 Jia-Qi Li
Jia-Qi Li Jia-Yan Feng2
Jia-Yan Feng2- 1The Center for Pediatric Liver Diseases, Children’s Hospital of Fudan University, Shanghai, China
- 2Department of Pathology, Children’s Hospital of Fudan University, Shanghai, China
- 3Department of Radiology, Children’s Hospital of Fudan University, Shanghai, China
- 4Department of Infectious Diseases, Anhui Provincial Children’s Hospital, Hefei, China
DGUOK deficiency has primarily been associated with lethal hepatic failure with or without hypotonia, nystagmus, and psychomotor retardation, features typical of mitochondrial disease. A study in 3 Turkish children identified homozygosity for a variant in DGUOK as associated with idiopathic non-cirrhotic portal hypertension (INCPH). However, no further instances of INCPH associated with DGUOK variants have been reported. We here describe a fourth patient with DGUOK variants and childhood-onset INCPH, a 12-year-old Han Chinese boy, reporting clinical manifestations, histopathologic findings, and results of genetic studies. The child presented with hepatosplenomegaly; portal hypertension and hypersplenism were found. Vascular changes with hepatic fibrosis (Scheuer score 3) were observed on liver biopsy. Whole-exome sequencing and family analyses revealed compound heterozygosity for the DGUOK (NM_080916.3) variants c.778_781dup, (p.Thr261Serfs*28) and c.831_832del, (p.*278Thrfs*9) in the proband. These observations support ascription of instances of INCPH in children to variation in DGUOK.
Introduction
Deoxyguanosine kinase, encoded by the nuclear gene DGUOK (1), mediates the first step in the phosphorylation of purine nucleosides in the mitochondrial matrix and is essential for the purine nucleoside salvage pathway (2, 3). Variants in DGUOK lead to impaired synthesis of mitochondrial dNTPs, resulting in decreased levels of mitochondrial DNA and depletion of mitochondrial DNA (2). More than 100 individuals with DGUOK-related mitochondrial DNA depletion syndrome (MDS) have been reported thus far (4). The typical characteristics of this disorder include significant hepatic failure with or without hypotonia, nystagmus, and psychomotor retardation (2, 5).
Three children from 2 Turkish kindreds with the recurrent recessive homozygous p.N46S mutation in DGUOK, leading to idiopathic non-cirrhotic portal hypertension (INCPH), are described (6). INCPH is characterized by intrahepatic portal hypertension when cirrhosis and other causes of liver disease or splanchnic venous thrombosis are absent (7). Findings in the 3 Turkish children thus expanded the phenotypic spectrum of DGUOK deficiency and suggested a new cause of INCPH. However, no further cases have been reported. We here describe a fourth patient, a 12-year-old Han Chinese boy, in whom pediatric-onset INCPH is associated with DGUOK variation.
Case description
Patient
The proband was a 12-year-old boy, the first child of a non-consanguineous healthy Han Chinese couple. He was born vaginally at term (3,150 g) following an uncomplicated pregnancy. Early growth and development were unremarkable. On examination at age 4 years 9 months, occasioned by bronchopneumonia, hepatosplenomegaly was noted incidentally (liver edge 3.3 cm and spleen tip 3.2 cm below costal margin). Aside from a slightly elevated alpha-fetoprotein value [183 ng/ml; reference 0–13.6], laboratory results were within expected ranges (Table 1). Abdominal sonography and computerized tomography found only nonspecific liver heterogeneity. Laboratory findings during an episode of upper respiratory infection, aged 8 years, were within expected ranges. Aged 12 years, during another bout of respiratory-tract infection, leukopenia and thrombocytopenia were identified, with mild unconjugated hyperbilirubinemia and slight hypocoagulability. Aside from slightly elevated serum γ-glutamyl transpeptidase activity, values for biomarkers of hepatobiliary injury and hepatic synthetic function were within expected ranges (Table 1). Abdominal contrast-enhanced computerized tomography imaging found enlargement of the spleen, accompanied by thickening of the splenic meridians and varicose gastric veins, indicating portal hypertension (Supplementary Figure S1A). The patient's parents refused determination of portal venous system and inferior vena cava pressures. Esophageal and gastric varices were not found on endoscopy (Supplementary Figure S1B). Liver stiffness, measured using FibroScan, was 14.6 kPa (reference value: <7.3 kPa; fibrosis stage 3–4) (8). Bone marrow biopsy found hyperplasia with normal megakaryocytes. After liver biopsy (v.i.), the patient was referred in consultation. Repeat evaluation found that normal growth and development were normal, with height 165 cm, 2 standard deviations above the mean, and weight 52.9 kg, 1–2 standard deviations above the mean. Hepatosplenomegaly was found (liver edge 2 cm and spleen tip 4 cm below costal margin), without other feature of note. No abnormality was identified on specialist neurological examination. Routine tests of urine and feces found no abnormality. A reticulocyte count yielded normal results. Values for serum creatine phosphokinase activity and for biomarkers of thyroid function were within expected ranges, as were those for glucose, lactate, ketones, triglycerides, blood ammonia, cholesterol, homocysteine, ferritin, folic acid, ceruloplasmin, complement 3, complement 4 and CD series. The distribution and amounts of urinary organic acids and plasma amino acids were normal on assessment by mass spectrophotometry. No serologic evidence was found for infection by hepatotropic viruses, Epstein-Barr virus, toxoplasma, rubella, cytomegalovirus, herpes, human immunodeficiency virus, syphilis, or mycoplasma. Autoantibodies could not be demonstrated (anti-smooth muscle, anti-dsDNA, anti-liver kidney microsomal, anti-soluble liver pancreas, anti-cytosolic, anti-tissue transglutaminase) except for anti-nuclear antibody (positive at 1:160 titer). No abnormalities were found on cardiac sonography, Wechsler intelligence scale testing, brain magnetic resonance imaging, or electroencephalography. The boy's sister, aged 5 years, is without abnormality on physical examination and clinical-laboratory assessment; findings on abdominal sonography were normal. Questioning elicited no history of neurologic or hepatobiliary disease in the proband's family and relatives.
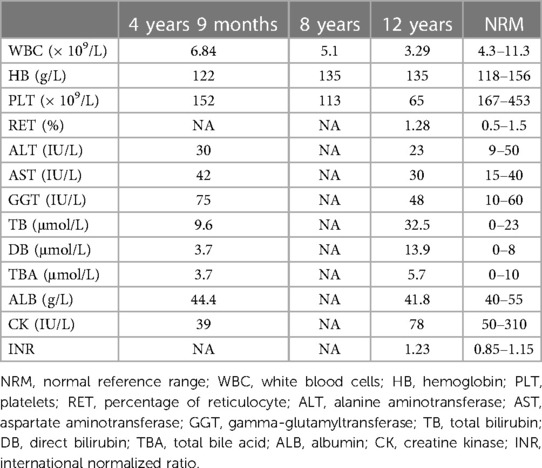
Table 1. Results, laboratory testing.
Liver biopsy
An ultrasound-guided percutaneous liver puncture was performed when the patient was 12 years old. Figure 1 and Supplementary Figure S2 show vascular changes with hepatic fibrosis (Staging 3, Scheuer's system) (9). However, cirrhosis was not present. Dilatation of hepatic sinusoids with irregular blood vessels also was seen. Some hepatocytes showed mild swelling with small fat droplets/microsteatosis. Portal tracts were not inflamed.
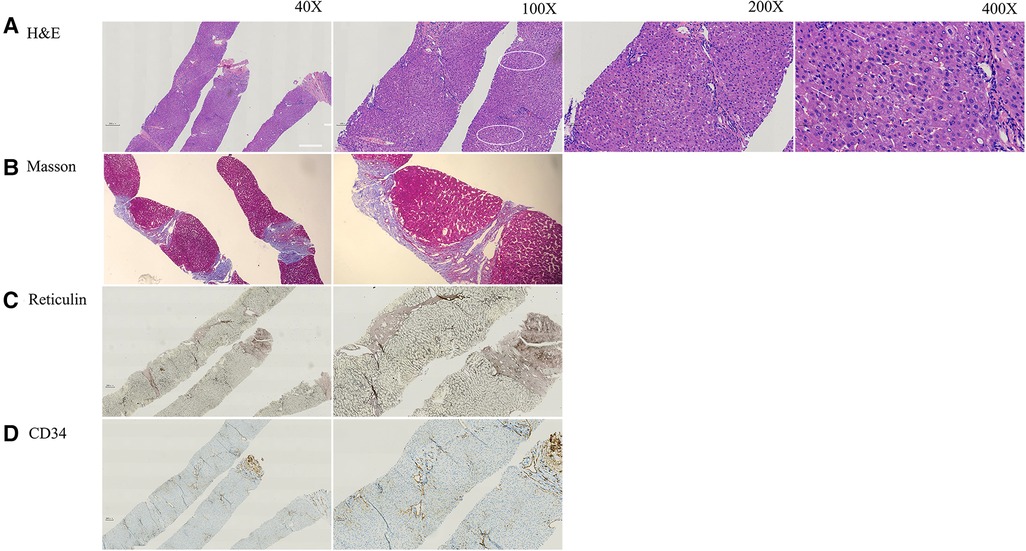
Figure 1. The pathology of current patient at age of 12 years. (A) H&E staining indicated mild hepatocyte swelling (arrow) and areas of mild steatosis with mild sinusoidal dilatation around central vein (circle) with no inflammation or cirrhosis. (B) Masson staining showed fibrosis (Scheuer score 3). (C) Reticular fiber staining showed the exist of reticular scaffold structure without obvious collapse. (D) CD34 antibody marks the irregular outlines of the portal venules and narrow lumen.
Genetic finding
Whole-exome sequencing revealed compound heterozygosity for the novel variants in DGUOK (NM_080916.3) c.778_781dup, (p.Thr261Serfs*28) and c.831_832del, (p.*278Thrfs*9). These are not recorded in the public databases Exome Aggregation Consortium Server (http://exac.broadinstitute.org/), Genome Aggregation Database (https://gnomad.broadinstitute.org/), NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), Thousand Genomes Project (http://www.1000genomes.org/home), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), DECIPHER database (https://www.deciphergenomics.org/), or Leiden Open Variation Database (https://www.lovd.nl/). The distribution of variants in the proband and, as confirmed by Sanger sequencing, in his sister and parents indicated a recessive mode of inheritance (Figure 2). American College of Medical Genetics and Genomics guidelines (10) classified the c.778_781dup and c.831_832del as “likely pathogenic” and “of uncertain significance”, respectively. No other pathogenic variants consistent with the mode of inheritance were identified.
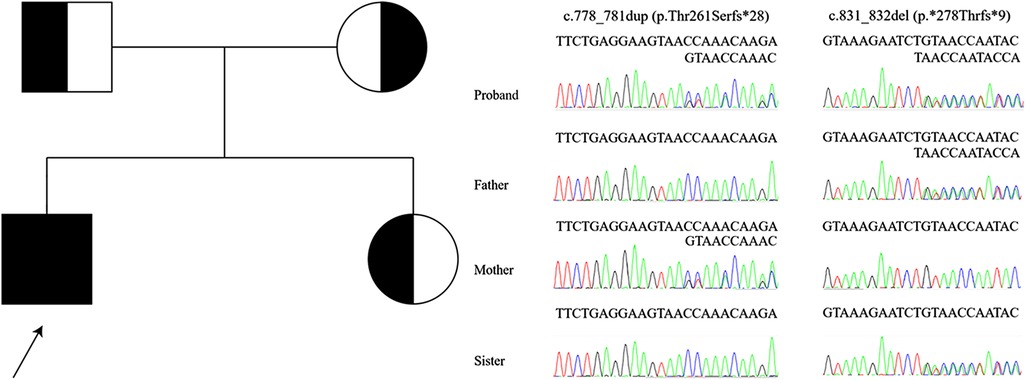
Figure 2. Mutated region, DGUOK, in proband, sister, and parents, with pedigree.
Discussion
DGUOK deficiency has primarily been associated with clinical features that typify mitochondrial diseases, with liver, neurological, and muscular systems involvement. In most cases, DGUOK related MDS is characterized by onset in infancy or childhood of progressive liver disease (hepatomegaly, cholestasis, elevated transaminase activities, and liver failure/cirrhosis), with neuromuscular manifestations (hypotonia, nystagmus, and psychomotor retardation), hypoglycemia, hyperlactatemia, and hypertyrosinemia (11, 12). Death in liver failure generally occurs before age 4 years (13). A single report has appeared of 3 Turkish children with INCPH and homozygosity for a DGUOK variant, c.137A > G, predicted to yield the substitution p.N46S (6). INCPH is characterized by portal hypertension in the absence of cirrhosis, hepatic synthetic dysfunction, myopathy, or neurological impairment (6).
Hepatosplenomegaly and sonographically heterogeneous liver were found incidentally in our patient at age 4 years 9 months. Biomarker values were then unremarkable, as they were again at age 8 years. At age 12 years hypersplenism was identified, with slight unconjugated hyperbilirubinemia and mild impairment of coagulation. Seijo et al. had reported that the mean liver stiffness values in INCPH patients was 8.4 ± 3.3 kPa (14). The liver stiffness value was relatively higher in our patient (14.6 kPa). The value of hepatic pressure gradient was not performed as rejected by the child's parents. Studies with more cases are still needed to evaluate the values of liver stiffness and hepatic pressure gradient in DGUOK related INCPH. Liver biopsy found architectural changes that comported with INCPH. This prompted genetic studies that identified compound heterozygosity for 2 unreported DGUOK variants, both predicted to yield frameshifts. As with the 3 Turkish children (Table 2), illness was clinically mild and progression was slow. Factors affecting penetrance of DGUOK disease remain to be identified.
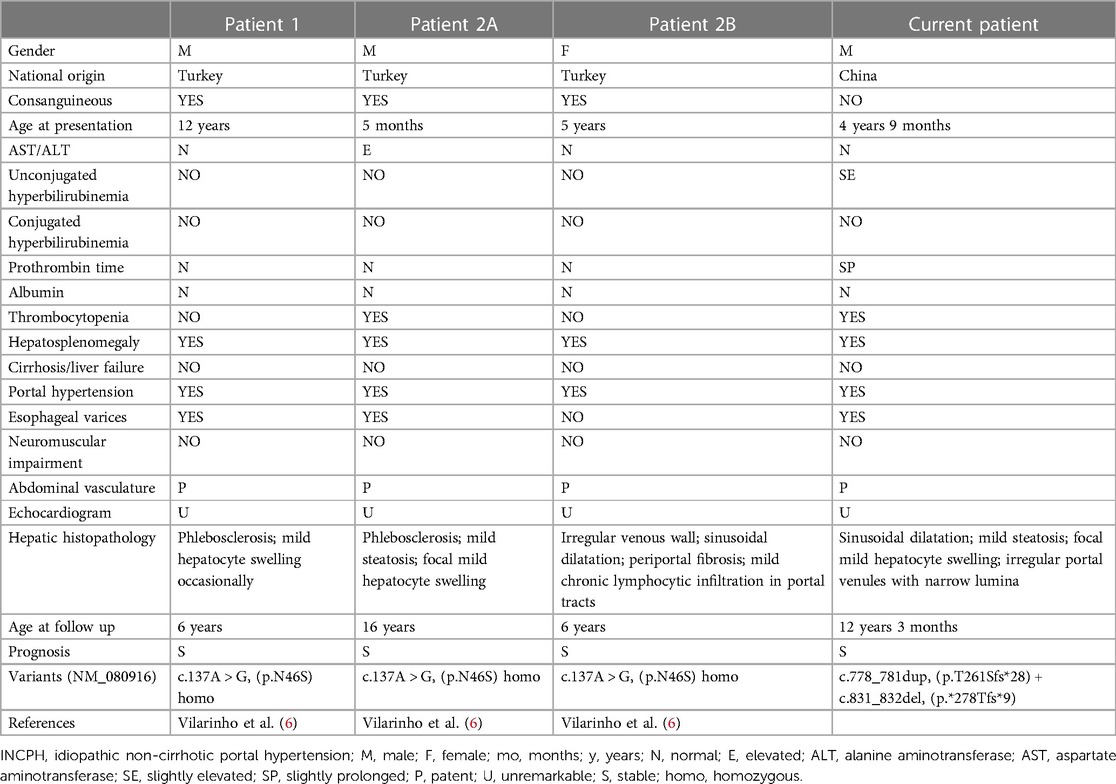
Table 2. Clinical features and molecular genetics of individuals with INCPH associated with variants in DGUOK.
Isolated liver involvement has been found in our patient as well as in the 3 Turkish children reported previously (6). Characteristic histopathologic abnormalities on liver biopsy in DGUOK-related MDS include cholestasis, microsteatosis, giant cell hepatitis, fibrosis, and cirrhosis (5, 11–13, 15–17). Unlike DGUOK-related MDS, liver biopsy in the 3 Turkish children found subtle vascular changes in the absence of significant cirrhosis (6), including portal changes with irregular portal-venule profiles, lumen narrowing, smooth-muscle proliferation, and mural fibrosis. One patient had mild focal microsteatosis and hepatocyte swelling, while another patient showed sinusoidal dilatation and mild chronic lymphocytic infiltration in portal tracts without significant interface activity. Our patient had mild focal hepatocyte swelling and microsteatosis with sinusoidal dilatation and subtle vascular changes with fibrosis but without cirrhosis (Scheuer's score 3), suggesting a chronic liver disease with concomitant signs of “INCPH”.
The diagnosis of INCPH is mainly based on the presence of portal hypertension in the absence of cirrhosis or other causes of non-cirrhotic portal hypertension, and include the histologic diagnosis of obliterative portal venopathy (18). At the Baveno VII Consensus workshop on portal hypertension, the term porto-sinusoidal vascular disorder (PVSD) was described (19). PSVD is a broad clinico-pathological entity encompassing INCPH, and various overlapping histological patterns including nodular regenerative hyperplasia, obliterative portal venopathy, hepatoportal sclerosis, incomplete septal cirrhosis with or without portal hypertension (19, 20). The pathology in the current case is consistent with the diagnosis of INCPH/PVSD. Known causes of INCPH or PVSD include immunological disorders, chronic infections, exposure to medications or toxins, prothrombotic conditions, and genetic diseases (7, 20, 21). On the limited basis of the 4 cases identified to date, DGUOK-related INCPH (MIM617068) seems to be an autosomal recessive disorder characterized by the onset of portal hypertension and hepatosplenomegaly in the first or second decades of life with no extrahepatic manifestations aside from hypersplenism. The exact pathogenesis of DGUOK-related INCPH is unknown. However, the disease is relatively benign, with slow disease progression, unlike typical MDS.
The two frameshift variants identified in the patient were novel. Each lies near the end of the coding region; each is predicted to extend the coding sequence and to produce an elongated protein, changes that may affect both protein function and mitochondrial metabolism. Only homozygosity for the c.137A > G variant in DGUOK predicted to yield the substitution p.N46S has before been associated with INCPH. However, homozygosity for the same variant was also associated with cirrhosis and liver failure in an infant aged 10 months (22). In 4 infants who were compound heterozygotes for that variant in DGUOK and another, severe liver dysfunction with variable progression was observed (5, 22–24), with one undergoing liver transplantation at an early age. As the variants in our patient both are novel, conclusions on their behavior in other settings and combinations are premature. It is not known whether the two novel variants might be associate with mild liver involvement or not. However, the link between genotype and phenotype in DGUOK disease appears complex.
We have above described a Han Chinese child whose INCPH/PVSD we attribute to novel variants in DGUOK. His case provides new evidence that DGUOK variants may be associated with INCPH/PVSD in children.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Research ethics of children's hospital of Fudan university. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
Conceived and designed experiments: TL Drafted manuscript: J-QL Critical revision of manuscript: TL Collected data and samples: J-QL, J-YF, YG, W-QL. All authors contributed to the article and approved the submitted version.
Funding
The project was funded by the National Natural Science Foundation of China (82201898 to TL). The work was also supported by the Shanghai Sailing Program (20YF1402900 to TL), Health Industry Clinical Research Project of Shanghai Municipal Health Commission (20224Y0281 to TL), Chan-Xue-Yan Program of Fudan University (FDEKCXY17 to TL).
Acknowledgment
The authors are grateful for the support of the family of the proband and to A S Knisely and Jian-She Wang for manuscript review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1236239/full#supplementary-material
Supplement Figure S1
Results of abdominal contrast-enhanced computed tomography scan and gastroscopy. (A) Abdominal contrast-enhanced computed tomography scan reveals lobular contour of the liver, hepertrophy of the caudate lobe, widened hepatic fissures, splenomegaly (arrow), accompanied by thickening of the splenic meridians (arrow) and varicose gastric veins (arrow). (B) Esophageal and gastric varices were not found from gastroscopy.
Supplement Figure S2
The MASSON staining of the overview liver biopsy.
References
1. Johansson M, Bajalica-Lagercrantz S, Lagercrantz J, Karlsson A. Localization of the human deoxyguanosine kinase gene (DGUOK) to chromosome 2p13. Genomics. (1996) 38:450–1. doi: 10.1006/geno.1996.0654
2. Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet. (2001) 29:337–41. doi: 10.1038/ng746
3. Johansson M, Karlsson A. Cloning and expression of human deoxyguanosine kinase cDNA. Proc Natl Acad Sci U S A. (1996) 93:7258–62. doi: 10.1073/pnas.93.14.7258
4. El-Hattab AW, Craigen WJ, Scaglia F. Mitochondrial DNA maintenance defects. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1539–55. doi: 10.1016/j.bbadis.2017.02.017
5. Dimmock DP, Zhang Q, Dionisi-Vici C, Carrozzo R, Shieh J, Tang LY, et al. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum Mutat. (2008) 29:330–1. doi: 10.1002/humu.9519
6. Vilarinho S, Sari S, Yilmaz G, Stiegler AL, Boggon TJ, Jain D, et al. Recurrent recessive mutation in deoxyguanosine kinase causes idiopathic noncirrhotic portal hypertension. Hepatology. (2016) 63:1977–86. doi: 10.1002/hep.28499
7. Schouten JN, Verheij J, Seijo S. Idiopathic non-cirrhotic portal hypertension: a review. Orphanet J Rare Dis. (2015) 10:67. doi: 10.1186/s13023-015-0288-8
8. Corpechot C, El NA, Poujol-Robert A, Ziol M, Wendum D, Chazouilleres O, et al. Assessment of biliary fibrosis by transient elastography in patients with PBC and PSC. Hepatology. (2006) 43:1118–24. doi: 10.1002/hep.21151
9. Desmet VJ, Gerber M, Hoofnagle JH, Manns M, Scheuer PJ. Classification of chronic hepatitis: diagnosis, grading and staging. Hepatology. (1994) 19:1513–20. doi: 10.1002/hep.1840190629
10. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
11. Salviati L, Sacconi S, Mancuso M, Otaegui D, Camano P, Marina A, et al. Mitochondrial DNA depletion and dGK gene mutations. Ann Neurol. (2002) 52:311–7. doi: 10.1002/ana.10284
12. Freisinger P, Futterer N, Lankes E, Gempel K, Berger TM, Spalinger J, et al. Hepatocerebral mitochondrial DNA depletion syndrome caused by deoxyguanosine kinase (DGUOK) mutations. Arch Neurol. (2006) 63:1129–34. doi: 10.1001/archneur.63.8.1129
13. Sezer T, Ozcay F, Balci O, Alehan F. Novel deoxyguanosine kinase gene mutations in the hepatocerebral form of mitochondrial DNA depletion syndrome. J Child Neurol. (2015) 30:124–8. doi: 10.1177/0883073813517000
14. Seijo S, Reverter E, Miquel R, Berzigotti A, Abraldes JG, Bosch J, et al. Role of hepatic vein catheterisation and transient elastography in the diagnosis of idiopathic portal hypertension. Dig Liver Dis. (2012) 44:855–60. doi: 10.1016/j.dld.2012.05.005
15. Brahimi N, Jambou M, Sarzi E, Serre V, Boddaert N, Romano S, et al. The first founder DGUOK mutation associated with hepatocerebral mitochondrial DNA depletion syndrome. Mol Genet Metab. (2009) 97:221–6. doi: 10.1016/j.ymgme.2009.03.007
16. Waich S, Roscher A, Brunner-Krainz M, Cortina G, Kostl G, Feichtinger RG, et al. Severe deoxyguanosine kinase deficiency in Austria: a 6-patient series. J Pediatr Gastroenterol Nutr. (2019) 68:e1–6. doi: 10.1097/MPG.0000000000002149
17. Pronicki M, Piekutowska-Abramczuk D, Rokicki D, Iwanicka-Pronicka K, Grajkowska W. Histopathological liver findings in patients with hepatocerebral mitochondrial depletion syndrome with defined molecular basis. Pol J Pathol. (2018) 69:292–8. doi: 10.5114/pjp.2018.79549
18. De Franchis R, Baveno VI Faculty. Expanding consensus in portal hypertension: report of the baveno VI consensus workshop: stratifying risk and individualizing care for portal hypertension. J Hepatol. (2015) 63:743–52. doi: 10.1016/j.jhep.2015.05.022
19. De Franchis R, Bosch J, Garcia-Tsao G, Reiberger T, Ripoll C, Baveno VII Faculty. Baveno VII—renewing consensus in portal hypertension. J Hepatol. (2022) 76:959–74. doi: 10.1016/j.jhep.2021.12.022
20. De Gottardi A, Rautou PE, Schouten J, Rubbia-Brandt L, Leebeek F, Trebicka J, et al. Porto-sinusoidal vascular disease: proposal and description of a novel entity. Lancet Gastroenterol Hepatol. (2019) 4:399–411. doi: 10.1016/S2468-1253(19)30047-0
21. Schouten JN, Garcia-Pagan JC, Valla DC, Janssen HL. Idiopathic noncirrhotic portal hypertension. Hepatology. (2011) 54:1071–81. doi: 10.1002/hep.24422
22. Sarzi E, Bourdon A, Chretien D, Zarhrate M, Corcos J, Slama A, et al. Mitochondrial DNA depletion is a prevalent cause of multiple respiratory chain deficiency in childhood. J Pediatr. (2007) 150:531–4. doi: 10.1016/j.jpeds.2007.01.044
23. Mousson DCB, Taanman JW, Padet S, Chassagne M, Mayencon M, Clerc-Renaud P, et al. Kinetic properties of mutant deoxyguanosine kinase in a case of reversible hepatic mtDNA depletion. Biochem J. (2007) 402:377–85. doi: 10.1042/BJ20060705
Keywords: case report, DGUOK, genetic variant, idiopathic non-cirrhotic portal hypertension, mitochondrial depletion syndrome, porto-sinusoidal vascular disorder
Citation: Li J-Q, Feng J-Y, Gong Y, Li W-Q and Liu T (2023) Case report: Novel DGUOK variants associated with idiopathic non-cirrhotic portal hypertension in a Han Chinese child. Front. Pediatr. 11:1236239. doi: 10.3389/fped.2023.1236239
Received: 7 June 2023; Accepted: 11 September 2023;
Published: 27 September 2023.
Edited by:
Pietro Vajro, University of Salerno, ItalyReviewed by:
Andrea Pietrobattista, Bambino Gesù Children’s Hospital (IRCCS), ItalyAleksandra Jezela-Stanek, National Institute of Tuberculosis and Lung Diseases, Poland
© 2023 Li, Feng, Gong, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teng Liu bGl1dGVuZ0BmdWRhbi5lZHUuY24=
Abbreviations INCPH, idiopathic non-cirrhotic portal hypertension; MDS, mitochondrial DNA depletion syndrome; PVSD, porto-sinusoidal vascular disorder.