai N, Eltze C, et al. Centromedian thalamic nuclei deep brain stimulation and anakinra treatment for FIRES—two different outcomes. Eur J Paediatr Neurol. (2019) 23(5):749–754. doi: 10.1016/j.ejpn.2019.08.001
153. Aledo-Serrano A, Hariramani R, Gonzalez-Martinez A, Álvarez-Troncoso J, Toledano R, Bayat A, et al. Anakinra and tocilizumab in the chronic phase of febrile infection-related epilepsy syndrome (FIRES): effectiveness and safety from a case-series. Seizure. (2022) 100:51–55. doi: 10.1016/j.seizure.2022.06.012
154. Shrestha A, Wood EL, Berrios-Siervo G, Stredny CM, Boyer K, Vega C, et al. Long-term neuropsychological outcomes in children with febrile infection-related epilepsy syndrome (FIRES) treated with anakinra. Front Neurol. (2023) 14:1100551. doi: 10.3389/fneur.2023.1100551
155. Girardin ML, Flamand T, Roignot O, Abi Warde MT, Mutschler V, Voulleminot P, et al. Treatment of new onset refractory status epilepticus/febrile infection-related epilepsy syndrome with tocilizumab in a child and a young adult. Epilepsia. (2023) 64(6):e87–92. doi: 10.1111/epi.17591
156. Kizilkilic EK, Unkun R, Uygunoglu U, Delil S, Ozkara C. Treatment of COVID-19-induced refractory status epilepticus by tocilizumab. Eur J Neurol. (2022) 29(9):2861–2863. doi: 10.1111/ene.15440
157. Reveron-Thornton R, Scott BJ, Post D, Caulfield AF, Werbaneth K, Hovsepian DA, et al. Recurrent Status epilepticus in the setting of chimeric antigen receptor (CAR)-T cell therapy. Neurohospitalist. (2022) 12(1):74–79. doi: 10.1177/19418744211000980
158. Lagarde S, Villeneuve N, Trébuchon A, Kaphan E, Lepine A, McGonigal A, et al. Anti-tumor necrosis factor alpha therapy (Adalimumab) in rasmussen’s encephalitis: an open pilot study. Epilepsia. (2016) 57(6):956–66. doi: 10.1111/epi.13387
159. Orsini A, Foiadelli T, Carli N, Costagliola G, Masini B, Bonuccelli A, et al. Rasmussen’s encephalitis: from immune pathogenesis towards targeted-therapy. Seizure. (2020) 81:76–83. https://doi.org/10.1016/j.seizure.2020.07.02332769034
160. Byun JI, Lee ST, Jung KH, Sunwoo JS, Moon J, Lim JA, et al. Effect of immunotherapy on seizure outcome in patients with autoimmune encephalitis: a prospective observational registry study. PLoS One. (2016) 11(1):e0146455. doi: 10.1371/journal.pone
Keywords: epilepsy, status epilepticus, neuroinflammation, inflammation, cytokines
Citation: Foiadelli T, Santangelo A, Costagliola G, Costa E, Scacciati M, Riva A, Volpedo G, Smaldone M, Bonuccelli A, Clemente AM, Ferretti A, Savasta S, Striano P and Orsini A (2023) Neuroinflammation and status epilepticus: a narrative review unraveling a complex interplay. Front. Pediatr. 11:1251914. doi: 10.3389/fped.2023.1251914
Received: 2 July 2023; Accepted: 11 October 2023;
Published: 21 November 2023.
Edited by:
Sara Matricardi, University of Studies G. d’Annunzio Chieti and Pescara, ItalyReviewed by:
Eleonora Palma, Sapienza University of Rome, Italy© 2023 Foiadelli, Santangelo, Costagliola, Costa, Scacciati, Riva, Volpedo, Smaldone, Bonuccelli, Clemente, Ferretti, Savasta, Striano and Orsini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: A. Santangelo YW5kcm9zYW50YW5nZWxvQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.