 Nicola Tovaglieri1
Nicola Tovaglieri1 Emanuele Micaglio
Emanuele Micaglio Simona Lobasso
Simona Lobasso
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 16 August 2023
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1250772
Background: Barth syndrome is a rare genetic disease characterized by cardiomyopathy, skeletal muscle weakness, neutropenia, growth retardation and organic aciduria. This variable phenotype is caused by pathogenic hemizygous variants of the TAFAZZIN gene on the X chromosome, which impair metabolism of the mitochondrial phospholipid cardiolipin. Although most patients are usually diagnosed in the first years of life, the extremely variable clinical picture and the wide range of clinical presentations may both delay diagnosis. This is the case reported here of a man affected with severe neutropenia, who was not diagnosed with Barth syndrome until adulthood.
Case presentation: We describe herein a family case, specifically two Caucasian male cousins sharing the same mutation in the TAFAZZIN gene with a wide phenotypic variability: an infant who was early diagnosed with Barth syndrome due to heart failure, and his maternal cousin with milder and extremely different clinical features who has received the same diagnosis only at 33 years of age.
Conclusions: Our report supports the underestimation of the prevalence of Barth syndrome, which should be always considered in the differential diagnosis of male patients with recurrent neutropenia with or without signs and symptoms of cardiomyopathy.
Barth syndrome (BTHS, MIM 302060) is a rare X-linked disorder associated with a wide spectrum of effects, particularly including early-onset cardiomyopathy, skeletal muscle myopathy, neutropenia, growth delay, organic aciduria and variable mitochondrial dysfunction (1–4). Loss-of-function mutations in the TAFAZZIN gene, located on chromosome Xq28.12, cause defective acyl-chain remodeling of the phospholipid cardiolipin (CL), resulting in mitochondrial bioenergetics and metabolic abnormalities in patient tissues (5–8). CL is a dimeric phospholipid with four acyl chains localized almost exclusively in the inner mitochondrial membrane, where it plays a key role in maintaining both structural and functional integrity of many mitochondrial proteins, including respiratory chain protein complexes and their assembly into supercomplexes (9–12). Loss of function of the protein tafazzin leads to an altered CL transacylation, resulting in depletion of remodeled CL forms (mainly tetra-linoleoyl CL in the heart) with accumulation of immature CL forms (containing more saturated acyl-chains) and monolysocardiolipin (MLCL), a phospholipid containing three acyl chains which is practically absent in healthy mitochondria. Therefore, alterations of CL remodeling causes a dramatic increase of the MLCL/CL ratio in BTHS mitochondria (13).
Diagnosis of BTHS can be tricky because the clinical and biochemical features of the syndrome are highly variable and may differ between affected individuals from the same family and within a patient over time (2). Many patients with BTHS have mildly to severely elevated levels of 3-methylglutaconic acid (3-MGCA) on quantitative analysis of urinary organic acids, so that the disorder is also referred to “type II 3-methylglutaconic aciduria” (2). However, the urinary 3-MGCA levels can be elevated in a number of other metabolic disorders (14), so this test shows poor diagnostic specificity.
In contrast, the measurement of the MLCL/CL ratio is the only biochemical test characterized by high diagnostic sensitivity and specificity and it can be performed in bloodspots, fibroblasts and nucleated blood cells (15–17). The elevated MLCL/CL ratio is considered the biochemical hallmark of BTHS. Molecular genetic testing for mutations in the TAFAZZIN gene is also used for the diagnosis of BTHS. More than 120 pathogenic mutations spanning all 11 exons and introns of the TAFAZZIN gene are known (4). Nevertheless, no correlation between genotype/phenotype in BTHS has been demonstrated to date.
Here we describe the wide clinical variability in age of presentation, severity, and type of symptoms of two male relatives with the same genetic mutation in the TAFAZZIN gene: a child with classic cardiac features (Patient 1) and an oldest patient presenting mainly neutropenia without heart failure (Patient 2).
Patients 1 and 2 belong to the same family, whose pedigree is shown in Figure 1.
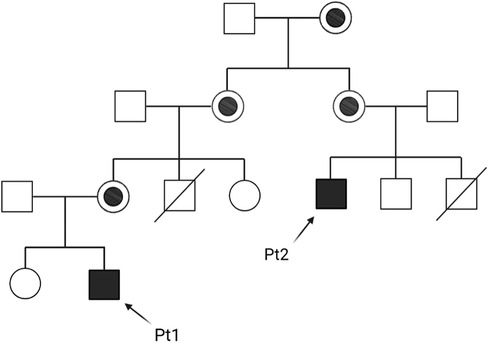
Figure 1. Family pedigree of patients 1 and 2 (Pt1 and Pt2).
We conducted this case study to aid in the diagnosis of BTHS and to highlight the need for a re-evaluation of diagnostic approaches, particularly with reference to patterns of neutropenia in male patients.
Patient 1 is a Caucasian child born full-term and with normal growth parameters after an uneventful pregnancy; as his maternal uncle died for complications of dilated cardiomyopathy (DCM) at age 7 months, the patient's cardiac function was evaluated by transthoracic echocardiography at age 2 months. Cardiac examinations revealed DCM with hypertrophied and hypertrabeculated walls, a mild-moderate mitral insufficiency and impaired left ventricular systolic function with an ejection fraction (EF) of about 38%.
Based on these findings, BTHS was clinically suspected and diagnosed by the genetic evaluation which identified a hemizygous DNA mutation (c.777 + 1G > A) in intron 10 of the TAFAZZIN gene, which is a splice donor variant classified as likely pathogenic (18). The patient started pharmacological treatments for heart failure with paediatric dosage of beta-blockers, ACE inhibitors, diuretics, and ivabradine.
At age 3, his EF was about 45%, while the Holter QTc was 448 msec with abnormal ventricular repolarization. His therapy was modified by quitting the diuretic. From then on, the EF has been around 52%–55% and the Holter QTc normal; however, left ventricular noncompaction (LVNC), a cardiomyopathy characterized by loose and trabeculated myocardium that is often associated with BTHS (2, 3), was observed.
Currently, the patient is 6 years old. His last cardiac evaluation showed increased wall thicknesses on the inferior wall and on all mid-apical segments (baseline IVS 5.5 mm, PW 8 mm), accentuation of the trabecular meshwork, a slight reduction of systolic function overall (LVEF 47%–48%) together with mild reduction of the mitral annular plane systolic excursion (MAPSE 9 mm).
Regarding neutropenia, another pathological condition associated with BTHS (2, 3), the patient showed a normal neutrophil count with episodic moderate neutropenia. At age 2, during an infection episode accompanied by severe neutropenia (neutrophil count 350/µl), therapy with Granulocyte Colony-Stimulating Factor (G-CSF) was started. However, the patient had remarkable neutrophilia, so therapy was stopped a few months later, and he never again experienced severe bacterial infections.
As regards metabolic screening, the first evaluation of plasmatic amino acids and organic urinary acids was performed at age 2, when the baby came to our attention, and showed augmented excretion of 3-MGCA. The patient was monitored for metabolic alterations from then on, and low citrulline and arginine levels were detected during the most recent follow-up, which have been corrected by increasing supplementation therapy (citrulline, arginine and vitamin B12).
No significant episodes of hypoglycaemia occurred, except for at 3 years of age, during a viral gastroenteritis. He is now under therapy with Glycosade, a slow-release starch to keep blood sugar levels consistent and prevent hypoglycaemia.
Neither skeletal muscle weakness nor growth retardation have been observed in Patient 1 until now.
Patient 2 is a maternal cousin of Patient 1 (see Figure 1), a 33-year-old Caucasian male with recurrent severe neutropenia since early childhood, who was not diagnosed with BTHS until adulthood. This patient had two brothers, the first one died due to congenital cardiomyopathy at age 1 month, while the second one is in good health (but without genetic investigation yet to date). Cardiac function parameters have always been normal throughout his life until now. However, neutropenia was observed at birth after an acute urinary tract infection, and then the patient was hospitalized at 1 year of age for septic shock due to a severe Pseudomonas aeruginosa infection. This event was followed by frequent and important infectious episodes (mainly either middle ear or inner ear infections) with hospitalization during the first years of life. After repeated neutrophil count examinations, which often indicated a value <500/µl, the patient started therapy with 4 µg/Kg G-CSF on alternate days at 4 years of age, with good recovery of neutrophil count and no other infectious episodes.
At age 11, after discontinuation of G-CSF therapy, the patient was hospitalized again for febrile gastroenteritis with severe dehydration. During this time, he suffered from cardiac arrest and then septic shock after surgical hemi-colectomy for two colic perforations. After this event, the G-CSF therapy was reinstated with a contextual normalization of the neutrophil count and only sporadic leucocytosis.
During his adolescence, the patient showed delayed growth in stature and weight and a bone mineral density that was lower than expected for gender and age (calculated T-score—2.9 at spinal level at 15 years of age). This finding is indicative for the presence of moderate osteopenia. Because of the associated presence of a hypovitaminosis D condition, supplemental therapy with calcifediol was introduced.
At age 23, the T-score at spinal level improved slightly to −2.2, but vertebral fractures (at D11, D12, and L1) were observed. For this reason, the patient was also treated with bisphosphonates by infusion.
At age 25, the patient was evaluated by a geneticist for hematopoietic system disorders by DNA sequencing and mutation analysis in a panel of candidate genes, including ELANE, HAX1, G6PC3, and WAS. However, the sequences were found to be normal, and no signs of immunodeficiency nor Wiskott-Aldrich syndrome has been noted after a careful genetic counselling. The patient had electrocardiogram and echocardiogram for physical activity normal.
At age 33, given his clinical history and over all the familiarity, the diagnosis of BTHS was finally made, 5 years later than the cousin's diagnosis. First, an elevated MLCL/CL ratio was detected in the leukocytes by MALDI-TOF/MS (17, 19). Finally, DNA sequencing of the TAFAZZIN gene showed that Patient 2 shared the same genetic mutation with his cousin, Patient 1.
After diagnosis, cardiac evaluation of Patient 2 showed a mild EF reduction that did not require therapy. Patient 2 waits for the evaluation of the amino acids to possibly fill in the missing ones.
Table 1 summarises the most important clinical, biochemical, and genetic findings of the two BTHS patients, and shows a considerable phenotypic variability.
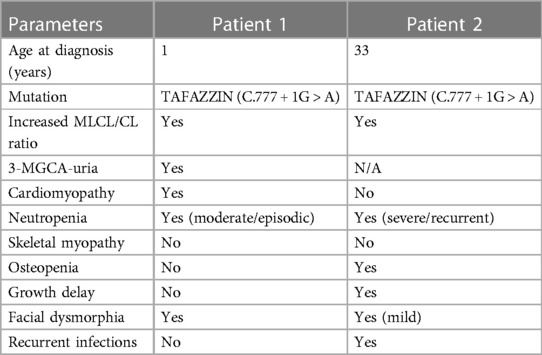
Table 1. Most important clinical, biochemical and genetic findings in the two patients.
BTHS is commonly classified as a paediatric disorder because most patients are diagnosed in the first years of life. The estimated prevalence of BTHS is 1 case per million males, with about 15 patients currently known in Italy and 250 worldwide, although it is generally accepted that the disease is underdiagnosed (20).
There are increasing reports of patients diagnosed with BTHS in adulthood (21, 22), suggesting that knowledge about this rare disorder has been growing among physicians in recent years. In light of the case study here reported, it should be considered that many more adult patients with idiopathic cardiomyopathy or recurrent neutropenia might be possible unidentified diagnoses of BTHS.
Although heterogeneous clinical features and intrafamilial variability have been previously reported in affected members harbouring the same genetic mutation in TAFAZZIN (4, 23), the significant variability characterizing the two cousins of this family is striking. BTHS patients without paediatric cardiac symptoms are rare (23); indeed, cardiomyopathy and heart failure are the primary reasons for diagnosis in infantile period and death (2, 4). The most common cardiomyopathies described in BTHS patients are DCM and LVNC (2, 4).
In contrast to Patient 1, Patient 2 of this study did not show significant cardiac disease as a child, but frequent infectious events due to severe and recurrent neutropenia were the main features of his disorder. Neutropenia (absolute neutrophil count less than 1,500/mcl) is one of the most frequently observed findings in BTHS patients and one of the earliest manifestations of the disease, which can range from absent to severe, intermittent, or recurrent (2, 24). The aetiology of neutropenia in BTHS is not yet understood (24).
As described above, Patients 1 and 2 have a different clinical presentation, although the same genetic mutation in the TAFAZZIN gene and similar increased levels of MLCL/CL ratio have been detected.
Of course, it can be also considered that many clinical parameters are different because of the vast age difference in patients. Of note, both patients presented with typical cranial-facial appearance, characterized by full cheeks, deep set eyes and prominent ears (in some patients with fleshy lobes) (2, 23).
To date, it remains unclear whether additional modifying genetic factors might contribute to the differential and pleiotropic phenotypic expression of BTHS patients. A recent study has demonstrated that genetic background in a mouse BTHS model powerfully impacted phenotypic expression of tafazzin loss-of-function possibly altering mitochondrial quality control (25). These data together with many reports describing no correlation between the type of mutation and clinical presentation in BTHS patients clearly indicate the existence of additional modifying factors that act as key determinants of pathogenesis of the syndrome (26).
In addition, splicing diversity may be responsible for the variability of BTHS phenotypes. Recently a case of infantile BTHS patient with severe heart failure and LVNC has been reported, in which amino acid substitution associated with a missense variant in TAFAZZIN gene was not sufficient to explain the syndrome severity, but it exhibited various splicing variants and contributed to the worse clinical course (27). These findings suggest that it is important to determine whether missense variants in TAFAZZIN are related to splicing abnormalities in BTHS.
Patient 2 here described did not receive the correct diagnosis of BTHS until adulthood and was probably diagnosed only due to his family history. As with other rare genetic diseases, it is important to identify the underlying genetic pathology in patients, although there is no specific therapy for BTHS to date. Early diagnosis is critical to allow rapid intervention for heart failure and antibiotic treatment for bacterial infections, thereby improving patients' quality of life. In addition, treatment of such patients requires a multidisciplinary team approach due to the variety of organ systems that could be involved.
In conclusion, the study here reported corroborates the underestimation of the prevalence of this rare genetic disease, which should always be considered in the differential diagnosis of male patients with unexplained neutropenia. Therefore, we suggest including TAFAZZIN mutation test in the next generation sequencing analysis designed to investigate idiopathic neutropenia.
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Milano Area 3—ASST GOM NIGUARDA (nr. 16-19012022). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Conception and design: NT and SL. Data collection: NT, SR, and SL. Data analysis: NT, SR, EM, AC, and SL. Drafting of the article: SR and SL. All authors contributed to the article and approved the submitted version.
The study was funded by a project grant from Consiglio Regionale Puglia awarded to SL.
We thank Patients and Families for helping us for the drafting of the manuscript. We also acknowledge Barth Italia Onlus for the constant support and the deep attention towards the organizational aspects of our study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Barth PG, Scholte HR, Berden JA, Van der Klei-Van Moorsel JM, Luyt-Houwen IE, Van’t Veer-Korthof ET, et al. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci. (1983) 62:327–55. doi: 10.1016/0022-510X(83)90209-5
2. Clarke SLN, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N, et al. Barth syndrome. Orphanet J Rare Dis. (2013) 8:23. doi: 10.1186/1750-1172-8-23
3. Zegallai HM, Hatch GM. Barth syndrome: cardiolipin, cellular pathophysiology, management, and novel therapeutic targets. Mol Cell Biochem. (2021) 476:1605–29. doi: 10.1007/s11010-020-04021-0
4. Taylor C, Rao ES, Pierre G, Chronopoulou E, Hornby B, Heyman A, et al. Clinical presentation and natural history of Barth syndrome: an overview. J Inherit Metab Dis. (2022) 45:7–16. doi: 10.1002/jimd.12422
5. Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet. (1996) 12:385–9. doi: 10.1038/ng0496-385
6. Whited K, Baile MG, Currier P, Claypool SM. Seven functional classes of Barth syndrome mutation. Hum Mol Genet. (2013) 22:483–92. doi: 10.1093/hmg/dds447
7. Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJ, et al. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun. (2000) 279:378–82. doi: 10.1006/bbrc.2000.3952
8. Schlame M, Xu Y. The function of tafazzin, a mitochondrial phospholipid-lysophospholipid acyltransferase. J Mol Biol. (2020) 432:5043–51. doi: 10.1016/j.jmb.2020.03.026
9. Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol. (2007) 292:C33–44. doi: 10.1152/ajpcell.00243.2006
10. Claypool SM, Koehler CM. The complexity of cardiolipin in health and disease. Trends Biochem Sci. (2012) 37:32–41. doi: 10.1016/j.tibs.2011.09.003
11. Ren M, Phoon CKL, Schlame M. Metabolism and function of mitochondrial cardiolipin. Prog Lipid Res. (2014) 55:1–16. doi: 10.1016/j.plipres.2014.04.001
12. Russo S, De Rasmo D, Signorile A, Corcelli A, Lobasso S. Beneficial effects of SS-31 peptide on cardiac mitochondrial dysfunction in tafazzin knockdown mice. Sci Rep. (2022) 12:19847. doi: 10.1038/s41598-022-24231-4
13. Schlame M, Kelley RI, Feigenbaum A, Towbin JA, Heerdt PM, Schieble T, et al. Phospholipid abnormalities in children with Barth syndrome. J Am Coll Cardiol. (2003) 42:1994–9. doi: 10.1016/j.jacc.2003.06.015
14. Wortmann SB, Kluijtmans LA, Engelke UFH, Wevers RA, Morava E. The 3-methylglutaconic acidurias: what’s new? J Inherit Metab Dis. (2012) 35:13–22. doi: 10.1007/s10545-010-9210-7
15. Vaz FM, van Lenthe H, Vervaart MAT, Stet FS, Klinkspoor JH, Vernon HJ, et al. An improved functional assay in blood spot to diagnose Barth syndrome using the monolysocardiolipin/cardiolipin ratio. J Inherit Metab Dis. (2022) 45:29–37. doi: 10.1002/jimd.12425
16. Bowron A, Frost R, Powers VEC, Thomas PH, Heales SJR, Steward CG. Diagnosis of Barth syndrome using a novel LC-MS/MS method for leukocyte cardiolipin analysis. J Inherit Metab Dis. (2013) 36:741–6. doi: 10.1007/s10545-012-9552-4
17. Angelini R, Lobasso S, Gorgoglione R, Bowron A, Steward CG, Corcelli A. Cardiolipin fingerprinting of leukocytes by MALDI-TOF/MS as a screening tool for Barth syndrome. J Lipid Res. (2015) 56:1787–94. doi: 10.1194/jlr.D059824
18. VCV000928890.1—ClinVar—NCBI. Available at: https://www.ncbi.nlm.nih.gov/clinvar/variation/928890/?new_evidence=true (Cited 2023 Jan 9).
19. Angelini R, Russo S, Corcelli A, Lobasso S. Fingerprinting cardiolipin in leukocytes by mass spectrometry for a rapid diagnosis of Barth syndrome. J Vis Exp. (2022) (181). doi: 10.3791/63552
20. Miller PC, Ren M, Schlame M, Toth MJ, Phoon CKL. A Bayesian analysis to determine the prevalence of Barth syndrome in the pediatric population. J Pediatr. (2020) 217:139–44. doi: 10.1016/j.jpeds.2019.09.074
21. Rao S, Kanwal A, Padmanabhan S. Case report of Barth syndrome: a forgotten cause of cardiomyopathy. Eur Heart J Case Rep. (2021) 5:ytab195. doi: 10.1093/ehjcr/ytab195
22. Seitz A, Hinck A, Bekeredjian R, Sechtem U. Late diagnosis of Barth syndrome in a 39-year-old patient with non-compaction cardiomyopathy and neutropenia. ESC Heart Fail. (2020) 7:697–701. doi: 10.1002/ehf2.12588
23. Spencer CT, Bryant RM, Day J, Gonzalez IL, Colan SD, Thompson WR, et al. Cardiac and clinical phenotype in Barth syndrome. Pediatrics. (2006) 118:e337–46. doi: 10.1542/peds.2005-2667
24. Steward CG, Groves SJ, Taylor CT, Maisenbacher MK, Versluys B, Newbury-Ecob RA, et al. Neutropenia in Barth syndrome: characteristics, risks, and management. Curr Opin Hematol. (2019) 26:6–15. doi: 10.1097/MOH.0000000000000472
25. Wang S, Yazawa E, Keating EM, Mazumdar N, Hauschild A, Ma Q, et al. Genetic modifiers modulate phenotypic expression of tafazzin deficiency in a mouse model of Barth syndrome. Hum Mol Genet. (2023) 32:2055–67. doi: 10.1093/hmg/ddad041
26. Ghosh S, Iadarola DM, Ball WB, Gohil VM. Mitochondrial dysfunctions in Barth syndrome. IUBMB Life. (2019) 71:791–801. doi: 10.1002/iub.2018
Keywords: rare X-linked disease, TAFAZZIN, cardiolipin remodeling, cardiomyopathy, neutropenia
Citation: Tovaglieri N, Russo S, Micaglio E, Corcelli A and Lobasso S (2023) Case report: Variability in clinical features as a potential pitfall for the diagnosis of Barth syndrome. Front. Pediatr. 11:1250772. doi: 10.3389/fped.2023.1250772
Received: 30 June 2023; Accepted: 7 August 2023;
Published: 16 August 2023.
Edited by:
Khurshid Ahmad, Yeungnam University, Republic of KoreaReviewed by:
Atsuhito Takeda, Hokkaido University Hospital, Japan© 2023 Tovaglieri, Russo, Micaglio, Corcelli and Lobasso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simona Lobasso c2ltb25hLmxvYmFzc29AdW5pYmEuaXQ=
Abbreviations ACE, Angiotensin-converting enzyme; BTHS, Barth syndrome; CL, cardiolipin; DCM, dilated cardiomyopathy; EF, ejection fraction; G-CSF, Granulocyte Colony-Stimulating Factors; IVS, interventricular septum; LVEF, left ventricular ejection fraction; LVNC, left ventricular noncompaction; MAPSE, mitral annular plane systolic excursion; 3-MGCA, 3-methylglutaconic acid; MLCL, monolysocardiolipin; PW, pulsed-wave.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.