 Zhibin Li
Zhibin Li Yubo Wang
Yubo Wang Yuanhao Liu
Yuanhao Liu Yining Jiang
Yining Jiang Xuefei Han1
Xuefei Han1- 1Department of Neurosurgery, First Hospital of Jilin University, Changchun, China
- 2Department of Clinical Laboratory, Second Hospital of Jilin University, Changchun, China
Atypical teratoid/rhabdoid tumours (AT/RTs) are rare central nervous system neoplasms that frequently occur in infants and children and have a very poor prognosis. In recent years, molecular analysis of AT/RTs has shown that biallelic inactivation of SMARCB1 (INI1, SNF5, BAF47) or SMARCA4 (BRG1) frequently occurs. Here, we present a case of basal ganglia AT/RT with SMARCB1 gene deficiency and CDK6 gene amplification in a 5-year-old child. A 5-year-old boy was hospitalized due to a 1-week history of frontal and parietal headache. Magnetic resonance imaging (MRI) demonstrated a 3 cm × 2 cm × 1.5 cm heterogeneous enhanced mass located at the right basal ganglia that partially protruded into the right lateral ventricle. The lesion was successfully resected under electrophysiological monitoring and neuronavigation. The postoperative pathological examination implied an AT/RT diagnosis, with loss of SMARCB1 protein, SMARCB1 gene deficiency and CDK6 gene amplification. Unfortunately, the patient died due to respiratory and circulatory failure at 5 weeks after the operation. To date, standard regimens have not yet been established due to the lack of large-scale prospective studies for AT/RT. The p16-RB signalling pathway should be considered as a potential target for AT/RT treatment modalities. Apart from traditional regimens, targeted therapies, especially CDK4/6 inhibitors, are likely a promising therapeutic option for AT/RT treatment.
Introduction
Atypical teratoid/rhabdoid tumours (AT/RTs) are rare, aggressive, embryonal tumours of the central nervous system (CNS) that are characterized by rhabdoid cells and loss of SMARCB1 (INI1, SNF5, BAF47) or rare SMARCA4 (BRG1) protein (1–5). Rhabdoid tumours that occur in the CNS are defined as AT/RTs (6). They were initially reported in 1987 but recognized as a distinct tumour entity by the World Health Organization in 1993 (7, 8). The overall prognosis of AT/RT patients is poor, with a median survival time of approximately 17 months (9). AT/RT can occur in both infratentorial and supratentorial locations, but predominantly occurs in the posterior fossa, especially in the cerebellum and cerebellopontine angle (5, 10, 11). Biallelic SMARCB1 or rare SMARCA4 mutations, encoding core subunits of the SWI/SNF chromatin-remodelling complexes, are thought to be frequent genetic alterations in AT/RT (2, 12). Here, we present a case of AT/RT, which resulted in CDK6 gene amplification and, independently, SMARCB1 gene deficiency. Unfortunately, the patient died because of respiratory failure at 5 weeks after the operation. To the best of our knowledge, this study is the first report of a case of atypical teratoid/rhabdoid tumour with CDK6 amplification.
Case report
A 5-year-old boy was hospitalized due to a 1-week history of frontal and parietal intermittent headache. Based on physical examination, the right limbs were scored as a 5 and the left limbs as a 4 on the Lovett scale muscle grade. MRI showed a 3 cm × 2 cm × 1.5 cm large irregular cyst-solid lesion invading the right basal ganglia and thalamus and protruding into the right lateral ventricle. Uneven hypointense-to-isointense signals were observed on T1-weighted imaging (T1WI). Mixed isointense-to-hyperintense signals were shown on T2-weighted imaging (T2WI) (Figure 1A) and T2 dark-fluid imaging (Figure 1B), and hyperintense signals were observed on diffusion-weighted imaging (DWI) (Figure 1C); inverse signals were indicated by the apparent diffusion coefficient (ADC) (Figure 1D). After gadolinium administration, heterogeneous enhancement was observed, and a cyst was detected in the centre of the mass (Figures 1E–G).
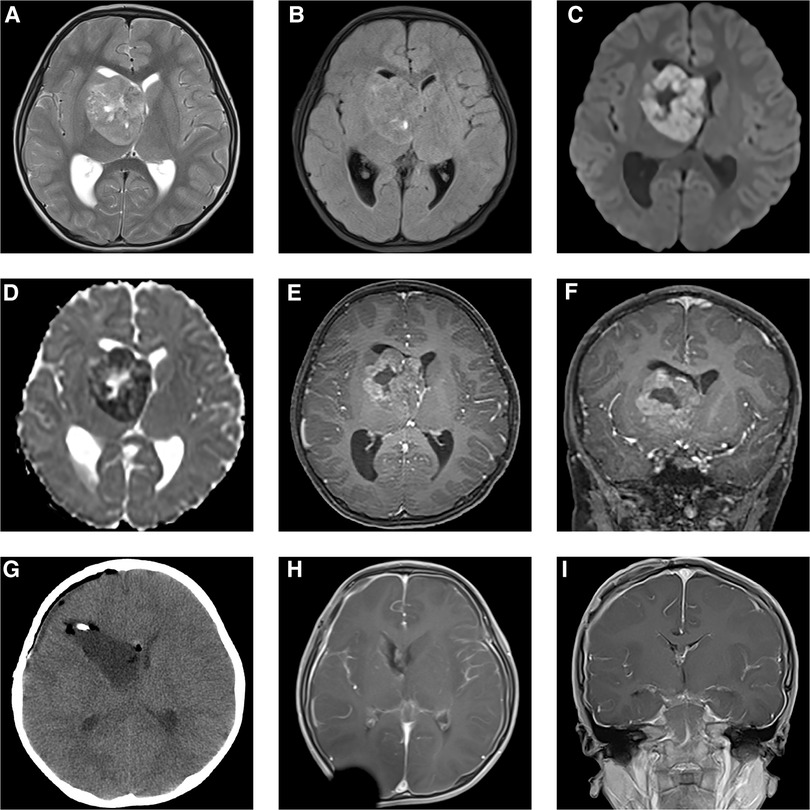
Figure 1. Preoperative MRI (A–F) demonstrated a 3 cm × 2 cm × 1.5 cm mass located in the right basal ganglia and thalamus, protruding into the right lateral ventricle. It was mixed isointense-to-hyperintense on T2WI (A) and T2 dark-fluid imaging (B). Hyperintense signals were shown by DWI (C), and inverse signals were indicated by the ADC (D). The tumour showed heterogeneous enhancement after gadolinium administration (E,F). Postoperative CT (G) and MRI 4 weeks after transfrontal-temporal craniotomy (H,I) suggested that the tumour was completely resected without signs of recurrence.
A preoperative diagnosis of glioblastoma was made. However, the diagnosis of AT/RT could not be excluded. Transfrontal-temporal craniotomy was performed under preoperative and intraoperative neuronavigation and intraoperative electrophysiological monitoring of the supratentorial functional area. The tumour was pinkish, cyst-solid, and bloody. Postoperative MRI showed gross total resection (Figures 1H,I).
The postoperative pathological analysis was suggestive of a diagnosis of AT/RT. Microscopic examination showed that the tumour comprised rhabdoid cells with eccentric nuclei, prominent nucleoli and eosinophilic cytoplasm (Figures 2A,B). Immunohistochemical examination indicated that the Ki-67 index was 80%, and INI-1 protein expression was deficient (Figure 2C). We also implemented next-generation sequencing (NGS) for diagnosis. Surprisingly, we not only found 2 categories of SMARCB1 gene mutations but also CDK6 gene amplification. One SMARCB1 gene mutation was exon 9 c.1130G > A p.R377H; the mutation abundance was 41.52%, which would dramatically reduce the combination of the C-terminal domain (CTD) of the SMARCB1 subunit, resulting in loss of functional proteins (13). SMARCB1 gene deficiency was also detected, and the copy number was 0.84 (Figure 3A). Moreover, we found that the CDK6 gene was amplified, with a copy number of 3.03 (Figure 3B). Integrating the histological, immunohistochemical and molecular examination, the AT/RT diagnosis was confirmed.
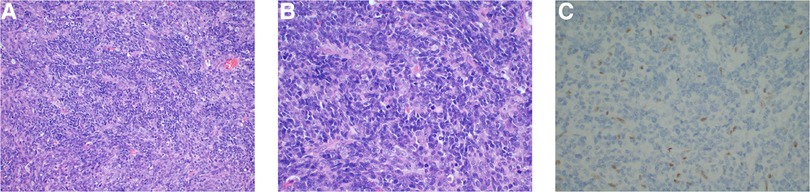
Figure 2. Haematoxylin and eosin-staining showed a large number of rhabdoid cells with eccentric nuclei, prominent nucleoli and eosinophilic cytoplasm (A,B). Immunohistochemical examination demonstrated INI-1 loss (C).
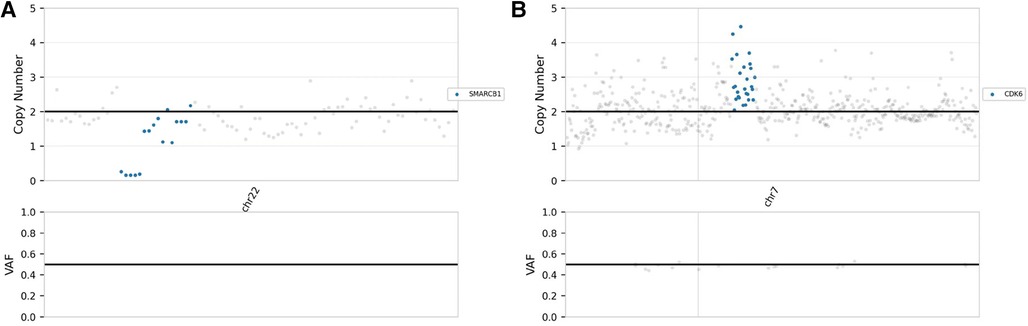
Figure 3. NGS demonstrated that the copy numbers of the SMARCB1 gene (A) and CDK6 gene (B) were 0.84 and 3.03, respectively.
A ventricular peritoneal shunt was implanted at 19 days after transfrontal-temporal craniotomy because of hydrocephalus. Nevertheless, this patient had frequent seizures and fever and entered a coma. Unfortunately, the patient died owing to respiratory and circulatory failure arising from pulmonary infection and hydrocephalus 5 weeks after the operation (Figure 4).
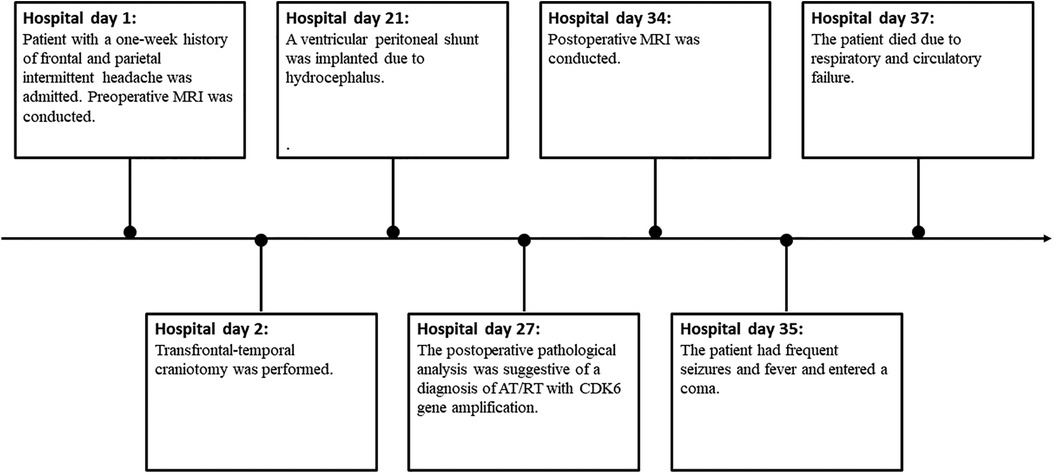
Figure 4. Timeline.
Discussion
The first time that a tumour entity was defined by molecular parameters other than histology was in the 2016 World Health Organization (WHO) classification of CNS tumours. The diagnosis of AT/RT required validation of specific molecular alterations, including inactivation of the SMARCB1 or SMARCA4 gene (14). Five years later, the fifth edition of the WHO classification of CNS tumours recognized 3 molecular subtypes of AT/RT, AT/RT-SHH, AT/RT-TYR and AT/RT-MYC, according to DNA methylation profiling and gene expression profiling. Using the WHO CNS tumour classification, the study by D'Arco et al. demonstrated that neuroimaging can help identify the tumour subtypes, providing information about prognosis and further management (15). Most AT/RTs demonstrate a hypointense signal on T1WI and variable signal on T2WI with heterogeneous contrast enhancement (15, 16). For one of the most important differential diagnoses, medulloblastoma, D'Arco et al. suggested that neuroimaging combined with clinical presentation could play an important role in differential diagnosis, as medulloblastoma occurred in an older age group than AT/RT (15). Other differential diagnoses should include choroid plexus tumours, other CNS embryonal tumours, high-grade gliomas, astrocytoma and ependymal tumours (17–19). For this patient, the preoperative diagnosis was glioblastoma. However, the diagnosis of AT/RT could not be excluded, and pathological examination indicated a diagnosis of AT/RT. To further confirm the diagnosis, we conducted NGS to validate the specific molecular alterations. Overall, integrating the clinical presentation, neuroimaging, pathological examination and NGS results, a diagnosis of AT/RT was made, which implied a very poor prognosis. After referring to the literature, we considered that selective CDK4/6 inhibitors might improve the prognosis of this child because of the CDK6 gene amplification detected by NGS. Unfortunately, the patient succumbed to respiratory and circulatory failure about 5 weeks after the operation, and the intervention of selective CDK4/6 inhibitors was not able to be administered.
AT/RT is a category of rare, aggressive, embryonal brain neoplasms. A study examining data from 13 European countries reported that the median age at AT/RT diagnosis is 29.5 months (20). The 5-year overall survival (OS) and event-free survival (EFS) are 34.7 ± 4.5% and 30.5 ± 4.2%, respectively (20). AT/RT comprises only 1%–2% of all paediatric CNS neoplasms but approximately 10%–20% of brain neoplasms in patients under 3 years old (21). Approximately 70% of cases occur in patients younger than 1 year old, with more than 90% in those younger than 3 years old (11).
Mutation of subunits of SWI/SNF chromatin-remodelling complexes is the central event in AT/RT. Studies in genetically engineered mouse models show that SMARCB1 is a genuine tumour suppressor gene (22–24). Biallelic mutations of SMARCB1 or SMARCA4 are frequent genetic alterations in AT/RTs (2, 25). The tumorigenesis of AT/RT follows a classical two-hit model (2). Approximately 35% of cases are identified as heterozygous germline mutations of SMARCB1 or SMARCA4, which constitutes the first hit (2). Another study demonstrated that germline mutations in AT/RTs can be identified at any age, and 60% of children under the age of 6 months are prone to multiple rhabdoid tumours (26). Therefore, genetic counselling is recommended for surveillance and identifying implications in pregnancies. In addition, the presence of germline mutations requires AT/RT patients to undergo whole-body examinations to detect potential extracranial lesions.
Due to the lack of large-scale prospective studies for AT/RT, a standard treatment regimen has not yet been established (27). Total tumour resection forms the typical basis for therapy (28–30). Although the implementation of radiotherapy in AT/RT patients remains controversial due to severe neurocognitive complications, the lack of alternative treatments has necessitated the continued use of radiotherapy as an important modality in AT/RT therapeutic regimens (27, 30). The efficacy of intensive chemotherapy and focal radiotherapy have been validated, with 37% 4-year EFS and 43% overall survival (31). This study, the first AT/RT-specific cooperative group trial, included 65 patients and identified high-dose chemotherapy with peripheral blood stem cell rescue (HDC/SCR) and involved-field radiotherapy as an important treatment modality for AT/RT.
SMARCB1 is implicated in many pathways, particularly the p16-RB pathway (32). The p16 tumour suppressor protein inhibits CDK4/6 (32). SMARCB1 is critical for regulating the expression of CDKN2A, which encodes p16 (33, 34). A principal consequence of SMARCB1 genetic inactivation is the inhibition of p16 expression, which correspondingly increases the activation of CDK4/6, facilitating the phosphorylation and inactivation of retinoblastoma tumour suppressor protein (RB) (35). Phosphorylated RB cannot suppress the E2F protein, which is required for progression from G1 to S phase, and thus, cell division is launched (32–34). The amplification or overexpression of CDK4/6 has been observed in several malignancies, including breast neoplasms, sarcoma, glioma, lymphoma and melanoma (36). In brain neoplasms, CDK6 amplification is frequently found in medulloblastoma glioma and glioblastoma (37–40). The NGS result of the present case is the first to show CDK6 amplification in AT/RT. The amplification of the CDK6 gene is related to high levels of CDK6 protein, which may increase the constitutive phosphorylation of RB, overriding p16 inhibition (41). Studies by Mendrzyk et al. and Ruano et al. suggested that CDK6 may promote the prognostic evaluation of medulloblastoma and glioblastoma, respectively (39, 40). This finding indicates that CDK6 may serve as a prognostic marker in AT/RT.
The use of CDK4/6 inhibitors palbociclib, ribociclib, and abemaciclib represents a major breakthrough in breast cancer therapeutics (42). More recently, CDK4/6 inhibitors have attracted attention as a potential option for AT/RT therapeutic regimens. Palbociclib is an oral selective inhibitor of CDK4/6, resulting in reduced RB phosphorylation and G1 phase arrest (43). A preclinical study by Hashizume et al. showed that palbociclib combined with irradiation therapy distinctly outperformed both therapies alone in terms of survival benefit to animal subjects. Furthermore, the study suggested an additional mechanism of palbociclib against tumours in which palbociclib expanded the period during which tumour cell irradiation impairs DNA double-strand breaks, supporting the application of palbociclib as a potential adjuvant to radiotherapy for AT/RT patients (35). Anderson et al. reported a case of asymptomatic nonsecreting pituitary adenoma and metastatic breast cancer treated with palbociclib and found regression of pituitary adenoma 1 year after initiating palbociclib (44). Although it is uncertain whether palbociclib contributes to the regression of pituitary adenoma, CDK6 was identified by another study by Zhang et al. as a potential therapeutic target of pituitary neuroendocrine tumours (45). In addition, palbociclib has been identified to be effective against several types of CNS neoplasms, including glioma, glioblastoma, medulloblastoma and breast cancer brain metastases (35, 46–48). Ribociclib is another CDK4/6 inhibitor, resembling palbociclib (49). A phase I study by Geoerger et al. on malignant rhabdoid tumours revealed prolonged stable disease in paediatric patients with an oral single-agent ribociclib regimen (50). One other phase 0 trial suggested that ribociclib demonstrated good CNS penetration across not only a disrupted blood-brain barrier (BBB) but also an intact BBB in human glioblastoma regions, which is a promising result and encourages us to explore ribociclib for treating AT/RT (49). Overall, selective CDK4/6 inhibitors may improve the prognosis of AT/RT patients. Palbociclib and ribociclib may represent promising molecular agents against AT/RT, especially for the present case with CDK6 gene amplification. Nevertheless, further clinical trials should be conducted to determine the validity of these agents for AT/RT treatment and their permeability of the BBB.
Conclusion
Since AT/RT is a category of CNS neoplasm with very poor prognosis, long-term follow-up, prospective studies and clinical trials are warranted to fully understand this disease. We highlighted the function of CDK4/6 in the tumorigenesis of AT/RT and the importance of CDK4/6 inhibitors as a potential option for AT/RT therapeutic regimens, serving as a potential guide for clinicians and clinical studies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/sra, PRJNA936291.
Ethics statement
The studies involving humans were approved by Ethics Committee of the First Hospital of Jilin University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
ZL, YW, and YLiu made study design, data collection, data analysis and interpretation, and composed the manuscript and literature review. YLi was the surgeon that performed the surgery and did data collection, data analysis, and interpretation. YJ and XH made English and grammar corrections, critical revisions, and approved final version. LZ and YLi had the acquisition, analysis or interpretation of data for the work, revising it critically for important intellectual content, final approval of the version to be published, and agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy, or integrity of any part of the work are appropriately investigated and resolved. All authors contributed to the article and approved the submitted version.
Acknowledgments
The authors sincerely thank the patient for his participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cacciotti C, Fleming A, Ramaswamy V. Advances in the molecular classification of pediatric brain tumors: a guide to the galaxy. J Pathol. (2020) 251(3):249–61. doi: 10.1002/path.5457
2. Frühwald MC, Biegel JA, Bourdeaut F, Roberts CW, Chi SN. Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro Oncol. (2016) 18(6):764–78. doi: 10.1093/neuonc/nov264
3. Thomas C, Knerlich-Lukoschus F, Reinhard H, Johann PD, Sturm D, Sahm F, et al. Two molecularly distinct atypical teratoid/rhabdoid tumors (or tumor components) occurring in an infant with rhabdoid tumor predisposition syndrome 1. Acta Neuropathol. (2019) 137(5):847–50. doi: 10.1007/s00401-019-02001-3
4. Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med. (2020) 26(5):712–9. doi: 10.1038/s41591-020-0821-8
5. Takei H, Adesina AM, Mehta V, Powell SZ, Langford LA. Atypical teratoid/rhabdoid tumor of the pineal region in an adult. J Neurosurg. (2010) 113(2):374–9. doi: 10.3171/2009.10.JNS09964
6. Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. (1996) 85(1):56–65. doi: 10.3171/jns.1996.85.1.0056
7. Biggs PJ, Garen PD, Powers JM, Garvin AJ. Malignant rhabdoid tumor of the central nervous system. Hum Pathol. (1987) 18(4):332–7. doi: 10.1016/S0046-8177(87)80161-2
8. Kleihues P, Burger PC, Scheithauer BW. The new WHO classification of brain tumours. Brain Pathol. (1993) 3(3):255–68. doi: 10.1111/j.1750-3639.1993.tb00752.x
9. Ginn KF, Gajjar A. Atypical teratoid rhabdoid tumor: current therapy and future directions. Front Oncol. (2012) 2:114. doi: 10.3389/fonc.2012.00114
10. Allen JC, Judkins AR, Rosenblum MK, Biegel JA. Atypical teratoid/rhabdoid tumor evolving from an optic pathway ganglioglioma: case study. Neuro Oncol. (2006) 8(1):79–82. doi: 10.1215/S1522851705000347
11. Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. (2016) 29(3):379–93. doi: 10.1016/j.ccell.2016.02.001
12. Mittal P, Roberts CWM. The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat Rev Clin Oncol. (2020) 17(7):435–48. doi: 10.1038/s41571-020-0357-3
13. Valencia AM, Collings CK, Dao HT, St Pierre R, Cheng YC, Huang J, et al. Recurrent SMARCB1 mutations reveal a nucleosome acidic patch interaction site that potentiates mSWI/SNF complex chromatin remodeling. Cell. (2019) 179(6):1342–56.e23. doi: 10.1016/j.cell.2019.10.044
14. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
15. D'Arco F, Khan F, Mankad K, Ganau M, Caro-Dominguez P, Bisdas S. Differential diagnosis of posterior fossa tumours in children: new insights. Pediatr Radiol. (2018) 48(13):1955–63. doi: 10.1007/s00247-018-4224-7
16. Au Yong KJ, Jaremko JL, Jans L, Bhargava R, Coleman LT, Mehta V, et al. How specific is the MRI appearance of supratentorial atypical teratoid rhabdoid tumors? Pediatr Radiol. (2013) 43(3):347–54. doi: 10.1007/s00247-012-2530-z
17. Calandrelli R, Massimi L, Pilato F, Verdolotti T, Ruggiero A, Attinà G, et al. Atypical teratoid rhabdoid tumor: proposal of a diagnostic pathway based on clinical features and neuroimaging findings. Diagnostics. (2023) 13(3):475. doi: 10.3390/diagnostics13030475
18. Zuccoli G, Izzi G, Bacchini E, Tondelli MT, Ferrozzi F, Bellomi M. Central nervous system atypical teratoid/rhabdoid tumour of infancy. CT and mr findings. Clin Imaging. (1999) 23(6):356–60. doi: 10.1016/S0899-7071(99)00165-5
19. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
20. Frühwald MC, Hasselblatt M, Nemes K, Bens S, Steinbügl M, Johann PD, et al. Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol. (2020) 22(7):1006–17. doi: 10.1093/neuonc/noz244
21. Udaka YT, Packer RJ. Pediatric brain tumors. Neurol Clin. (2018) 36(3):533–56. doi: 10.1016/j.ncl.2018.04.009
22. Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci U S A. (2000) 97(25):13796–800. doi: 10.1073/pnas.250492697
23. Klochendler-Yeivin A, Fiette L, Barra J, Muchardt C, Babinet C, Yaniv M. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. (2000) 1(6):500–6. doi: 10.1093/embo-reports/kvd129
24. Guidi CJ, Sands AT, Zambrowicz BP, Turner TK, Demers DA, Webster W, et al. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol. (2001) 21(10):3598–603. doi: 10.1128/MCB.21.10.3598-3603.2001
25. Hasselblatt M, Isken S, Linge A, Eikmeier K, Jeibmann A, Oyen F, et al. High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer. (2013) 52(2):185–90. doi: 10.1002/gcc.22018
26. Bourdeaut F, Lequin D, Brugières L, Reynaud S, Dufour C, Doz F, et al. Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res. (2011) 17(1):31–8. doi: 10.1158/1078-0432.CCR-10-1795
27. Hoffman LM, Richardson EA, Ho B, Margol A, Reddy A, Lafay-Cousin L, et al. Advancing biology-based therapeutic approaches for atypical teratoid rhabdoid tumors. Neuro Oncol. (2020) 22(7):944–54. doi: 10.1093/neuonc/noaa046
28. Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, et al. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. (2004) 22(14):2877–84. doi: 10.1200/JCO.2004.07.073
29. Lafay-Cousin L, Hawkins C, Carret AS, Johnston D, Zelcer S, Wilson B, et al. Central nervous system atypical teratoid rhabdoid tumours: the Canadian paediatric brain tumour consortium experience. Eur J Cancer. (2012) 48(3):353–9. doi: 10.1016/j.ejca.2011.09.005
30. Nesvick CL, Lafay-Cousin L, Raghunathan A, Bouffet E, Huang AA, Daniels DJ. Atypical teratoid rhabdoid tumor: molecular insights and translation to novel therapeutics. J Neurooncol. (2020) 150(1):47–56. doi: 10.1007/s11060-020-03639-w
31. Reddy AT, Strother DR, Judkins AR, Burger PC, Pollack IF, Krailo MD, et al. Efficacy of high-dose chemotherapy and three-dimensional conformal radiation for atypical teratoid/rhabdoid tumor: a report from the children’s oncology group trial ACNS0333. J Clin Oncol. (2020) 38(11):1175–85. doi: 10.1200/JCO.19.01776
32. Kohashi K, Oda Y. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Sci. (2017) 108(4):547–52. doi: 10.1111/cas.13173
33. Oruetxebarria I, Venturini F, Kekarainen T, Houweling A, Zuijderduijn LM, Mohd-Sarip A, et al. P16INK4a is required for hSNF5 chromatin remodeler-induced cellular senescence in malignant rhabdoid tumor cells. J Biol Chem. (2004) 279(5):3807–16. doi: 10.1074/jbc.M309333200
34. Betz BL, Strobeck MW, Reisman DN, Knudsen ES, Weissman BE. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene. (2002) 21(34):5193–203. doi: 10.1038/sj.onc.1205706
35. Hashizume R, Zhang A, Mueller S, Prados MD, Lulla RR, Goldman S, et al. Inhibition of DNA damage repair by the CDK4/6 inhibitor palbociclib delays irradiated intracranial atypical teratoid rhabdoid tumor and glioblastoma xenograft regrowth. Neuro Oncol. (2016) 18(11):1519–28. doi: 10.1093/neuonc/now106
36. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. (2009) 9(3):153–66. doi: 10.1038/nrc2602
37. Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. (2016) 45:129–38. doi: 10.1016/j.ctrv.2016.03.002
38. Wiedemeyer WR, Dunn IF, Quayle SN, Zhang J, Chheda MG, Dunn GP, et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc Natl Acad Sci U S A. (2010) 107(25):11501–6. doi: 10.1073/pnas.1001613107
39. Mendrzyk F, Radlwimmer B, Joos S, Kokocinski F, Benner A, Stange DE, et al. Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. J Clin Oncol. (2005) 23(34):8853–62. doi: 10.1200/JCO.2005.02.8589
40. Ruano Y, Mollejo M, Ribalta T, Fiaño C, Camacho FI, Gómez E, et al. Identification of novel candidate target genes in amplicons of glioblastoma multiforme tumors detected by expression and CGH microarray profiling. Mol Cancer. (2006) 5:39. doi: 10.1186/1476-4598-5-39
41. Faast R, White J, Cartwright P, Crocker L, Sarcevic B, Dalton S. Cdk6-cyclin D3 activity in murine ES cells is resistant to inhibition by p16(INK4a). Oncogene. (2004) 23(2):491–502. doi: 10.1038/sj.onc.1207133
42. Spring LM, Wander SA, Andre F, Moy B, Turner NC, Bardia A. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. Lancet. (2020) 395(10226):817–27. doi: 10.1016/S0140-6736(20)30165-3
43. Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. (2004) 3(11):1427–38. doi: 10.1158/1535-7163.1427.3.11
44. Anderson E, Heller RS, Lechan RM, Heilman CB. Regression of a nonfunctioning pituitary macroadenoma on the CDK4/6 inhibitor palbociclib: case report. Neurosurg Focus. (2018) 44(6):E9. doi: 10.3171/2018.2.FOCUS17660
45. Zhang F, Zhang Q, Zhu J, Yao B, Ma C, Qiao N, et al. Integrated proteogenomic characterization across major histological types of pituitary neuroendocrine tumors. Cell Res. (2022) 32(12):1047–67. doi: 10.1038/s41422-022-00736-5
46. Whiteway SL, Harris PS, Venkataraman S, Alimova I, Birks DK, Donson AM, et al. Inhibition of cyclin-dependent kinase 6 suppresses cell proliferation and enhances radiation sensitivity in medulloblastoma cells. J Neurooncol. (2013) 111(2):113–21. doi: 10.1007/s11060-012-1000-7
47. Sun Y, Sun Y, Yan K, Li Z, Xu C, Geng Y, et al. Potent anti-tumor efficacy of palbociclib in treatment-naïve H3.3K27M-mutant diffuse intrinsic pontine glioma. EBioMedicine. (2019) 43:171–9. doi: 10.1016/j.ebiom.2019.04.043
48. Figura NB, Potluri TK, Mohammadi H, Oliver DE, Arrington JA, Robinson TJ, et al. CDK 4/6 inhibitors and stereotactic radiation in the management of hormone receptor positive breast cancer brain metastases. J Neurooncol. (2019) 144(3):583–9. doi: 10.1007/s11060-019-03260-6
49. Tien AC, Li J, Bao X, Derogatis A, Kim S, Mehta S, et al. A phase 0 trial of ribociclib in recurrent glioblastoma patients incorporating a tumor pharmacodynamic- and pharmacokinetic-guided expansion cohort. Clin Cancer Res. (2019) 25(19):5777–86. doi: 10.1158/1078-0432.CCR-19-0133
50. Geoerger B, Bourdeaut F, DuBois SG, Fischer M, Geller JI, Gottardo NG, et al. A phase I study of the CDK4/6 inhibitor ribociclib (LEE011) in pediatric patients with malignant rhabdoid tumors, neuroblastoma, and other solid tumors. Clin Cancer Res. (2017) 23(10):2433–41. doi: 10.1158/1078-0432.CCR-16-2898
Keywords: atypical teratoid/rhabdoid tumour, case report, central nervous system neoplasms, CDK6, treatment, CDK4/6 inhibitors
Citation: Li Z, Wang Y, Liu Y, Jiang Y, Han X, Zhao L and Li Y (2023) Atypical teratoid/rhabdoid tumour with CDK6 amplification in a child: a case report and literature review. Front. Pediatr. 11:1237572. doi: 10.3389/fped.2023.1237572
Received: 9 June 2023; Accepted: 8 August 2023;
Published: 22 August 2023.
Edited by:
Flavio Giordano, University of Florence, ItalyReviewed by:
Xiaojuan Wu, Huazhong University of Science and Technology, ChinaMario Ganau, Oxford University Hospitals NHS Trust, United Kingdom
© 2023 Li, Wang, Liu, Jiang, Han, Zhao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liyan Zhao emhhb2xpeUBqbHUuZWR1LmNu Yunqian Li eXVucWlhbkBqbHUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship